

Abstract
Importance
Studies in experimental and human heart failure suggest that phosphodiesterase type-5 inhibitors may enhance cardiovascular function, and thus, exercise capacity in heart failure with preserved ejection fraction.
Objective
To determine the effect of the phosphodiesterase type-5 inhibitor, sildenafil, in comparison to placebo on exercise capacity and clinical status in heart failure with preserved ejection fraction.
Design, setting, and patients
Multicenter, double-blind, placebo-controlled, parallel design, randomized clinical trial of 216 stable outpatients with heart failure, ejection fraction ≥ 50%, elevated N-terminal pro-brain natriuretic peptide or elevated invasively-measured filling pressures, and reduced exercise capacity. Participants were randomized from October 2008 through February 2012 at 26 centers in the United States and Canada.
Intervention
Sildenafil (n=113) or placebo (n=103) administered orally at 20 mg three times daily for 12 weeks followed by 60 mg three times daily for 12 weeks.
Main outcome measures
Primary endpoint was change in peak oxygen consumption after 24 weeks of therapy. Secondary endpoints included change in six-minute walk distance and a three tier hierarchical composite clinical status score where patients were ranked (range 1-N) based on time to death, time to cardiovascular or cardiorenal hospitalization and change in quality of life for participants alive without cardiovascular or cardiorenal hospitalization at 24 weeks.
Results
Median age was 69 years and 48% of patients were female. At baseline, median peak oxygen consumption (11.7 ml/kg/min) and six-minute walk distance (308 meters) were reduced and median E/e′ (16), left atrial volume index (44 ml/m2) and pulmonary artery systolic pressure (41 mmHg) were consistent with chronically-elevated left ventricular filling pressures. At 24 weeks, median (interquartile range) changes in peak oxygen consumption (ml/kg/min) in patients who received placebo [−0.20 (−0.70, 1.00)] or sildenafil [−0.20 (−1.20, 1.10); p=0.90] were not significantly different. The mean clinical status rank score (higher value indicates better status; expected value with no treatment effect = 95) was not significantly different (p=0.85) at 24 weeks in patients who received placebo (95.8) or sildenafil (94.2). Changes in six-minute walk distance (meters) at 24 weeks in patients who received placebo [15.0 (−26.0, 45.0)] or sildenafil [5.0 (−37.0, 55.0); p=0.92] were also not significantly different. Adverse events occurred in 78 (76%) of patients who received placebo and 90 (80%) of patients who received sildenafil. Serious adverse events occurred in 16 (16%) of patients who received placebo and 25 (22%) of patients who received sildenafil.
Conclusion
Chronic phosphodiesterase type-5 inhibitor therapy with sildenafil for 24 weeks did not alter exercise capacity or clinical status compared to placebo in patients with heart failure and preserved ejection fraction.
Trial registration
clinicaltrials.gov number, NCT00763867
Heart failure (HF) with preserved ejection fraction (HFpEF) or “diastolic HF” is a common and highly-morbid condition.1 Clinical trials of renin-angiotensin system (RAS) antagonists have not demonstrated improvement in outcomes or clinical status in HFpEF, and effective therapies for HFpEF are needed.2 Phosphodiesterase type-5 (PDE-5) metabolizes the nitric oxide (NO) and natriuretic peptide (NP) systems’ second messenger cyclic guanosine monophosphate (cGMP), and thus may limit beneficial NO and NP actions in the heart, vasculature and kidney. Pre-clinical studies suggest that inhibition of PDE-5 reverses adverse cardiac structural and functional remodeling and enhances vascular, neuroendocrine and renal function.3 In clinical studies, PDE-5 inhibitor therapy improved exercise tolerance and clinical status in patients with idiopathic pulmonary arterial hypertension and in patients with HF and reduced EF (HFrEF).4–7 A small, single-center study in HFpEF observed improved hemodynamics, left ventricular (LV) diastolic function, right ventricular (RV) systolic function, LV hypertrophy (LVH) and lung function with chronic PDE-5 inhibition as compared to placebo.8 In aggregate, these studies suggest the potential for PDE-5 inhibition to ameliorate several key pathophysiological perturbations in HFpEF, and thus improve exercise capacity and clinical status. Accordingly, the Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure with Preserved Ejection Fraction (RELAX) trial was designed to test the hypothesis that as compared to placebo, chronic therapy with the PDE-5 inhibitor sildenafil would improve exercise capacity in HFpEF after 24 weeks of therapy as assessed by the change in peak oxygen consumption (VO2).
METHODS
Study oversight
The National Heart, Lung, and Blood Institute (NHLBI)–sponsored Heart Failure Clinical Research Network (HFN) conceived, designed, and conducted the RELAX trial. The trial protocol was approved by an NHLBI-appointed protocol review committee and a data and safety monitoring board and by the institutional review board at each participating site. The Duke Clinical Research Institute served as the data coordinating center.
Study design
The rationale for and design of the RELAX trial have been previously described.3 Patients with normal (≥50%) EF and HF with New York Heart Association (NYHA) functional class II–IV symptoms on stable medical therapy were eligible to participate if they had objective evidence of HF (previous HF hospitalization, or acute HF therapy with intravenous diuretic, or chronic loop diuretic therapy for HF with left atrial enlargement, or invasively documented elevation in LV filling pressures). All study participants provided written informed consent prior to screening. A peak VO2 ≤ 60% of the age and sex specific normal value9 with a respiratory exchange ratio (RER) ≥ 1.0 at screening cardiopulmonary exercise test (CPXT), and either elevated (≥400 pg/ml) N-terminal pro-brain natriuretic peptide (NT-proBNP) level or elevation in LV filling pressures at the time of an NT-proBNP level < 400 pg/ml were required for study entry. A complete list of the trial inclusion and exclusion criteria is provided in the online supplement (e-Table 1). As required in federally-funded trials, a self-identification of investigator-defined race/ethnicity option was recorded. Participants who met screening criteria underwent baseline studies [history and physical examination, CPXT, six-minute walk distance (6MWD), Minnesota Living with Heart Failure Questionnaire (MLHFQ), echocardiography, cardiac magnetic resonance imaging (CMR, if sinus rhythm) and phlebotomy for biomarkers], and were then randomly assigned, in a 1:1 ratio, to either sildenafil or placebo with the use of an automated Web-based system. A permuted-block randomization scheme was used with stratification according to clinical site and the presence of atrial fibrillation.
Study drug was administered orally at 20 mg three times daily (TID) for 12 weeks after which history and physical examination, CPXT, 6MWD, MLHFQ, and phlebotomy for biomarkers and sildenafil levels 2 hours after a scheduled dose of study drug were obtained. The dose was then increased to 60 mg TID for 12 weeks after which baseline studies were repeated including phlebotomy for biomarkers and sildenafil levels 2 hours after study drug. If side effects developed, study staff could recommend discontinuation or return to a lower or previously-tolerated dose of study drug.
Blinded core laboratories assessed biomarkers (University of Vermont), CPXT (Massachusetts General Hospital, Harvard University), CMR (Duke University) and echocardiograms (Mayo Clinic).
Study endpoints
The primary endpoint was the change in peak VO2 after 24 weeks of therapy. A number of subgroup analyses were pre-specified. Secondary endpoints included a composite hierarchical-rank clinical score where patients were ranked (range 1-N with data) based on time-to-death (tier 1), time-to-hospitalization for cardiovascular or cardiorenal causes (tier 2), and change in MLHFQ from baseline (tier 3) for patients alive without cardiovascular or cardiorenal hospitalization after 24 weeks of therapy.10 As 189 patients had data for this endpoint, the anchor value (mean value in each group indicating no treatment effect) was 95. Other secondary endpoints included change in 6MWD at 24 weeks and change in peak VO2 and 6MWD after 12 weeks of therapy. Peak sildenafil levels at 12 and 24 weeks and coinciding plasma cGMP levels at 24 weeks were assessed. Using other pre-specified endpoints, we assessed the effect of PDE-5 inhibition on LV structure and vascular function by CMR, Doppler-estimated diastolic function parameters and pulmonary artery systolic pressure (PASP), and biomarkers that reflect renal and neuroendocrine function, oxidative stress and collagen metabolism.
The percent-predicted peak VO2, 6MWD, and the presence of chronotropic incompetence and LVH were calculated using published criteria (e-Methods).11–14
Statistical analysis
Power calculations were based on the standard deviation (SD) for change in peak VO2 and the magnitude of the change in peak VO2 associated with improvements in other markers of clinical status (NYHA class, 6MWD and quality of life scores) in HF trials.5,14,15 Based on these studies, a difference between treatment groups of 1.2 ml/kg/min in the change in peak VO2 was considered clinically significant. We estimated a 20% rate of incomplete primary endpoint data due to death, withdrawal or incidence of new factors limiting ability to exercise. Using a two-sample t-test and a two-sided alpha of 0.05, a sample size of 190 patients had 85% power to detect a difference of 1.2 ml/kg/min in change in peak VO2 assuming 20% missing data and a SD of change in peak VO2 of 2.5 ml/kg/min. As an early-blinded interim analysis of aggregated primary endpoint completeness indicated that the missingness rate approached 20%, the blinded investigators recommended increasing the sample size to 215 patients. The primary analyses were two-sided, and patients’ without-week data were excluded. Sensitivity analysis for the primary end-point was based on the intention-to-treat principle and utilized multiple imputations with 100 imputed datasets to account for missing 24-week data. Finally, a pre-specified “last observation carried forward” sensitivity analysis utilized “carry-forward” of 12-week data if 24-week data were missing.
Data are presented as median (interquartile range, IQR). For the comparison of treatment groups in the primary analysis, a multivariable linear-regression model was used adjusting for baseline peak VO2. A similar approach was used for the secondary endpoint of change in 6MWD, adjusting for baseline 6MWD. For the composite hierarchal-rank clinical score, patients were ranked independent of treatment assignment from 1 (worst outcome—the earliest death) to N (best outcome—survival with no cardiovascular or cardiorenal hospitalization, and the most favorable improvement in MLHFQ), and treatments compared using the Wilcoxon rank-sum test. For primary and secondary endpoints, a p-value less than 0.05 was considered significant. For subgroup analyses, a treatment by subgroup interaction p-value less than 0.001 was considered significant.
All analyses were conducted with the use of SAS software (version 9.2).
RESULTS
Patient Population
Patients (n=216) were enrolled in the trial between October 13, 2008, and February 21, 2012 at 26 sites in the United States and Canada (Figure 1). The baseline characteristics are shown in Table 1 and were not significantly different between treatment groups except for a lower prevalence of hypertension in the sildenafil group. On average, patients in this study were elderly, 48% female and obese with controlled blood pressure and multiple comorbidities including hypertension, ischemic heart disease, atrial fibrillation, diabetes, anemia, and chronic kidney disease. The MLHFQ score was consistent with their NYHA class II/III status. Evidence of volume overload (elevated jugular venous pressure or edema) and hospitalization for HF in the previous year were common. The majority of patients were on diuretics, RAS antagonists, beta blockers and statins. There was evidence of neuroendocrine activation and altered collagen metabolism as levels of NT-proBNP, aldosterone, endothelin-1 and NT-procollagen III were above reference ranges (e-Table 2). Both peak VO2 and 6MWD were reduced and chronotropic incompetence was common.
Figure 1. CONSORT Diagram.

Reasons for study drug discontinuation included withdrawal of consent (placebo n=4, sildenafil n=5), adverse events (placebo n=1, sildenafil n=9), physician decision (placebo n=1, sildenafil n=1) and other (placebo n=2, sildenafil n=5). Note that some patients discontinued study drug for a specific reason but then subsequently withdrew from the study.
Table 1.
Baseline Characteristics of the Patients
| No. (%) of Patientsa | |||
|---|---|---|---|
|
| |||
| All (n=216) | Placebo (n=103) | Sildenafil (n=113) | |
| Age, median (IQR), years | 69 (62,77) | 69 (62, 77) | 68 (62,77) |
| Female sex | 104 (48) | 55 (53) | 49 (43) |
| Self-reported white race | 197 (91) | 95 (92) | 102 (90) |
| Body mass index (kg/m2) - median (IQR) | 32.9 (28.3,39.1) | 32.8 (28.6,38.6) | 33.3 (28.2, 40.0) |
| NYHA Functional Classification | |||
| Class II | 101(47) | 46 (45) | 55 (49) |
| Class III | 115 (53) | 57 (55) | 58 (51) |
| MLHFQ total score, median (IQR) | 43 (30, 62) | 43 (29, 58) | 44 (30, 63) |
| Systolic BP, median (IQR), mmHg | 126 (113, 138) | 127 (113, 138) | 124 (113, 138) |
| Heart rate, median (IQR), bpm | 69 (61, 78) | 68 (62, 77) | 70 (61, 80) |
| Jugular venous pressure ≥ 8 cmb | 95/209 (45) | 48/99 (48) | 47/110 (43) |
| Edema | 125 (58) | 61 (59) | 64 (57) |
| ≥ 1 HF hospitalizations in previous year | 79 (37) | 40 (39) | 39 (35) |
| Qualifying ejection fraction, median (IQR),% | 60 (55, 66) | 60 (55, 65) | 60 (55, 66) |
| History of hypertension | 183 (85) | 93 (90) | 90 (80) |
| History of ischemic heart disease | 84 (39) | 37 (36) | 47 (42) |
| History of atrial fibrillation or flutter | 111 (51) | 52 (50) | 59 (52) |
| Diabetes mellitus | 93 (43) | 45 (44) | 48 (42) |
| Chronic obstructive pulmonary disease | 42 (19) | 20 (19) | 22 (19) |
| Anemia (Hgb <13 (male),< 12 (female) g/dl) | 76 (35) | 34 (33) | 42 (38) |
| Local Lab Creatinine, median (IQR), mg/dl | 1.2 (0.9, 1.5) | 1.1 (0.9, 1.5) | 1.3 (1.0, 1.5) |
| Local Lab GFR, median (IQR), ml/min/1.73m2 | 57 (43, 75) | 59 (43, 76) | 57 (43, 70) |
| Stage 3 or 4 chronic kidney disease | 119 (55) | 56 (54) | 63 (56) |
| Medications at enrollment | |||
| Loop diuretic | 166 (77) | 79 (77) | 87 (77) |
| Any diuretic | 186 (86) | 86 (83) | 100 (88) |
| ACE inhibitor or Angiotensin receptor blocker | 152 (70) | 78 (76) | 74 (65) |
| Beta-blocker | 164 (76) | 77 (75) | 87 (77) |
| Aldosterone antagonist | 23 (11) | 9 (9) | 14 (12) |
| Calcium channel blocker | 66 (31) | 35 (34) | 31 (27) |
| Statin | 138 (64) | 67 (65) | 71 (63) |
| Core Laboratory Biomarker data | |||
| Cystatin Cc, median (IQR), mg/L | 1.31 (1.07, 1.74) | 1.34 (1.08, 1.76) | 1.29 (1.04,1.70) |
| NT-proBNPd, median (IQR), pg/ml | 700 (283,1553) | 648 (247,1679) | 757 (318,1465) |
| Aldosteronec, median (IQR), pg/ml | 189 (118, 283) | 180 (114, 294) | 207 (130, 275) |
| Endothelin-1c, median (IQR), pg/ml | 2.36 (1.95, 3.20) | 2.37 (2.03, 3.14) | 2.35 (1.85, 3.22) |
| NT-procollagen IIIc, median (IQR), ug/L | 7.7 (6.1, 10.0) | 7.9 (6.4, 11.2) | 7.5 (5.5, 9.3) |
| Uric acide, median (IQR), mg/dL | 7.4 (5.8, 8.5) | 7.1 (5.8, 8.5) | 7.5 (5.8, 8.5) |
| Functional status | |||
| Peak VO2f, median (IQR), ml/min/kg | 11.7 (10.2, 14.4) | 11.9 (10.1, 14.4) | 11.7 (10.4, 14.5) |
| % predicted peak VO2, median (IQR), | 41 (35, 49) | 41 (36, 49) | 41 (34, 48) |
| Peak Respiratory exchange ratiof, median | 1.09 (1.02, 1.15) | 1.10 (1.03, 1.15) | 1.09 (1.02, 1.16) |
| Peak systolic BPg, median(IQR), mmHg | 156 (132, 170) | 154 (134, 168) | 156 (130, 170) |
| Chronotropic incompetencef | 164 (77) | 79 (78) | 85 (76) |
| 6MWD, median (IQR), m | 308 (229, 383) | 305 (213, 372) | 308 (241, 392) |
| % predicted 6MWD, median (IQR), % | 69 (51, 83) | 68 (48, 83) | 70 (54, 83) |
ACE, angiotensin converting enzyme; bpm, beats per minute; BP, blood pressure; GFR, glomerular filtration rate; Hgb, hemoglobin; IQR, interquartile range; MLHFQ, Minnesota Living with Heart Failure Questionnaire; 6MWD, six-minute walk distance
unless otherwise indicated
indicates the number of patients/number of patients with non-missing data for the variable (percentage)
available in 214 (102 placebo and 112 sildenafil treated) patients
available in 213 (101 placebo and 112 sildenafil treated) patients
available in 212 (101 placebo and 111 sildenafil treated) patients
available in 215 (103 placebo and 112 sildenafil treated) patients due to technical limitation precluding core laboratory measurement of peak VO2.
available in 209 (99 placebo and 110 sildenafil treated) patients
Table 2.
Baseline core laboratory echocardiographic and CMR characteristics of the patients
| All (n=216) | Placebo (n=103) | n† | Sildenafil (n=113) | n† | |
|---|---|---|---|---|---|
| Doppler echocardiographic data | |||||
| Ejection fraction, median (IQR),% | 60 (56, 65) | 60 (56, 65) | 103 | 60 (55, 65) | 110 |
| Cardiac index, median (IQR), L/min/m2 | 2.47 (2.07, 2.92) | 2.48 (2.06, 2.86) | 89 | 2.47 (2.09, 2.92) | 86 |
| LV end diastolic dimension, median (IQR), cm | 4.6 (4.2, 5.2) | 4.6 (4.3, 5.1) | 76 | 4.6 (4.2, 5.2) | 88 |
| LV mass/BSA, median (IQR), g/m2 | 77 (62, 96) | 77 (63, 90) | 73 | 78 (61, 97) | 85 |
| LV hypertrophy, n (%) | 76 (48) | 36 (49) | 73 | 40 (47) | 85 |
| Relative wall thickness ≥ 0.42, n (%) | 75 (48) | 32 (44) | 73 | 43 (51) | 85 |
| E/A ratio, median (IQR) | 1.5 (1.0, 2.0) | 1.3 (1.0, 2.0) | 70 | 1.6 (1.0, 2.1) | 72 |
| Deceleration time, median (IQR), ms | 185 (155, 218) | 190 (156, 228) | 95 | 182 (153, 212) | 98 |
| Medial e′, median (IQR), m/sec | 0.06 (0.05, 0.08) | 0.06 (0.05, 0.07) | 92 | 0.06 (0.05, 0.08) | 105 |
| Medial E/e′, median (IQR) | 16 (11, 23) | 17 (12, 24) | 90 | 15 (10, 23) | 98 |
| Left atrial volume index, median (IQR), ml/m2 | 44 (35, 59) | 43 (38, 59) | 74 | 44 (33, 59) | 75 |
| PA systolic pressure, median (IQR), mmHg | 41 (33, 51) | 41 (34, 54) | 75 | 41 (32, 51) | 63 |
| Cardiac magnetic resonance imaging data | |||||
| Ejection fraction, median (IQR), % | 66 (58, 70) | 66 (59, 71) | 56 | 66 (55, 70) | 61 |
| Cardiac index, median (IQR), L/min/m2 | 2.35 (1.98, 2.76) | 2.38 (1.94, 2.80) | 56 | 2.32 (2.11, 2.74) | 59 |
| LV end-diastolic volume/BSA, median (IQR), ml/m2 | 56 (47, 67) | 58 (49, 68) | 56 | 53 (46, 66) | 61 |
| LV mass/BSA, median (IQR), g/m2 | 63 (54, 77) | 61 (53, 73) | 56 | 65 (54, 78) | 61 |
| LV hypertrophy, n (%) | 30 (26) | 14 (25%) | 56 | 16 (26%) | 61 |
| Aortic distensibility, median (IQR),10−3mmHg−1 | 1.17 (0.67, 1.76) | 1.08 (0.63,1.64) | 41 | 1.18 (0.74, 2.07) | 45 |
BSA, body surface area; IQR, interquartile range; LV, left ventricular; PA, pulmonary artery
number of patients in placebo or sildenafil treatment groups with data for the variable
Baseline echocardiographic and CMR characteristics of the patients were not significantly different between treatment groups (Table 2). At echocardiography, ejection fraction (EF) and LV diastolic dimension were within normal limits, while cardiac index was reduced. Nearly 50% of patients had LVH or evidence of concentric remodeling/hypertrophy (relative wall thickness ≥ 0.4216). There was Doppler evidence of diastolic dysfunction and elevated LV filling pressures with reduced e′ and elevated E/A ratio, E/e′ ratio, left atrial volume and PASP.
Overall, 132 (61%) patients were eligible for CMR and 117 (89%) of these underwent CMR. EF and LV end-diastolic volume index were within normal limits while cardiac index was reduced. In the CMR cohort, 25% of patients had LVH. Aortic distensibility was reduced as compared to published values in elderly normal subjects.17
Primary endpoint
At 24 weeks, the change in peak VO2 from baseline was not significantly different in placebo and sildenafil-treated patients (Figure 2). Using multiple imputation to account for missing 24-week data, the mean difference between sildenafil and placebo is 0.01 ml/min/kg (favoring sildenafil) with a 95% confidence interval of (−0.60, 0.61) and a p-value of 0.98. Carrying forward 12-week peak VO2 when 24-week data were missing (n=5 placebo and n=9 sildenafil), median (IQR) change in peak VO2 (ml/kg/min) from baseline was −0.20 (−0.83, 1.10) in placebo and −0.13 (−1.50, 1.16; p=0.98) in sildenafil-treated patients. In subgroup analyses (e-Table 3), the change in peak VO2 was not significantly different between treatment groups when analysis was restricted to those patients still taking study drug at week 24, in patients with or without LVH by CMR, PASP < or ≥ 40 mmHg, NT-proBNP < or ≥ 400 pg/ml, with or without atrial fibrillation or treated or not with RAS antagonists, beta blockers or statins.
Figure 2. Change in peak VO2 from baseline to 24 weeks in placebo and sildenafil-treated patients.
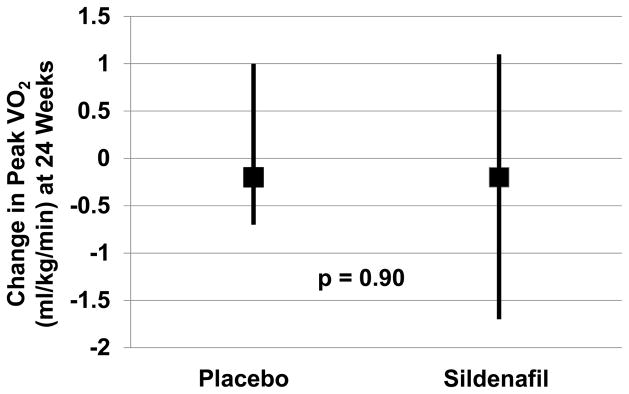
Median and interquartile range for the change in peak VO2 from baseline to 24 weeks are shown.
Table 3.
Secondary and safety endpoints
| Placebo (n=103) | n† | Sildenafil (n=113) | n† | p value | |
|---|---|---|---|---|---|
| Clinical rank score, mean* | 95.8 | 94 | 94.2 | 95 | 0.85 |
| Change in 6MWD at 24 weeks, median (IQR), m | 15.0 (−26.0, 45.0) | 95 | 5.0 (−37.0, 55.0) | 90 | 0.92 |
| Change in Peak VO2 at 12 weeks, median (IQR), ml/kg/min | 0.0 (−1.1, 0.7) | 96 | 0.1 (−1.4, 1.3) | 97 | 0.98 |
| Change in 6MWD at 12 weeks, median (IQR), m | 18.0 (−14.5, 48.0) | 96 | 10.0 (−25.0, 36.0) | 99 | 0.13 |
| Components of clinical rank score at 24 weeks | |||||
| Death‡, no. (%) | 0 (0) | 103 | 3 (3) | 113 | 0.25 |
| Hospitalization for CV or renal cause, no. (%) | 13 (13) | 103 | 15 (13) | 113 | 0.89 |
| Change in MLHFQ, median (IQR) | −8 (−21, 5) | 91 | −8 (−19, 0) | 91 | 0.44 |
| Safety endpoints | |||||
| Adverse events, no. (%) | 78 (76) | 103 | 90 (80) | 113 | 0.49 |
| Serious adverse events, no. (%) | 16 (16) | 103 | 25 (22) | 113 | 0.22 |
6MWD, six-minute walk distance; CV, cardiovascular; IQR, interquartile range; MLHFQ, Minnesota Living with Heart Failure Questionnaire; VO2, oxygen consumption
A mean value of 95 in each group is expected under the null hypothesis of no treatment effect
number of patients in placebo or sildenafil treatment groups with data for the variable
Site investigator identified causes of death were sudden death (n=1), progressive cardiorenal failure (n=1) and non-cardiovascular (n=1)
Secondary endpoints and safety data
There were no significant differences in the clinical rank score, change in 6MWD at 24 weeks or change in peak VO2 or 6MWD at 12 weeks between treatment groups (Table 3). There were no significant differences in the components of the clinical rank score at 24 weeks or in the overall incidence of adverse or serious adverse events in the treatment groups. Adverse events occurring in ≥ 5% of either study group are listed in e-Table 4. Sildenafil-treated patients had a higher incidence of “vascular” adverse events, which included (but were not limited to) headache, flushing and hypotension, although the change in mean arterial pressure from baseline to 24 weeks was not significantly different in sildenafil-treated [−1(−8, 6)] and placebo [−2(−10, 7)], p = 0.45) patients. All serious adverse events exclusive of death or cardiovascular or cardiorenal hospitalization (shown in table 3) are listed in e-Table 5. There were no other notable differences in the incidence of specific serious adverse events between study groups.
Table 4.
Additional pre-specified endpoints
| Placebo (n=103) | n† | Sildenafil (n=113) | n† | ||
|---|---|---|---|---|---|
| Change in LV structure by CMR at 24 weeks | |||||
| LV mass by CMR, g | 0.6 (−5.7, 7.9)* | 47 | −1.5 (−5.9, 7.1) | 49 | 0.93 |
| LV end-diastolic volume by CMR, ml | −4.3 (−15.5, 8.1) | 47 | 3.7 (−4.9, 14.5) | 49 | 0.13 |
| Change in diastolic function parameters at 24 weeks | |||||
| Medial e′, m/sec | 0.00 (−0.01, 0.01) | 83 | 0.00 (−0.01, 0.01) | 77 | 0.88 |
| E/e′ | −1.6 (−4.7, 2.2) | 80 | 0.2 (−2.4, 3.1) | 75 | 0.16 |
| PA systolic pressure, mmHg | −2 (−8 − 8) | 58 | 2 (−5, 7) | 45 | 0.94 |
| Change in vascular function by CMR at 24 weeks | |||||
| Arterial elastance, mmHg/ml | 0.03 (−0.14, 0.23) | 45 | −0.08 (−0.41, 0.12) | 47 | 0.02 |
| SVR, wood units | 0.13 (−0.15, 0.43) | 45 | −0.04 (−0.38, 0.21) | 47 | 0.09 |
| Aortic distensibility, 10−3mmHg−1 | 0.13 (−0.17, 0.42) | 31 | 0.18 (−0.12, 0.67) | 29 | 0.38 |
| Change in core laboratory biomarkers at 24 weeks | |||||
| Creatinine, mg/dl | 0.01 (−0.10, 0.09) | 94 | 0.05 (−0.04, 0.15) | 94 | 0.047 |
| Cystatin C, mg/L | 0.01 (−0.08, 0.11) | 95 | 0.05 (−0.04, 0.16) | 95 | 0.01 |
| NT-proBNP, pg/ml | −23 (−198, 139) | 94 | 15 (−90, 372) | 95 | 0.03 |
| Endothelin-1, pg/ml | −0.01 (−0.48, 0.47) | 95 | 0.38 (−0.10, 0.97) | 95 | 0.046 |
| Aldosterone, pg/ml | 0 (−70, 48) | 95 | −11 (−77, 30) | 95 | 0.85 |
| NT-procollagen III, ug/L | −0.03 (−1.49, 1.54) | 93 | 0.07 (−1.17, 1.42) | 95 | 0.77 |
| Uric acid, mg/dl | −0.1 (−0.7, 0.7) | 94 | 0.3 (−0.4, 1.4) | 94 | 0.02 |
number of patients in placebo or sildenafil groups with data for the variable.
results shown as median (IQR)
Study drug and cGMP levels
Median (IQR) sildenafil concentrations measured approximately 2 hours after the last dose at 12 and 24 weeks were 78 (35, 130) and 200 (92, 330) ng/ml, respectively. At week 24, there was a weak correlation between sildenafil dose and sildenafil level (r=0.29, p=0.008). In paired analysis, plasma cGMP levels increased significantly from baseline to 24 weeks in patients randomized to sildenafil [mean increase 8.72 pmol/ml, 95% confidence interval (CI) (2.56, 14.87; p=0.006)], but not in patients randomized to placebo [mean increase 1.28 pmol/ml (95% CI; −6.27, 8.83) p=0.74]; although, the change in cGMP was not significantly different between groups (p=0.11).
Additional endpoints
At CMR, there was no difference in change in LV mass or LV end-diastolic volume between treatment groups (Table 4). There was also no difference in change in Doppler-assessed LV diastolic function parameters or PASP between treatment groups. By CMR, arterial elastance decreased more and systemic vascular resistance tended (p=0.09) to decrease more in sildenafil-treated patients. However, the change in mean arterial pressure in the entire study population was not significantly different between groups as noted above. More patients had missing data for aortic distensibility at 24 weeks than baseline, but there was no difference in change in distensibility between groups. Sildenafil-treated patients had a greater increase in creatinine, cystatin C, NT-proBNP, uric acid and endothelin-1 than placebo-treated patients, while changes in aldosterone and NT-procollagen III were not significantly different between groups.
DISCUSSION
To our knowledge, RELAX is the first multicenter study to investigate the effect of PDE-5 inhibition in HFpEF. Contrary to our hypothesis, chronic PDE-5 inhibition in HFpEF had no effect on maximal or submaximal exercise capacity, clinical status, quality of life, LV remodeling, diastolic function parameters or PASP. Indeed, renal function worsened more and NT-proBNP, endothelin-1 and uric acid levels increased more in sildenafil-treated patients. Further, there were numerically more patients in the sildenafil arm who withdrew consent, died, or were too sick to perform CPXT, and sildenafil-treated patients had a higher incidence of vascular adverse events. The findings of RELAX do not suggest that chronic therapy with the PDE-5 inhibitor sildenafil provides clinical benefit in the general HFpEF population.
Given the strong rationale for testing PDE-5 inhibition in HFpEF,3 and the lack of benefit observed in RELAX, it is important to consider whether the study population, specifics of the therapeutic intervention and endpoints were appropriate.
The clinical characteristics of patients enrolled in major ongoing or completed clinical trials in HFpEF patients have been summarized.18 The RELAX study population was similar to others in terms of age, sex distribution and body size. Severity of HF (NYHA functional class, quality of life score, physical findings of volume overload and NT-proBNP levels) in RELAX was similar to or greater than other HFpEF trials; although, comorbidity burden (diabetes, atrial fibrillation, kidney disease) may have been greater in the RELAX study population. Concentric remodeling and hypertrophy were common but not severe, and Doppler evidence of elevated filling pressures and pulmonary hypertension were present as was neuroendocrine activation consistent with the HF state. However, the characteristics of the study population were notably different from the only other study that evaluated the effect of PDE-5 inhibition in HFpEF. In the study of Guazzi et al., sildenafil had a number of beneficial effects as outlined above although effect on exercise capacity was not tested.8 Importantly, in the Guazzi study, HFpEF patients had fewer comorbidities and much higher blood pressure, LV mass, and PASP than in RELAX and catheterization documented pulmonary arterial hypertension, profound RV systolic dysfunction and RV failure were present. This profile is somewhat atypical for HFpEF cohorts.19 It may be that the primary therapeutic effects of PDE-5 inhibitors in HF reside in their ability to dilate the pulmonary vascular bed, enhance RV contractility, and reduce ventricular interdependence,4,19–22 and that pulmonary arterial hypertension and RV failure must be significant in order to observe clinical benefit in HFpEF. The subgroup analysis in RELAX did not show any trends towards improvement in peak VO2 in patients with higher PASP, but the presence of pulmonary arterial hypertension or RV dysfunction was not assessed in RELAX.
While LVH was common in RELAX, it was far less severe than noted in the study of Guazzi et al.8 In murine-pressure overload studies, PDE-5 inhibition did not have anti-hypertrophic effects in mice with less severe pressure overload and compensated LVH with relatively preserved EF, whereas dramatic anti-remodeling benefits were observed in mice with severe pressure overload, eccentric LVH, reduced EF and pulmonary congestion.23,24 Conceivably, activation of PDE-5 or of cGMP-sensitive downstream pathways in the LV or other organs may occur only in HF associated with advanced LV remodeling.
In a randomized clinical trial of PDE-5 inhibition in pulmonary arterial hypertension, the effect of sildenafil on exercise capacity was not dose related as improvement in 6MWD was seen with 20 mg TID after just four weeks of therapy with no further improvement with higher doses or longer duration of therapy.4 Sildenafil levels at 12 and 24 weeks were variable, but on average, similar to random levels observed with similar doses in HFrEF where improvement in exercise capacity with chronic sildenafil was observed (G. Lewis, personal communication).5 In HFrEF, the benefit of PDE-5 inhibition on exercise capacity has been demonstrated acutely after a single 50 mg dose,25,26 and with 75 mg TID (dose up-titrated over 6 weeks) for 12 weeks.5 While only 73% of patients in the sildenafil group attending the 24-week visit were taking the per-protocol dose, 92% of patients were on at least 20 mg TID. While studies in pulmonary arterial hypertension and HFrEF have observed effects on exercise capacity with similar doses and duration of therapy, we cannot exclude the possibility that inadequate dose or duration of PDE-5 inhibition contributed to our findings.
Therapeutic sildenafil levels were associated with minimal increases in plasma cGMP. HFpEF is characterized by endothelial dysfunction27 and by lower NP levels than observed in HFrEF,28 which may suggest limited NO and NP activity in HFpEF. Inability to enhance cGMP with PDE-5 inhibition in HFpEF may have contributed to our findings.
As previously described,3 change in peak VO2 was chosen as the primary endpoint in RELAX based on previous preclinical and clinical studies, and because non-cardiovascular comorbidities and motivational factors can influence measures of submaximal exercise performance in HFpEF. The trial was powered to detect a clinically significant difference in the change in peak VO2 between groups, and the estimate of variability (standard deviation of 2.5 ml/kg/min) in change in peak VO2 used in the power calculations was consistent with the standard deviation of change in peak VO2 observed in the placebo-treated patients in RELAX (2.0 ml/kg/min). The lack of treatment effect on submaximal exercise, clinical status and physiologic endpoints supports the validity of the observed lack of treatment effect on maximal exercise capacity.
The high prevalence of chronotropic incompetence in the study population is noteworthy. Chronotropic incompetence may contribute to exercise limitation in HFpEF, and may not be improved by PDE-5 inhibition.
While numerous studies in animal models of renal dysfunction suggest that PDE-5 inhibition ameliorates progression of renal dysfunction of various etiologies,3,29–31 in this trial, modest but statistically significant worsening of renal function was observed in sildenafil-treated patients, and was associated with concordant increases in NT-proBNP, uric acid and endothelin-1 suggesting that the decline in renal function was physiologically significant. Studies in pulmonary arterial hypertension and erectile dysfunction have not reported worsening of renal function with PDE-5 inhibitor therapy, but little is known of the effect of PDE-5 inhibition on renal function in HFpEF.
There were numerically more patients who withdrew consent, died or were too sick to perform CPXT in the sildenafil treatment group, potentially accentuating the lack of benefit observed, particularly if those who withdrew did so due to side effects or poor clinical status.
A modest decrease in arterial elastance was noted in sildenafil-treated patients in the CMR cohort. This may have been related to an effect of sildenafil on resistance that tended to decrease more in sildenafil-treated patients, but in the entire cohort, there were no differences in change in mean arterial pressure between treatment groups.
The RELAX findings must be interpreted in the context of other potential limitations. Multicenter trials using peak VO2 as a primary endpoint are challenging, but rigorous methodologies were used in the design and execution of the CPXT study protocol.3 Patients were selected who could perform CPXT, and who had significant reduction in peak VO2—these entry criteria may have selected for a unique HFpEF phenotype. The trial was not powered to address differences in clinical outcomes.
Conclusion
Chronic therapy with the PDE-5 inhibitor sildenafil was not associated with clinical benefit in HFpEF. Continued efforts to identify key pathophysiologic perturbations and novel therapeutic targets in HFpEF are needed.
Supplementary Material
Supplementary material includes the following appendices and online-only material:
- Appendix A: RELAX Protocol
- Appendix B: RELAX Statistical Analysis Plan
- Appendix C: RELAX Trial Members, Investigators and Committees
- Appendix D: CONSORT trial checklist
- e-Table 1: RELAX trial entry criteria
- e-Methods and references
- e-Table 2: Biomarker reference ranges
- e-Table 3. Pre-specified subgroup analysis of change in peak VO2 at 24 weeks according to baseline characteristics or study drug use at 24 weeks
- e-Table 4. Adverse events occurring in at least 5% of patients in either treatment group
- e-Table 5. Serious adverse events exclusive of death and cardiovascular or cardiorenal hospitalization.
Acknowledgments
Pfizer provided study drug (sildenafil and matched placebo), but had no role in the design and conduct of the study, collection, management, analysis or interpretation of the data, or preparation, review or approval of the manuscript.
Drs. Lee and Anstrom had full access to all the data in the study, and take responsibility for the integrity of the data and the accuracy of the data analysis.
Supported by grants from the NHLBI: U10HL084904 (for the data coordinating center), and U10HL084861, U10HL084875, U10HL084877, U10HL084889, U10HL084890, U10HL084891, U10HL084899, U10HL084907, and U10HL084931 (for the clinical centers). This work is also supported by the National Center for Advancing Translational Sciences (NCATS): UL1TR000454; and the National Institute on Minority Health and Health Disparities (NIMHD): 8 U54 MD007588.
We thank the patients who participated in this study, the HFN site investigators and coordinators, the members of the HFN data and safety monitoring board and protocol review committee, and the NHLBI representatives. For a complete listing of the HFN members, see Appendix C.
The authors are solely responsible for the content of this article, which does not necessarily represent the official views of the NHLBI or the National Institutes of Health.
Footnotes
Conflicts of Interest
Potential conflicts of interest including financial disclosures are listed below:
MMR: Receives financial support from the NIH and royalties from Annexon.
HHC: Has received funding for patents that his institution has licensed to Nile Therapeutics and Annexon with other patents pending at the U.S. patent office; has royalties with Nile Therapeutics, Annexon and Up To Date.
BAB: Consults for Medscape, Amgen and GlaxoSmithKline; and receives financial support from Atcor Medical and Gilead.
MJS: Receives financial support from the NIH for participation in the HFN, and from Bayer, Inc.
KLL: Receives financial support from the NIH for participation in the HFN.
GDL: None reported.
MML: Receives financial support from the NIH for participation in the HFN.
JLR: None reported.
DAB: Receives financial support from the NIH for participation in the HFN.
DLM: None reported.
AD: Receives financial support from the NIH for participation in the HFN.
LWS: Receives financial support from the NIH for participation in the HFN.
MMG: Receives financial support from the NIH for participation in the HFN.
EOO: Receives financial support from the NIH; consults for Merck, Novartis and Arbor Pharmaceuticals; currently employed by the Morehouse School of Medicine; has grants pending from the NHLBI and NIMHD; and received lecture fees from Arbor Pharmaceuticals.
CMO: Consults for Roche Diagnostics, Amgen, Novartis, Ikaria, Acetlion Pharma, Heartware, ResMed, Pozen, GE Healthcare, Johnson & Johnson, Gilead, Critical Diagnostics, BG Medicine, Otsuka, Astellas, Novella, Cytokinetics, Capricor; holds stock options with Neurotronik/Intervential Automatics Corporation; edits the journal, American College of Cardiology, and co-owns Cardiology Consulting Associates.
GMF: Receives financial support from the NIH; consults for Novartis, Amgen, Trevena, Roche Diagnostics, Merck, Singulex and BG Medicine; and receives funding from Amgen, Otsuka and Roche Diagnostics.
SRG: None reported.
BAB: Receives financial support from the NIH for participation in the HFN.
SEM: None reported.
JCI: Receives financial support from the NIH for participation in the HFN.
GL: None reported.
JKO: Receives financial support as the Echo Core Lab, royalties for an Echo Lab Manual, and payment for ECHO tutorial DVDs; has been invited as a visiting professor for the Korean Society of Echo, Japanese Society of Echo, Sheba Medical Center in Israel, and Columbia Medical Center in NY; received Echo Core Lab projects from Medtronic for Core Valve, Neochord for Mitral valve, NIH for the Heart Failure Network; and collaborates with Samsung Medical Center, Korea.
MP: Consults for Genzyme, Baxter, Jensen and Bayer; and his financial institution receives financial support from Astra Zeneca, Johnson & Johnson, and Maquet.
RJK: Receives financial support from the NIH as the HFN DCC and MRI Core Lab, and funding for a patent with Northwestern University; and his institution has received a grant from Siemens.
RT: Receives financial support from the NIH for participation in the HFN; consults for Merck, Tibotec—Johnson & Johnson, and Abbott; and has given expert testimony about biomarkers in blood clotting.
EJV: Receives financial support from the NIH for participation in the HFN.
KJA: Receives financial support from the NIH for participation in the HFN.
AFH: Receives financial support from the NIH for participation in the HFN; consults for Astra Zenceca, Corthera, Janseen and BMS; and his institution has received funding from Amylin, Johnson & Johnson and BMS.
AMM: None reported.
EB: Consults for Merck & Co., Daiichi Sankyo, Genzyme, Amorcyte, Medicines Co., MC Communications, Ikaria, CardioRentis, Sanofi Aventis; has received funding (or pending) from Astra Zeneca, Johnson & Johnson, Merck & Co., Sanofi Aventis, Daiichi Sankyo, Glaxo Smith Kline, Bristol Myers Squibb, Beckman Coulter, Roche Diagnostics and Pfizer; has been compensated for lectures for Eli Lilly, Merck, CVRx (no compensation), CV Therapeutics (now Gilead), Daiichi Sankyo, MC Communications, Manarini International, Medscape and Bayer; and has received payment for development of educational presentations from MC Communications.
References
- 1.Owan TE, Redfield MM. Epidemiology of diastolic heart failure. Prog Cardiovasc Dis. 2005 Mar-Apr;47(5):320–332. doi: 10.1016/j.pcad.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Shah RV, Desai AS, Givertz MM. The effect of renin-angiotensin system inhibitors on mortality and heart failure hospitalization in patients with heart failure and preserved ejection fraction: a systematic review and meta-analysis. J Card Fail. 2010 Mar;16(3):260–267. doi: 10.1016/j.cardfail.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Redfield MM, Borlaug BA, Lewis GD, et al. PhosphdiesteRasE-5 Inhibition to Improve CLinical Status and EXercise Capacity in Diastolic Heart Failure (RELAX) trial: rationale and design. Circ Heart Fail. 2012 Sep 1;5(5):653–659. doi: 10.1161/CIRCHEARTFAILURE.112.969071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005 Nov 17;353(20):2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 5.Lewis GD, Shah R, Shahzad K, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007 Oct 2;116(14):1555–1562. doi: 10.1161/CIRCULATIONAHA.107.716373. [DOI] [PubMed] [Google Scholar]
- 6.Guazzi M, Vicenzi M, Arena R. Phosphodiesterase 5 inhibition with sildenafil reverses exercise oscillatory breathing in chronic heart failure: a long-term cardiopulmonary exercise testing placebo-controlled study. Eur J Heart Fail. 2012 Jan;14(1):82–90. doi: 10.1093/eurjhf/hfr147. [DOI] [PubMed] [Google Scholar]
- 7.Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2010 Jan 1;4(1):8–17. doi: 10.1161/CIRCHEARTFAILURE.110.944694. [DOI] [PubMed] [Google Scholar]
- 8.Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011 Jul 12;124(2):164–174. doi: 10.1161/CIRCULATIONAHA.110.983866. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher GF, Balady G, Froelicher VF, Hartley LH, Haskell WL, Pollock ML. Exercise standards. A statement for healthcare professionals from the American Heart Association. Writing Group. Circulation. 1995 Jan 15;91(2):580–615. doi: 10.1161/01.cir.91.2.580. [DOI] [PubMed] [Google Scholar]
- 10.Packer M. Proposal for a new clinical end point to evaluate the efficacy of drugs and devices in the treatment of chronic heart failure. Journal of cardiac failure. 2001 Jun;7(2):176–182. doi: 10.1054/jcaf.2001.25652. [DOI] [PubMed] [Google Scholar]
- 11.Enright PL, Sherrill DL. Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med. 1998 Nov;158(5 Pt 1):1384–1387. doi: 10.1164/ajrccm.158.5.9710086. [DOI] [PubMed] [Google Scholar]
- 12.Khan MN, Pothier CE, Lauer MS. Chronotropic incompetence as a predictor of death among patients with normal electrograms taking beta blockers (metoprolol or atenolol) Am J Cardiol. 2005 Nov 1;96(9):1328–1333. doi: 10.1016/j.amjcard.2005.06.082. [DOI] [PubMed] [Google Scholar]
- 13.Chirinos JA, Segers P, De Buyzere ML, et al. Left ventricular mass: allometric scaling, normative values, effect of obesity, and prognostic performance. Hypertension. 2010 Jul;56(1):91–98. doi: 10.1161/HYPERTENSIONAHA.110.150250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman AM, Silver MA, Francis GS, et al. Enhanced external counterpulsation improves exercise tolerance in patients with chronic heart failure. J Am Coll Cardiol. 2006 Sep 19;48(6):1198–1205. doi: 10.1016/j.jacc.2005.10.079. [DOI] [PubMed] [Google Scholar]
- 15.Abraham WT, Fisher WG, Smith AL, et al. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002 Jun 13;346(24):1845–1853. doi: 10.1056/NEJMoa013168. [DOI] [PubMed] [Google Scholar]
- 16.Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005 Dec;18(12):1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Hundley WG, Kitzman DW, Morgan TM, et al. Cardiac cycle-dependent changes in aortic area and distensibility are reduced in older patients with isolated diastolic heart failure and correlate with exercise intolerance. J Am Coll Cardiol. 2001 Sep;38(3):796–802. doi: 10.1016/s0735-1097(01)01447-4. [DOI] [PubMed] [Google Scholar]
- 18.Shah SJ, Heitner JF, Sweitzer NK, et al. Baseline Characteristics of Patients in the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) Trial. Circ Heart Fail. 2012 Dec 20; doi: 10.1161/CIRCHEARTFAILURE.112.972794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forfia PR, Borlaug BA. Letter by Forfia and Borlaug regarding article, “Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study”. Circulation. 2012 Feb 28;125(8):e408. doi: 10.1161/CIRCULATIONAHA.111.064584. author reply e409–410. [DOI] [PubMed] [Google Scholar]
- 20.Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007 Jul 17;116(3):238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 21.Xie YP, Chen B, Sanders P, et al. Sildenafil prevents and reverses transverse-tubule remodeling and Ca(2+) handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension. 2012 Feb;59(2):355–362. doi: 10.1161/HYPERTENSIONAHA.111.180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borgdorff MA, Bartelds B, Dickinson MG, et al. Sildenafil enhances systolic adaptation, but does not prevent diastolic dysfunction, in the pressure-loaded right ventricle. Eur J Heart Fail. 2012 Sep;14(9):1067–1074. doi: 10.1093/eurjhf/hfs094. [DOI] [PubMed] [Google Scholar]
- 23.Nagayama T, Hsu S, Zhang M, et al. Pressure-overload magnitude-dependence of the anti-hypertrophic efficacy of PDE5A inhibition. J Mol Cell Cardiol. 2009 Apr;46(4):560–567. doi: 10.1016/j.yjmcc.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005 Feb;11(2):214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 25.Lewis GD, Lachmann J, Camuso J, et al. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation. 2007 Jan 2;115(1):59–66. doi: 10.1161/CIRCULATIONAHA.106.626226. [DOI] [PubMed] [Google Scholar]
- 26.Guazzi M, Tumminello G, Di Marco F, Fiorentini C, Guazzi MD. The effects of phosphodiesterase-5 inhibition with sildenafil on pulmonary hemodynamics and diffusion capacity, exercise ventilatory efficiency, and oxygen uptake kinetics in chronic heart failure. J Am Coll Cardiol. 2004 Dec 21;44(12):2339–2348. doi: 10.1016/j.jacc.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 27.Borlaug BA, Olson TP, Lam CS, et al. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2010 Sep 7;56(11):845–854. doi: 10.1016/j.jacc.2010.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishu K, Deswal A, Chen HH, et al. Biomarkers in acutely decompensated heart failure with preserved or reduced ejection fraction. Am Heart J. 2012 Nov;164(5):763–770. e763. doi: 10.1016/j.ahj.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tapia E, Sanchez-Lozada LG, Soto V, et al. Sildenafil treatment prevents glomerular hypertension and hyperfiltration in rats with renal ablation. Kidney Blood Press Res. 2012;35(4):273–280. doi: 10.1159/000334952. [DOI] [PubMed] [Google Scholar]
- 30.Medeiros PJ, Villarim Neto A, Lima FP, Azevedo IM, Leao LR, Medeiros AC. Effect of sildenafil in renal ischemia/reperfusion injury in rats. Acta Cir Bras. 2010 Dec;25(6):490–495. doi: 10.1590/s0102-86502010000600006. [DOI] [PubMed] [Google Scholar]
- 31.Choi DE, Jeong JY, Lim BJ, et al. Pretreatment of sildenafil attenuates ischemia-reperfusion renal injury in rats. Am J Physiol Renal Physiol. 2009 Aug;297(2):F362–370. doi: 10.1152/ajprenal.90609.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material includes the following appendices and online-only material:
- Appendix A: RELAX Protocol
- Appendix B: RELAX Statistical Analysis Plan
- Appendix C: RELAX Trial Members, Investigators and Committees
- Appendix D: CONSORT trial checklist
- e-Table 1: RELAX trial entry criteria
- e-Methods and references
- e-Table 2: Biomarker reference ranges
- e-Table 3. Pre-specified subgroup analysis of change in peak VO2 at 24 weeks according to baseline characteristics or study drug use at 24 weeks
- e-Table 4. Adverse events occurring in at least 5% of patients in either treatment group
- e-Table 5. Serious adverse events exclusive of death and cardiovascular or cardiorenal hospitalization.