

Abstract
Nucleotide-binding oligomerization domain-like receptors (NLRs) detect pathogens and danger-associated signals within the cell. Salmonella Typhimurium, an intracellular pathogen, activates caspase-1 required for the processing of the pro-inflammatory cytokines, pro-IL-1β and pro-IL-18, and pyroptosis. Here we show that Salmonella infection induces the formation of an ASC–Caspase-8–Caspase-1 inflammasome in macrophages. Caspase-8 and caspase-1 are recruited to the ASC focus independently of one other. Salmonella infection initiates caspase-8 proteolysis in a manner dependent on NLRC4 and ASC, but not NLRP3, caspase-1 or caspase-11. Caspase-8 primarily mediates the synthesis of pro-IL-1β, but is dispensable for Salmonella-induced cell death. Overall, our findings highlight that the ASC inflammasome can recruit different members of the caspase family to induce distinct effector functions in response to Salmonella infection.
INTRODUCTION
Members of the NLR family detect pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) and initiate the formation a multi-meric complex known as the inflammasome. NAIPs and NLRC4, for example, have been shown to recognize bacterial flagellin and certain Type III secretion system-associated rod or needle proteins (1-4), while NLRP3 is activated by a large repertoire of PAMPs and DAMPs, including ATP, uric acid crystals, silica, aluminium hydroxide, asbestos and bacterial or viral RNA (5-10). Formation of the inflammasome facilitates processing of the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their mature forms, which is critical for host defense during microbial infection (11). Conversely, sterile inflammation induced by dysregulated inflammasome activation in response to endogenous DAMPs could lead to autoinflammatory disorders (12).
The inflammasome consists of an NLR, such as NLRC4 or NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1. Salmonella enterica serovar Typhimurium (S. Typhimurium) activates caspase-1 via both NLRC4 and NLRP3 (13). Emerging evidence suggests that caspases other than caspase-1 play an important role in inflammasome signaling (14-18). Human caspase-4 or the mouse equivalent, caspase-11, activates caspase-1 in response to a subset of NLRP3 activators (14-18). During infection with Gram-negative bacteria including S. Typhimurium, LPS from these microorganisms induces TLR4-TRIF-mediated Type I interferon production, which up-regulates caspase-11 (16-18). Caspase-11 does not process pro-IL-1β directly (19), but serves as an activator of the NLRP3 inflammasome (16, 18, 20). Caspase-11, however, is not required when NLRP3 is activated independently of Gram-negative bacteria, for example by ATP, nigericin, silica and Gram-positive bacteria (15, 16). Caspase-11 is also dispensable for NLRC4 inflammasome activity induced by S. Typhimurium and Pseudomonas aeruginosa (16, 17). Other pathogens including Candida and Mycobacterium species trigger caspase-8 activation via dectin-1, where caspase-8 processes pro-IL-1β independently of the classical inflammasome (21). Evidence now suggests a central role for caspase-8 in the AIM2 and NLRP3 inflammasomes in driving cell death (22, 23). Francisella tularensis activates AIM2 to drive caspase-8–dependent cell death in the absence of caspase-1 (23). The role of caspase-8 during Salmonella infection is unclear.
Caspase-8 is an initiator caspase which can activate another initiator caspase, caspase-9, as well as downstream effector caspases, caspase-3 and caspase-7 (24). Caspase-8 is synthesized as a single-chain zymogen, pro-caspase-8, which consists of two death effector domains (DEDs) and two active domains, p18 and p10 (25). Caspase-8 can be activated by death receptors, CD95 (also known as FAS or Apo1) or TNF receptor 1 (TNFR1), both requiring the adaptor protein FAS-associated death domain protein (FADD) (26, 27). Binding of TNF-α to TNFR1 induces the formation of a Receptor Interacting Protein Kinase (RIPK)1-FADD-Pro-caspase-8 complex that mediates apoptosis (27). In response to CD95 signaling, the death-inducing signaling complex (DISC) is formed, which results in the recruitment and activation of pro-caspase-8 by FADD (27). CD95 activation of caspase-8 leads to pro-IL-1β and pro-IL-18 maturation independently of RIPK3 in TLR-stimulated macrophages and dendritic cells (28). A later report, in contrast, suggests that caspase-8 deficiency results in LPS-induced pro-IL-1β maturation in a RIPK3- and RIPK1-dependent manner and that caspase-8 has an inhibitory role in the NLRP3 inflammasome when dendritic cells are stimulated with LPS (29).
In this study we show that Salmonella infection activates a caspase-8-dependent pathway via NLRC4 that induces an ASC-Caspase-8-Caspase-1 inflammasome complex. Caspase-8 primarily contributes to pro-IL-1β synthesis, but not to cell death driven by NLRC4 and caspase-1. These results highlight a novel effector function of caspase-8 within the inflammasome which contributes to the host response against Salmonella infection.
MATERIALS AND METHODS
Mice
Wild-type C57BL/6 mice (Harlan, Loughborough, UK), Nlrc4−/− mice, Nlrp3−/− mice, Asc−/− mice and caspase-1−/− (caspase-11−/−) mice on the C57BL/6 background were housed in a specific pathogen-free facility according to the Animals Scientific Procedures outlined by the UK Home Office regulations. Caspase-8+/− ripk3−/−, Caspase-8−/− ripk3−/− and caspase-8+/− ripk3+/− mice were from D.R. Green (St Jude Children’s Research Hospital, USA).
Cell stimulation and analysis
Primary bone marrow-derived macrophages (BMMs) were infected with log-phase S. Typhimurium strain SL1344 using the indicated multiplicities of infection (MOI). For 2 h infections, supernatant was removed after 1 h and replaced with media containing 50 μg/ml gentamicin (Sigma) for 1 h to kill extracellular bacteria. For 6 and 24 h infections after 1 h incubation in media containing 50 μg/ml gentamicin, supernatant was removed and replaced with media containing 10 μg/ml gentamicin. In experiments that required LPS priming, BMMs were stimulated with 200 ng/ml of ultrapure LPS from Escherichia coli (InvivoGen) for 3 h. Ultrapure flagellin (60 ng) from S. Typhimurium (InvivoGen) was incubated with Profect-P1 reagent (Targeting Systems) for 20 min to promote complex formation, added to BMMs (in 40 μl volume per well in a 96 well plate) and centrifuged at 11 ×g for 10 min. For inhibitor experiments, cells were incubated with 30 or 50 μM Z-IETD-FMK, a caspase-8 inhibitor (21, 30) (Merck) at the same time as bacterial infection or ligand stimulation. Host cell viability was determined using the CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega). Cytokines secreted into cell culture supernatants were measured using the OptEIA™ Mouse IL-1β Set (BD), the mouse TNF-α DuoSet ELISA kit (R&D Systems) or the mouse IL-18 ELISA kit (MBL International).
Immunofluorescence staining
BMMs were washed twice with PBS, fixed in −30 °C methanol for 5 min or 4% (w/v) paraformaldehyde for 15 min. Blocking was performed using 10% normal goat serum or rabbit serum (Dako) in 0.1% saponin (Sigma) for 1 h. Cells were stained with primary and secondary antibodies for 50 min or overnight and 40 min, respectively. The primary antibodies used were rabbit anti-ASC antibody (AL177; ENZO), mouse anti-ASC antibody (clone 2EI-7; Millipore), rabbit anti-mouse cleaved caspase-8 (#8592; Cell Signaling Technology) and goat anti-mouse IL1-β (AF-401-NA; R&D Systems). The secondary antibodies used were Alexa Fluor 488, 568 or 647, anti-rabbit, anti-mouse or anti-rat IgG (Invitrogen). Cells were counterstained in DAPI mounting medium (Vecta Labs). To stain for active caspases, the macrophage supernatant was removed following stimulation and replaced with media containing 0.5× reconstituted FLICA active caspase-1 or caspase-8 reagents (ImmunoChemistry Technologies) and incubated for an additional 1 h. Cells were then fixed in 4% (w/v) paraformaldehyde and stained as described above. Cells and inflammasomes were visualized, counted and imaged using a Leica DM6000 B fluorescence microscope or Leica TCS SP5 confocal microscope.
Co-immunoprecipitation
BMMs were washed with PBS and a cross-linker reagent (5 mM DTBP) was added and incubated on ice for 30 min. Cells were lysed in lysis buffer (60 mM Tris pH8, 10 mM MgCl2, 60 mM NaCl, 1% Triton X-100, protease inhibitor and phosphatase inhibitor cocktails). Lysates were collected following centrifugation at 1,000 rpm for 8 min at 4 °C. Caspase-8 was immunoprecipitated overnight at 4 °C using an anti-caspase-8 antibody (IG12, ENZO) and Protein A/G PLUS-Agarose Gel (Santa Cruz).
Western blotting
Proteins from cell culture supernatants were precipitated using the methanol-chloroform method as described previously (7). Cell lysates were prepared by adding cell lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH8, 1% Triton X-100, 1 mM PMSF, 10 μg/ml leupeptin, 1 μg/ml aprotinin) to cells and incubated for 10 min on ice. Cell lysates were collected after centrifugation. Samples were separated by 4-20% gradient SDS-PAGE and transferred onto PVDF membranes. PVDF membranes were probed with primary antibodies overnight and with secondary antibodies for 2 h. The primary antibodies used were rabbit anti-mouse caspase-1 p10 (sc-514; Santa Cruz Biotechnology), rabbit anti-mouse cleaved caspase-8 (#8592; Cell Signaling Technology), rabbit anti-caspase-8 antibody (IG12, ENZO), mouse anti-NLRP3 antibody (ALX-804-881-C100; ENZO), rabbit anti-ASC antibody (AL177; ENZO), goat anti-mouse IL-1β (AF-401-NA; R&D Systems) and mouse anti-β-actin monoclonal antibody (ab3280; Abcam). The secondary antibodies used were goat anti-rabbit IgG-HRP, rabbit anti-goat IgG-HRP (sc-2004 and sc-2922, respectively; Santa Cruz Biotechnology) or polyclonal goat anti-mouse IgG-HRP (P0447; DAKO). Blots were developed using Amersham Hyperfilm ECL (GE Healthcare) and Curix 60 Tabletop processor (AGFA Healthcare).
Real-time PCR
RNA from BMMs was extracted using TRIzol® (Life Technologies) according to the manufacturers’ instructions. Synthesis of cDNA was performed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Levels of transcripts were quantified using SYBR-Green with an Applied Biosystems 7500 real-time PCR instrument. The primers used were: mIL-1βF 5′-GAT CCA CAC TCT CCA GCT GCA-3′; mIL-1βR 5′-CAA CCA ACA AGT GAT ATT CTC CAT G-3′; mGAPDH-RT-F 5′-TTG ATG GCA ACA ATC TCC AC-3′; and mGAPDH-RT-R 5′-CGT CCC GTA GAC AAA ATG GT-3′. Relative expression of pro-IL-1β was calculated using the ΔΔCt standardization method.
Statistical analysis
Statistical significance between values from two groups was determined using unpaired Student’s t-test and values between three or more groups was determined using the Kruskal–Wallis One-way ANOVA with all values corrected using a Dunnett’s multiple comparisons test. P<0.05 is considered significant.
RESULTS
Salmonella infection of macrophages induces caspase-8 recruitment to the ASC inflammasome
Recent work suggests that inflammasomes, in addition to recruiting caspase-1, can also recruit caspase-11 (15-18). Caspase-8 processes pro-IL-1β in response to dectin-1 stimulation independently of classical inflammasome formation (21). We wondered whether caspase-8 might be recruited to the Salmonella-induced inflammasome. Inflammasome activation results in ASC and caspase-1 redistribution in the host cytosol to form a single cytoplasmic focus (speck), which can be visualized using immuno-labeling and microscopy techniques (13, 31, 32). We infected unprimed primary bone marrow-derived macrophages (BMMs) with Salmonella pathogenicity Island-1 (SPI-1) competent S. Typhimurium to activate the NLRC4 inflammasome (1, 3, 33, 34) and used immuno-labeling and microscopy techniques to determine whether caspase-8 forms a distinct speck-like focus which colocalizes with ASC. We found caspase-8 colocalized with the ASC foci formed in wildtype and caspase-8+/− ripk3−/− control BMMs infected with S. Typhimurium, but not in the ASC foci of caspase-8−/− ripk3−/− BMMs (Figure 1A and 1B). Caspase-8−/− ripk3−/− BMMs were used because genetic ablation of caspase-8 in mice results in embryonic lethality, which can be rescued by ablation of RIPK3 (35, 36).
Figure 1. Salmonella infection induces recruitment of caspase-8 to the ASC inflammasome.
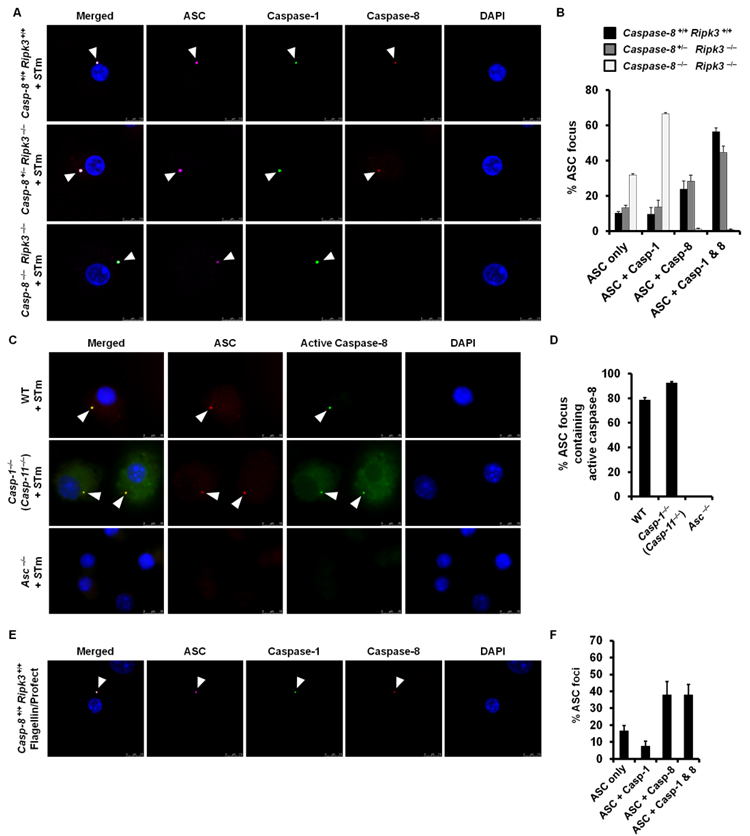
(A) Wildtype (Caspase-8 +/+ ripk3 +/+), Caspase-8 +/− ripk3 −/−, Caspase-8 −/− ripk3 −/− primary bone marrow–derived macrophages (BMMs) were infected with S. Typhimurium (STm, MOI 10) for 30 min and stained for ASC (magenta), active caspase-1 (green), caspase-8 (red) and DNA (blue). (B) Percentage of ASC foci which harbored a caspase-1 focus, caspase-8 focus or both caspase-1 and caspase-8 foci in A. (C) Wildtype, caspase-1 −/− (caspase-11 −/−) and Asc −/− BMMs were infected with STm for 30 min and stained for ASC (red), active caspase-8 (green) and DNA (blue). (D) Percentage of ASC foci containing a caspase-8 focus in C. (E) Wildtype BMMs were stimulated with flagellin from STm for 1 h and stained for ASC (magenta), active caspase-1 (green), caspase-8 (red) and DNA (blue). (F) Percentage of ASC foci which harbored a caspase-1 focus, caspase-8 focus or both caspase-1 and caspase-8 foci in E. Cells were fixed and stained with a rabbit anti-ASC antibody and a rat anti-caspase-8 antibody (A,E) or caspase-8 FLICA stain (C). At least 100 (B,F) or 200 (D) ASC–focus–containing BMMs were counted in each independent experiment. Data are from three independent experiments.
Both caspase-8 and caspase-1 redistributed to the same ASC focus in wildtype BMMs stimulated with S. Typhimurium (Figure 1A and 1B). We quantified the percentage of ASC focus containing no caspase, caspase-1, caspase-8 or caspase-1 and caspase-8 in wildtype, caspase-8+/− ripk3−/− and caspase-8−/− ripk3−/− BMMs infected S. Typhimurium. A large proportion of ASC foci found in wildtype and caspase-8+/− ripk3−/− BMMs contained both caspase-1 and caspase-8 (57% in wildtype and 45% in caspase-8+/− ripk3−/− BMMs), suggesting that the formation of the ASC–Caspase-8–Caspase-1 inflammasome in these cells is a common event (Figure 1B). Caspase-1 was still recruited to the ASC focus of caspase-8−/− ripk3−/− BMMs, indicating that caspase-8 deficiency does not impair the recruitment of caspase-1 (Figure 1A and 1B). Caspase-8 within the ASC focus was active, as shown by staining with FAM-IETD-FMK, a fluorescent compound that binds irreversibly to active caspase-8 (Figure 1C and 1D). We observed active caspase-8 in ASC foci of caspase-1−/− (caspase-11−/−) BMMs infected with S. Typhimurium, suggesting that caspase-8 is recruited independently of caspase-1 or caspase-11 (Figure 1C and 1D). Caspase-8 foci failed to form in Asc−/− BMMs (Figure 1C and 1D).
Previous studies have shown that S. Typhimurium activates caspase-1 via the inflammasome receptors NLRC4 (1, 3, 13, 37). To investigate whether NLRC4 is involved in Salmonella–induced ASC-Caspase-8 focus formation, we stimulated caspase-8 +/+ ripk3 +/+ (wildtype) BMMs with ultrapure flagellin from S. Typhimurium to activate NLRC4 and used immunofluorescence staining techniques to visualize the distribution of ASC, caspase-8 and caspase-1. We found that caspase-8 and caspase-1 were recruited to the ASC focus of BMMs stimulated with flagellin (Figure 1E). Quantification of the prevalence of caspase-8 and caspase-1 in the ASC focus revealed that 38% of the ASC foci were colocalized with both caspases (Figure 1F). We also infected unprimed wildtype or Nlrc4−/− BMMs with S. Typhimurium and found that wildtype, but not Nlrc4−/−, BMMs induced ASC focus formation (Supplementary Figure 1), indicating that Salmonella-induced ASC focus formation is NLRC4-dependent. These results collectively suggest that Salmonella-induced formation of the ASC-Caspase-8-Caspase-1 inflammasome is dependent on NLRC4. Finally we used co-immunoprecipitation techniques to confirm that caspase-8 and ASC are part of the same complex. We found that endogenous caspase-8 co-immunoprecipitated with ASC in BMMs infected with S. Typhimurium (MOI 10) after 30 min, but not in uninfected BMMs (Figure 2A).
Figure 2. Salmonella infection induces recruitment of caspase-8 into the inflammasome where it undergoes proteolysis.
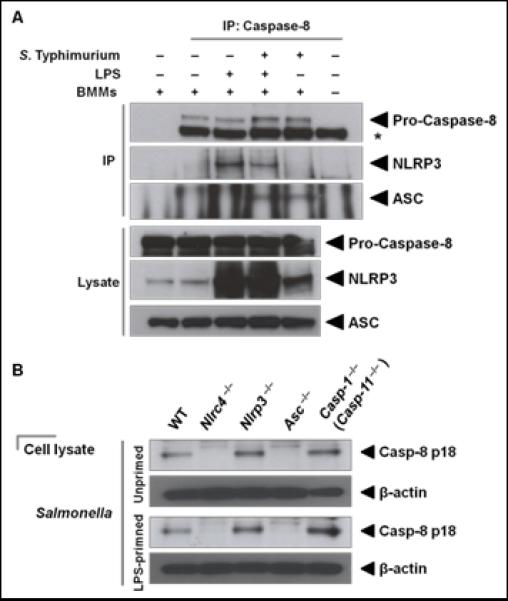
(A) Unprimed or LPS-primed wildtype primary bone marrow–derived macrophages (BMMs) were infected with S. Typhimurium (STm, MOI 10) for 30 min. Endogenous caspase-8 was immunoprecipitated using an anti-caspase-8 antibody. Western blotting was used to detect caspase-8, ASC and NLRP3. * indicates heavy chain of the IP antibody. (B) Western blot analysis of caspase-8 proteolysis in wildtype, Nlrc4 −/−, Nlrp3 −/−, Asc −/− and caspase-1−/− (caspase-11−/−) BMMs infected with STm for 30 min. Data are representative of two (B) or three (A) independent experiments.
In cells primed with LPS or at 6 h post-infection, Salmonella also activates the NLRP3 inflammasome (13). We wondered whether caspase 8 would also associate with this second Salmonella-driven inflammasome. Kang and colleagues have previously shown that caspase-8 inhibits the NLRP3 inflammasome following LPS treatment in dendritic cells (29). In LPS-primed BMMs we did not observe the formation of an ASC-Caspase-8 focus in the absence of Salmonella infection. When we pulled down caspase-8 in LPS-primed BMMs, we saw caspase-8 associated with NLRP3, but not with ASC (Figure 2A). Caspase-8 co-immunoprecipitated with both ASC and NLRP3 in LPS-primed BMMs infected with S. Typhimurium (Figure 2A). When unprimed BMMs were infected with S. Typhimurium, caspase-8 co-immunoprecipitated only with ASC, which could be due to insufficient levels of NLRP3 proteins in unprimed cells (Figure 2A). These results suggest that caspase-8 may play distinct roles depending on the contextual cue received by the cell. In cells stimulated with LPS alone (signal 1, priming), caspase-8 may interact with NLRP3 and inhibit IL-1β production as shown by Kang and colleagues (29), whereas Salmonella infection (signal 2, priming and inflammasome activation) induces the formation of the ASC inflammasome that contains caspase-8 and caspase-1, where these caspases can potentially undergo proteolysis.
Salmonella infection induces caspase-8 proteolysis via NLRC4 and ASC
To investigate whether Salmonella infection induces caspase-8 proteolysis, we infected BMMs with S. Typhimurium for 30 min and immunoblotted for the presence of the caspase-8 p18 subunits, which are yielded upon proteolysis of pro-caspase-8 (38). We found that Salmonella infection induced caspase-8 proteolysis in wildtype BMMs (Figure 2B). To investigate whether NLRC4 and NLRP3 (two NLRs involved in the recognition of S. Typhimurium) are involved in Salmonella–induced caspase-8 proteolysis, we compared caspase-8 proteolysis in wildtype, Nlrc4−/− and Nlrp3−/− BMMs infected with S. Typhimurium for 30 min and only found NLRC4–dependent caspase-8 proteolysis (Figure 2B). Previous studies have shown that LPS priming is required to induce NLRP3 expression (39), but even in LPS-primed BMMs, we could only observe NLRC4–induced caspase-8 proteolysis in response to Salmonella infection (Figure 2B).
Asc−/− BMMs stimulated with S. Typhimurium failed to induce caspase-8 proteolysis, indicating that ASC was essential in this process (Figure 2B). Caspase-1−/− (caspase-11−/−) BMMs retained the capacity to undergo caspase-8 proteolysis (Figure 2B). These results support the observation that recruitment of caspase-8 into a single focus requires ASC but not caspase-1 and caspase-11 (Figure 1). Taken together, these results demonstrate that NLRC4 initiates ASC-dependent, caspase-1- and caspase-11-independent proteolysis of caspase-8 in response to Salmonella infection.
Caspase-8 contributes to Salmonella–induced IL-1β production
To investigate the role of caspase-8 in the inflammasome in response to Salmonella infection, we infected caspase-8−/− ripk3−/− BMMs with S. Typhimurium and examined IL-1β production, pro-IL-1β processing, caspase-1 proteolysis and cell death. IL-1β production was significantly impaired in caspase-8−/− ripk3−/− BMMs compared to caspase-8+/− ripk3−/− BMMs or wildtype BMMs (caspase-8+/− ripk3+/− or caspase-8+/+ ripk3+/+) infected with S. Typhimurium (Figure 3A; P<0.001 for 2, 6 and 24 h). Reduced levels of cleaved IL-1β were also found in the supernatant of caspase-8−/− ripk3−/− BMMs compared to the corresponding controls (Figure 3B). Caspase-8−/− ripk3−/− BMMs stimulated with S. Typhimurium maintained their capacity to induce caspase-1 proteolysis, suggesting that caspase-1 proteolysis occurred independently of caspase-8 (Figure 3B) and confirmed our observation that caspase-8 deficiency did not impair recruitment of caspase-1 into the ASC focus (Figure 1A and 1B). Conversely, caspase-8 proteolysis was observed in caspase-1−/− (caspase-11−/−) BMMs (Figure 3B), supporting our findings that recruitment of caspase-8 into the ASC focus is independent of caspase-1 and 11 (Figure 1C and 1D). Taken together, these results indicate that caspase-8 plays a role in modulating IL-1β production during Salmonella infection.
Figure 3. Salmonella infection drives Caspase-8–dependent IL-1β production.
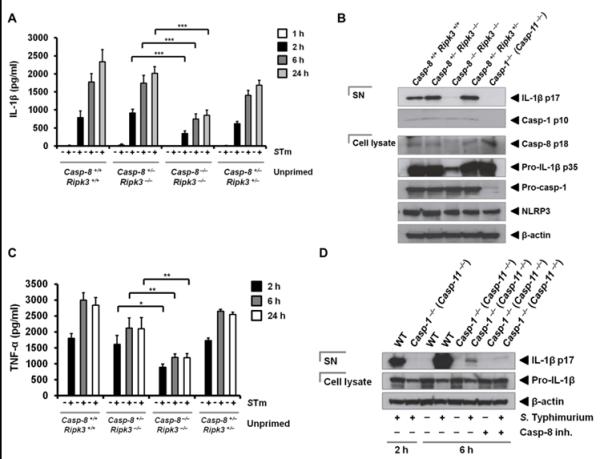
(A) Unprimed wildtype (Caspase-8 +/+ ripk3 +/+), caspase-8 +/− ripk3 −/−, caspase-8 −/− ripk3 −/− and caspase-8 +/− ripk3 +/− primary bone marrow–derived macrophages (BMMs) were infected with S. Typhimurium (STm, MOI 1) for 1, 2, 6 and 24 h and levels of IL-1β were measured. (B) Western blot analysis of the cleaved IL-1β p17 subunit (processed IL-1β), cleaved caspase-1 p10 subunit (active caspase-1), cleaved caspase-8 p18 subunit (active caspase-8), pro-caspase-1, NLRP3, pro-IL-1β and β-actin in the supernatant or cell lysate of LPS-primed BMMs. (C) BMMs were infected with STm (MOI 1) for 2, 6 and 24 h and levels of TNF-α were measured. (D) LPS-primed wildtype and caspase-1 −/− (caspase-11 −/−) BMMs were infected with STm in the presence or absence of a caspase-8 inhibitor (30 μM) for 2 or 6 h. Western blot analysis of the cleaved IL-1β p17 subunit (processed IL-1β), pro-IL-1β and β-actin in the supernatant or cell lysate. Data are representative of two (C,D) or three (A,B) independent experiments and error bars represent s.e.m. * P < 0.05; ** P < 0.01; *** P < 0.001.
Our data studying pro-IL-1β cleavage also showed a reduced level of total pro-IL-1β in the infected caspase-8−/− ripk3−/− BMMs, suggesting that pathways leading to the generation of the inflammasome signal 1 may be affected in these cells (Figure 3B). In agreement with this, qPCR analysis showed that caspase-8−/− ripk3−/− BMMs infected with S. Typhimurium produced a substantially lower level of pro-IL-1β transcripts than the corresponding controls (Supplementary Figure 2). The expression of NLRP3 and caspase-1, however, did not appear to be impaired in the absence of caspase-8 (Figure 3B). We investigated whether TLR signaling was intact in caspase-8−/− ripk3−/− BMMs infected with S. Typhimurium by measuring TNF-α production. The levels of TNF-α released by caspase-8−/− ripk3−/− BMMs stimulated with S. Typhimurium were significantly lower than the corresponding controls (Figure 3C; P=0.01 for 2 h, P<0.01 for 6 and 24 h). These results suggest that caspase-8 operates as a checkpoint at the level of pro-IL-1β synthesis to regulate the amount of pro-IL-1β available for caspase-1-dependent processing.
We then investigated whether caspase-8 has a role in processing of pro-IL-1β in BMMs infected for 2 and 6 h. Processing of pro-IL-1β after 2 h of infection with S. Typhimurium was abolished in caspase-1−/− (caspase-11−/−) BMMs, confirming that caspase-1 and -11 were essential for early processing of pro-IL-1β (Figure 3D). At 6 h post infection a low level of cleaved IL-1β (p17 subunit) was detected in the supernatant of these cells (Figure 3D). Inhibition of caspase-8 using Z-IETD-FMK in caspase-1−/− (caspase-11−/−) BMMs almost abolished caspase-1- and -11-independent pro-IL-1β processing, but further experiments are required to confirm the role of caspase-8 in delayed processing of pro-IL-1β (Figure 3D). We then investigated whether caspase-8 is involved in the processing of IL-18, a constitutively expressed inflammasome–dependent cytokine whose expression is not under the influence of TLRs. We infected BMMs from caspase-8+/+ ripk3+/+, caspase-8+/− ripk3−/−, caspase-8−/− ripk3−/− and caspase-8+/− ripk3+/− mice with S. Typhimurium for 2 and 24 h and measured IL-18 from the supernatant of these cells. Caspase-8−/− ripk3−/− BMMs did not produce less IL-18 compared to any of the controls after 2 h of infection, which confirms that caspase-8 may not be involved in early processing of IL-18 at this time point (Supplementary Figure 3). The levels of IL-18 released by caspase-8 +/+ ripk3 +/+ and caspase-8 −/− ripk3 −/− BMMs were similar, however, both of which were higher than the levels observed in caspase-8 +/− ripk3 −/− and caspase-8 +/− ripk3 +/− BMMs (not statically significant; P > 0.05). We were unable to detect any IL-18 from BMMs infected with S. Typhimurium for 24 h (data not shown), and therefore, it is unclear whether caspase-8 may play a role in delayed processing of IL-18.
Recruitment of pro-IL-1β to the inflammasome is an important event which mediates processing of this cytokine. We have shown that caspase-8 inhibition in wildtype BMMs did not prevent recruitment of pro-IL-1β to the inflammasome in response to Salmonella infection (Figure 4A). Caspase-1 was, however, required for efficient recruitment of pro-IL-1β to the ASC inflammasome, since 52% of the ASC foci in wildtype BMMs contained pro-IL-1β compared to only 15% in BMMs deficient in caspase-1 (Figure 4B). Taken together, these results demonstrate that caspase-1 and caspase-8 orchestrate distinct roles in the inflammasome. Caspase-1 recruits and processes pro-IL-β, while caspase-8 primarily controls the synthesis of pro-IL-1β.
Figure 4. Recruitment of pro-IL-1β to the ASC focus is dependent on caspase-1 and unaffected by inhibition of caspase-8.
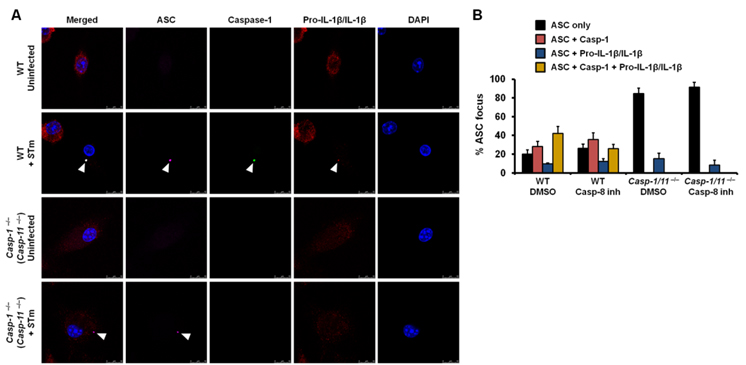
(A) LPS-primed wildtype or caspase-1 −/− (caspase-11 −/−) BMMs were infected with STm for 30 min in the presence of DMSO (vehicle control) or a caspase-8 inhibitor (50 μM) and immunostained for active caspase-1 (green), ASC (magenta), pro-IL-1β/IL-1β (red) and DNA (blue). (B) Percentage of ASC foci which harbored either caspase-1 or pro-IL-1β/IL-1β or both. At least 100 ASC–focus–containing BMMs were counted for each treatment in each independent experiment. Data are representative of three independent experiments and error bars represent s.e.m.
Salmonella activation of NLRC4, but not NLRP3 results in a rapid, caspase-1–dependent cell death, but this is independent of ASC (Supplementary Figure 4) (37). The proteolysis of caspase-8 dependent on ASC in response to Salmonella infection suggested that caspase-8 may not, therefore, be involved in the NLRC4-induced cell death pathway. We have shown that the levels of cell death in wildtype, caspase-8 +/− ripk3 −/− and caspase-8 −/− ripk3 −/− BMMs were similar following 2, 6 and 24 h of infection with S. Typhimurium, suggesting that caspase-8 had no effect on cell death induced by Salmonella infection (Figure 5A). LPS priming of BMMs to induce NLRP3 levels prior to Salmonella infection also show a lack of a role for caspase-8 in driving Salmonella-induced cell death (Figure 5B). These data confirm that caspase-8 does not contribute to Salmonella-induced cell death in the presence of caspase-1.
Figure 5. Salmonella infection does not induce early cell death via Caspase-8.
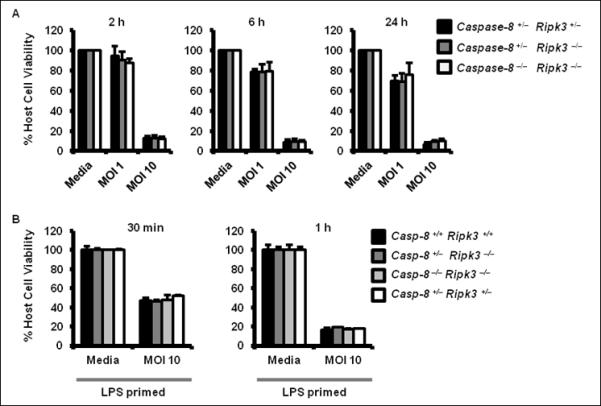
(A) Wildtype (Caspase-8 +/− ripk3 +/−), Caspase-8 +/− ripk3 −/−, Caspase-8 −/− ripk3 −/− primary bone marrow–derived macrophages (BMMs) were infected with S. Typhimurium (STm) for 2, 6 and 24 h and levels of lactate dehydrogenase were measured. (B) LPS-primed wildtype (caspase-8 +/+ ripk3 +/+), caspase-8 +/− ripk3 −/−, caspase-8 −/− ripk3 −/− and caspase-8 +/− ripk3 −/− BMMs were infected with STm (MOI 10) for 30 min or 1 h and levels of lactate dehydrogenase were measured in the lysate. Data are representative of three independent experiments and error bars represent s.e.m.
DISCUSSION
Caspase-8 is a multi-functional effector protein which is recruited to different complexes according to the stimulus received by the cell. Here we have shown that Salmonella infection induces caspase-8 proteolysis in macrophages infected with S. Typhimurium. Caspase-8 is clearly associated with ASC and caspase-1 within the inflammasome. It is likely that caspase-8 undergoes proteolysis in the assembled inflammasome complex, since the lack of ASC prevented caspase-8 proteolysis in response to Salmonella infection. Our observation showing that distinct members of the caspase family (caspase-1 and caspase-8) and pro-IL-1β are colocalized in the ASC structure is interesting and support the notion that only certain substrates specific for inflammasome processing, such as pro-IL-1β and pro-IL-18, would gain access into the ASC inflammasome. These results highlight that the inflammasome is a dynamic complex which has the ability to recruit distinct members of the caspase family. It will be interesting to investigate the spatial orientation of NLR proteins in the ASC-Caspase-8-Caspase-1 inflammasome in future studies to understand whether NLR proteins reside in the same ASC complex.
We and other have shown that caspase-8 has the capacity to induce pro-IL-1β processing in host cells infected with pathogenic bacteria (21). It is possible that the effect of caspase-8–mediated processing of pro-IL-1β may be more apparent in the absence of caspase-1 or caspase-11. Pharmacological inhibition affects the catalytic activity of caspase-8, however, it does not substantially affect caspase-8–mediated pro-IL-1β synthesis. This suggests that caspase-8 itself, rather than its proteolytic activity, may have a role in driving NF-kB signaling, possibly acting as a scaffolding protein. Caspase-8 has been shown to assemble a multi-meric protein complex in response to dectin-1 activation, which is distinct from the NLRP3-Caspase-1 inflammasome (21). It is possible that caspase-8 is preferentially recruited to other structures in a stimulus-dependent manner, for example, following dectin-1 activation by Candida and mycobacterial species. The tyrosine kinase, Syk, is downstream of dectin-1. A previous study has shown that inhibition of Syk does not affect IL-1β production in dendritic cells infected with S. Typhimurium (40), which suggests a lack of a role for Syk in the Salmonella-induced inflammasome. It is, therefore, likely that caspase-8 could be recruited to the ASC inflammasome during Salmonella infection in the absence of a competing complex induced by Dectin-1-Syk activation. Caspase-8 has been shown to assemble the DISC following CD95 activation, indicating that it is entirely possible for this effector protein to be recruited to a different complex in a stimulus-dependent manner (27).
Kang and colleagues have shown that caspase-8 deficiency in bone marrow dendritic cells results in LPS-induced pro-IL-1β maturation and suggest that caspase-8 has an inhibitory role in the NLRP3 inflammasome (29). Caspase-8, however, may play distinct roles during LPS stimulation and Salmonella infection. Co-immunoprecipitation of endogenous proteins in macrophages show that caspase-8 interacts with NLRP3 in cells stimulated with LPS alone (signal 1), which could contribute to inhibition of NLRP3-mediated IL-1β production (29). Salmonella infection (signal 2), however, activates NLRC4 and induces the formation of the ASC inflammasome that contains caspase-8 and caspase-1, where these caspases undergo proteolysis. These results suggest that caspase-8 may have an effector, rather than an inhibitory, function during microbial infection.
We did not observe IL-1β production in BMMs deficient in caspase-8 when they were primed with LPS alone, and instead, found reduced pro-IL-1β levels in these BMMs. The differences in findings may be due to differences in cell type and the type of mouse used. We used bone marrow-derived macrophages from caspase-8−/− ripk3−/− mice, whereas Kang et al. used bone marrow-derived dendritic cells from caspase-8fl/−:Itgax-Cre conditional knockout mice (29). Kang and colleagues did not observe a decrease in the levels of IL-1β and TNF-α in the serum of LPS-stimulated caspase-8fl/−:Itgax-Cre conditional knockout mice (29). These mice, however, lack caspase-8 specifically in the dendritic cell population and it is possible that other cell types which still express functional caspase-8, including macrophages, neutrophils and keratinocytes, release IL-1β and TNF-α in a caspase-8-dependent manner. It is, therefore, difficult to directly compare results from our study to the study by Kang and colleagues. Previous studies have shown that caspase-8 mediates NF-κB signaling following stimulation of TLRs in T and B cells (41, 42). B cells from caspase-8 conditional knockout mice have delayed ability to translocate NF-κB-p65 translocation into the nucleus, and therefore, display defective transcriptional activation of NF-κB genes, including IL-6, TNF-α and IFN-β (42). Caspase-8 deficiency in human T cells and NK cells reduces NF-κB transcription (41). These results firmly support our observation that caspase-8 deficiency results in reduced pro-IL-1β and TNF-α production. We have previously shown that TLR4 is the major TLR involved in the recognition of S. Typhimurium in macrophages (43, 44). It is, therefore, likely that caspase-8 may play a role in mediating TLR4-dependent NF-kB activation. It is also possible that caspase-8 may have cell-type specific functions in response to TLR activation.
While caspase-8 itself is important for pro-IL-1β synthesis, we hypothesize that the process of caspase-8 recruitment to the inflammasome is not critical for pro-IL-1β synthesis. This is based on the observations that ASC-deficient BMMs infected with S. Typhimurium retain the capacity to synthesize pro-IL-1β (45) despite their inability to form the ASC and caspase-8 inflammasome. We, therefore, propose that caspase-8 has distinct roles depending on its localization within the cell. In conclusion we have shown that Salmonella infection induces a dynamic inflammasome unit which comprises an ASC platform that recruits caspase-8 and caspase-1 for proteolysis.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to D.R. Green, C.P. Dillon and P. Fitzgerald for femurs from Caspase-8+/− ripk3−/−, Caspase-8−/− ripk3−/− and Caspase-8+/− ripk3+/− mice. We would like to acknowledge T-D Kanneganti and members of her lab for providing technical assistance; E. Creagh, V.A. Rathinam and members of the Bryant lab for providing critical review of the manuscript.
Financial Support: S.M.M was supported by a Cambridge International Scholarship. T.P.M was supported by a Wellcome Trust Research Career Development Fellowship (WT085090MA). This study was supported by a BBSRC grant and a BBSRC Research Development Fellowship awarded to C.E.B.
Abbreviations
- ASC
apoptosis–associated speck–like protein containing a CARD
- BMMs
bone marrow–derived macrophages
- DAMPs
danger–associated molecular patterns
- PAMPs
pathogen–associated molecular patterns
- SN
supernatant
REFERENCES
- 1.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 2.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A. 2013;110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 6.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 10.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Nunez G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 11.Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer HD. Caspase-4 is required for activation of inflammasomes. J Immunol. 2012;188:1992–2000. doi: 10.4049/jimmunol.1101620. [DOI] [PubMed] [Google Scholar]
- 15.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 16.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurung P, Malireddi RK, Anand PK, Demon D, Walle LV, Liu Z, Vogel P, Lamkanfi M, Kanneganti TD. Toll or Interleukin-1 Receptor (TIR) Domain-containing Adaptor Inducing Interferon-beta (TRIF)-mediated Caspase-11 Protease Production Integrates Toll-like Receptor 4 (TLR4) Protein- and Nlrp3 Inflammasome-mediated Host Defense against Enteropathogens. J Biol Chem. 2012;287:34474–34483. doi: 10.1074/jbc.M112.401406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, Greenberg AH, Yuan J. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 21.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TB. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012 doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 22.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013 doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, Henry T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19:1709–1721. doi: 10.1038/cdd.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19:36–41. doi: 10.1038/cdd.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Sui X, Ren H. From procaspase-8 to caspase-8: revisiting structural functions of caspase-8. J Cell Physiol. 2010;225:316–320. doi: 10.1002/jcp.22276. [DOI] [PubMed] [Google Scholar]
- 26.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 27.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2012;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, Mocarski ES, Subramanian D, Green DR, Silverman N, Fitzgerald KA, Marshak-Rothstein A, Latz E. Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1beta and IL-18 Maturation via Caspase-8 in an RIP3-Independent Manner. J Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity. doi: 10.1016/j.immuni.2012.09.015. doi: 10.1016/j.immuni.2012.1009.1015. [DOI] [PubMed] [Google Scholar]
- 30.Rytomaa M, Martins LM, Downward J. Involvement of FADD and caspase-8 signalling in detachment-induced apoptosis. Curr Biol. 1999;9:1043–1046. doi: 10.1016/s0960-9822(99)80454-0. [DOI] [PubMed] [Google Scholar]
- 31.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang MT, Taxman DJ, Holley-Guthrie EA, Moore CB, Willingham SB, Madden V, Parsons RK, Featherstone GL, Arnold RR, O’Connor BP, Ting JP. Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol. 2009;182:2395–2404. doi: 10.4049/jimmunol.0800909. [DOI] [PubMed] [Google Scholar]
- 33.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 35.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 38.Van de Craen M, Van Loo G, Declercq W, Schotte P, Van den brande I, Mandruzzato S, van der Bruggen P, Fiers W, Vandenabeele P. Molecular cloning and identification of murine caspase-8. J Mol Biol. 1998;284:1017–1026. doi: 10.1006/jmbi.1998.2226. [DOI] [PubMed] [Google Scholar]
- 39.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 41.Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 42.Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, Hakem A. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- 43.Royle MC, Totemeyer S, Alldridge LC, Maskell DJ, Bryant CE. Stimulation of Toll-like receptor 4 by lipopolysaccharide during cellular invasion by live Salmonella typhimurium is a critical but not exclusive event leading to macrophage responses. J Immunol. 2003;170:5445–5454. doi: 10.4049/jimmunol.170.11.5445. [DOI] [PubMed] [Google Scholar]
- 44.Talbot S, Totemeyer S, Yamamoto M, Akira S, Hughes K, Gray D, Barr T, Mastroeni P, Maskell DJ, Bryant CE. Toll-like receptor 4 signalling through MyD88 is essential to control Salmonella enterica serovar typhimurium infection, but not for the initiation of bacterial clearance. Immunology. 2009;128:472–483. doi: 10.1111/j.1365-2567.2009.03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.