

Abstract
Rationale
A stable 40 kD fragment is produced from cardiac myosin binding protein-C (cMyBP-C) when the heart is stressed, using a stimulus such as ischemia reperfusion injury. Elevated levels of the fragment can be detected in both the diseased mouse and human heart but its ability to interfere with normal cardiac function in the intact animal is unexplored.
Objective
To understand the potential pathogenicity of the 40 kD fragment in vivo and to investigate the molecular pathways that could be targeted for potential therapeutic intervention.
Methods and Results
We generated cardiac myocyte-specific transgenic mice (TG) using a Tet-Off inducible system to permit controlled expression of the 40 kD fragment in cardiomyocytes. When 40 kD protein expression is induced by crossing the responder animals with tetracycline transactivator (tTA) mice under conditions where substantial quantities approximating those observed in disease hearts are reached, the double TG (DTG) mice subsequently develop sarcomere dysgenesis, altered cardiac geometry and the heart fails between 3 to 17 weeks of age. The induced DTG mice developed cardiac hypertrophy with myofibrillar disarray and fibrosis, and activation of pathogenic MEK-ERK pathways. Inhibition of MEK-ERK signaling was achieved by injection of the MAPK/ERK kinase inhibitor U0126. The drug effectively improved cardiac function, decreased fibrosis, normalized heart size and increased probability of survival.
Conclusion
These results suggest that the 40 kD cMyBP-C fragment, which is produced at elevated levels during human cardiac disease, is a pathogenic fragment that is sufficient to cause hypertrophic cardiomyopathy and heart failure.
Keywords: Myosin-binding protein-C, cardiomyopathy, mitogen-activated protein kinase
Introduction
Myosin binding protein C (MyBP-C) is a thick filament-associated protein consisting of 1274 amino acid residues (149 kD), which is localized to the crossbridge-containing C zones in striated muscle sarcomeres.1 Three isoforms of MyBP-C, fast skeletal, slow skeletal and cardiac, are present in both the human and mouse.2,3 While the three isoforms have similarities, the cardiac isoform of MyBP-C (cMyBP-C) differs from the skeletal isoforms in that it contains an additional domain at the N-terminus (C0) and a phosphorylatable domain located between the C1 and C2 domains (Figure 1B). The actin and myosin heavy chain's ATPase -binding regions lie at the protein's amino terminus.4 The importance of understanding the structure-function relationships of cMyBP-C is underscored by the identification of numerous MYBPC3 mutations that are responsible for an estimated 20-35% of verified familial hypertrophic cardiomyopathy (FHC) cases.5,6 Although mutation of MYBPC3 is one of the most frequent causes of HCM on a per gene basis, the majority of these mutations (about 60%) result not in a full-length, mutated protein, but rather in truncated peptides.
Figure 1. Inducible transgene expression.
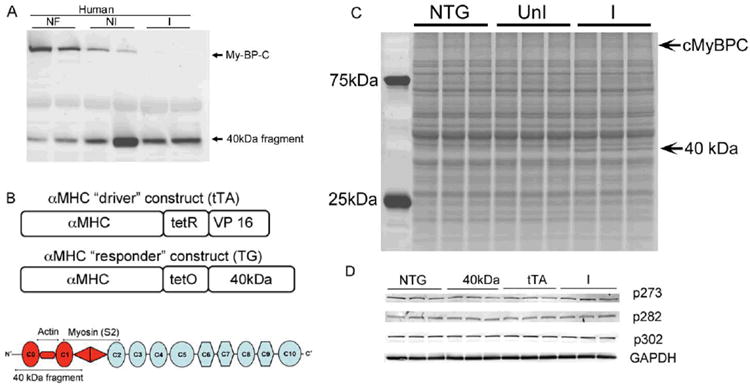
A, Human heart samples showing the levels of cMyBP-C and the 40 kD fragment in normal (non-failing, NF), diseased, non-ischemic (NI) and ischemic (I) samples. B, cMyBP-C structure and transgenic constructs. cMyBP-C is a 149 kD protein made up of eight immunoglobulin-type (ovals) and three fibronectin-like (octagon) domains. The protein binds multiple components of the thick and thin filaments and the location of the 40 kD fragment is shown. One mouse line contains the cardiomyocyte-specific promoter driving the fusion tet-VP16 protein (tTA) that, in the absence of tetracycline will bind to the responder promoter and drive expression of the 40 kDa fragment. We generated multiple lines of cardiomyocyte specific TG mice, choosing a line that showed reasonably tight control of the transgene and producing levels that approximated those in human disease. B, C, When 40 kD protein expression is induced by crossing the responder animals with the tTA mice, the double TG (DTG) mice show protein expression in the absence of Dox and almost no protein when Dox is present. For panel C, three independently isolated samples from each set of animals were prepared and electrophoresed. D, Phosphorylation status of c-MyBP-C at 273, 282 and 302 sites. Expression was examined at 12 weeks in NTG animals, single TG mice carrying the 40 kD fragment (40kD) or tTA transgene (tTA) and DTG mice expressing the 40 kD transgene (DTG).
We recently confirmed that cardiac stress can trigger the production and accumulation of a 40 kD truncated fragment derived from the amino terminus of cMyBP-C. The fragment appears to be generated as a result of dephosphorylation that unmasks a μ-calpain site, resulting in cleavage of intact cMyBP-C.7 A recent report using neonatal rat ventricular cardiomyocytes showed that hypoxic stress resulted in decreased levels of cMyBP-C phosphorylation, its specific cleavage, and the subsequent production of N′-terminal fragments.8 The 40 kD fragment can be detected in both diseased or stressed mouse and human hearts.7-9 The fragment is apparently stable, however, the functional consequences in terms of normal cardiac function are unknown. Appreciable quantities of a truncated fragment of cMyBP-C in the diseased human heart raises the possibility of potential pathogenic consequences, as the fragment has been shown to effectively compete for the normal protein's binding sites to the head regions of myosin and actin.10
Considering the frequency with which truncated cMyBP-C protein can serve as a “poison peptide,”2 we sought to determine the potential pathogenicity of the 40 kD fragment in vivo. We generated cardiac myocyte-specific transgenic mice (TG) using a Tet-Off inducible system to permit controlled expression in cardiomyocytes.11 When 40 kD protein expression is induced in the hearts by crossing the responder animals with tetracycline transactivator (tTA) mice (double transgenic; DTG) in the absence of doxycycline, the DTG mice undergo sarcomere dysgenesis, show altered cardiac geometry and display signs of heart failure by 3 weeks of age, even though intact cMyBP-C expression is unaffected. Expression of the 40 kD fragment in cardiomyocytes led to development of significant cardiac hypertrophy with myofibrillar disarray and fibrosis. Since hypertrophy appeared to due directly to this fragment's expression, we wished to determine if normal, pathogenic signaling was activated, or if some novel pathway was involved. Mitogen-activated protein kinase (MAPK) is one of the major signaling pathways involved in cardiac hypertrophy and heart failure and we subsequently explored the role of this pathway in the developing pathology. MEK-ERK hypertrophic signaling pathways were activated in the 40 kD mice; treating the animals with intraperitoneal injections of U0126, a MEK-ERK pathway inhibitor, effectively improved cardiac function and prolonged survival as compared to the untreated, control mice.
Methods
DNA constructs and TG mice
For cardiomyocyte specific inducible transgene expression, two lines of mice are needed. The driver line (tTA) contains the α myosin heavy chain (MHC) promoter fused to tet-VP16 protein, which was described previously.12 We generated the “responder” line containing the MHC promoter driving the 40 kD N-terminal fragment of cMyBP-C (amino acids 1-271).7,13,14 An N-terminal c-myc-tag encoding the human c-myc peptide (EQKLISEEDL), which has no effect on cMyBP-C function and stability, was inserted after the initiation methionine codon to differentiate TG from endogenous protein. Earlier studies confirmed that introduction of the c-myc epitope was benign as, when we fused c-myc to the wild type cMyBP-C, we did not see any effects on structural, functional or hemodynamic parameters.7,13,14 In the presence of doxycycline the protein was not expressed. Animals were handled in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee at Cincinnati Children's Hospital approved all experimental procedures.
Human sample collection
Tissue was harvested from the left ventricular free wall near the apex of male patients (aged 59.6 +/- 3.1 years) in end-stage (stage D) heart failure at the time of left ventricular assist device placement (HeartMate or HeartMate II; Thoratec, Pleasanton, CA) as either destination therapy or bridge to transplantation. Samples from patients with the diagnosis of ischemic (I) or non ischemic (NI) heart failure etiology were analyzed. Non-failing (NF) human heart tissue was obtained from the left ventricular free wall of male organ donor hearts rejected for transplantation because of physical incompatibility. Left ventricular tissue obtained from surgery was immediately frozen in liquid nitrogen and stored at -140°C. All harvest and use of human tissue procedures were performed in accordance with the National Institutes of Health, University of Rochester Medical Center and Cincinnati Children's Hospital Medical Center institutional review board guidelines.
Protein analyses
To identify modifications at the protein level, enriched myofibrillar proteins were isolated using F60 buffer (60 mmol/L KCl, 30 mmol/L imidazole, 7.2 mmol/L MgCl2, pH 7.0) with protease/phosphatase inhibitors (Cocktail I and II, Sigma) as described.15 The presence of 40 kD protein was confirmed by SDS-PAGE (12.5% criterion Tris-glycine pre-cast gels, Bio- Rad) followed by Western blots using an anti-c-myc monoclonal antibody (clone 9E10, Roche) and an anti-cMyBP-C rabbit polyclonal antibody raised against the C0-C1 domains.16,17
Evaluation of cMyBP-C phosphorylation
To define cMyBP-C phosphorylation status, myofibrillar proteins were purified from nontransgenic (NTG), 40 kD single TG and DTG mouse hearts, electrophoresed on SDS-PAGE and Western blots performed using phospho-specific cMyBP-C antibodies as described.18
Histochemistry, immunohistochemistry and transmission electron microscopy
For histopathological examinations, beating hearts were removed from anesthetized mice, the blood allowed to drain and the tissues fixed in 10% formalin. Hearts were bisected longitudinally, dehydrated through a graded series of alcohols, and laid open before being paraffin embedded. Step-serial sections (5 μm) were taken from 2-3 hearts per group. Sections were stained with hematoxylin-eosin or Masson's trichrome. Fibrosis, myocyte disarray and calcification were each evaluated by an expert pathologist who was blinded to genotype. Localization and integration of the 40 kD cMyBP-C fragment in the sarcomere was determined by immunohistochemistry with confocal microscopy as described.18
Ultrastructural analyses were performed by electron microscopy as described.14,19 Briefly, mice were anesthetized with isofluorane and the hearts fixed by perfusion with 1% paraformaldehyde and 2% glutaraldehyde in cardioplegic buffer (5% dextrose, 30 mmol/L KCl in PBS) for 2 minutes, followed by treatment with the fixative in 100 mmol/L cacodylate buffer (pH 7.3). The hearts were then excised and subsequently separated into the left ventricle and septum. Each region was then divided into small (1 mm) cubes and fixed in glutaraldehyde-cacodylate fixative overnight at 4°C. The tissue fragments were post-fixed in 1% OsO4 in cacodylate buffer, dehydrated in a series of acetone baths and embedded in a Poly/Bed 812 resin mixture. Thin sections, counterstained with uranium and lead salts, were examined using a Hitachi 7600 transmission electron microscope equipped with an AMT digital camera. Multiple sections were cut from 2-3 mice of mixed gender and >50 fields were observed by a blinded observer.
Cardiac function
To observe changes in left ventricular chamber size and fractional shortening, echocardiography was performed by noninvasive M-mode echocardiography.18
MEK inhibitor treatment
Mice were treated with U0126, a pharmacologic inhibitor of MAPK/ERK kinase that blocks phosphorylation of ERK1/2.20 We injected U0126 i.p. on a daily basis (5 mg per kg body weight per day) into the pregnant dams until delivery and continued the injections during the peri- and neonatal periods to the nursing females up to P9 and then to individual pups until P24 at 2 day intervals. After weaning, individual mice were treated up to 12 weeks of age.
Statistical analyses
All biochemical and functional assays were performed in mice with mixed-gender controls. For comparisons of data from two groups, Student's t test was used. For comparisons of multiple groups, one-way ANOVA with post hoc Tukey's multiple comparisons test were used. Paired data were evaluated by Student's t test. Results are shown as mean ± SE or ±SD. P<0.05 was considered significant.
Results
Inducible, cardiomyocyte-specific expression of the 40 kd fragment
Previously, we found that cleavage of cMyBP-C by μ−χαλπαιν produced a stable 40 kD fragment. This fragment, which either was undetectable or present in very small amounts in normal mouse hearts, was easily detectable in both mouse and human hearts that had been subjected to ischemia reperfusion injury and/or general cardiovascular stress.7 Using isolated systems, we found that this fragment was able to interact efficiently with the other filament systems in the sarcomere and, in fact, could compete effectively with endogenous cMyBP-C for binding to the thick and thin filaments.10,21 On the basis of those data and its presence in hearts collected from human heart failure patients, we wanted to determine if production of the 40 kD fragment was merely a consequence of cardiac disease or whether its expression was sufficient to cause cardiac pathology and dysfunction. It was therefore imperative to be able to rigorously control expression of the 40 kD fragment independent of exogenously induced cardiac stress. We therefore chose to express the 40 kD fragment inducibly using a “tet-off” system.
Before initiating the transgenic studies, we confirmed the levels of the 40 kD fragment in normal and failing human hearts in order to titrate transgenically-driven expression to the approximate levels observed in our human samples. While low levels can be detected in our non-failing samples, the amount of 40 kD fragment seen in hearts suffering from either ischemic or non-ischemic disease is substantially increased (Figure 1A). To investigate the role of cMyBP-C's 40 kD fragment in cardiac function and its intrinsic ability to cause cardiac disease, we generated cardiac-specific TG mice using our inducible transgenic system, which is bi-genic and consists of one line (the “driver”) producing the tetracycline transactivator (tTA) under the control of the α-MHC promoter and another line carrying cDNA encoding the 40 kD fragment under the control of the “responder” promoter, which is only active in the absence of doxycycline (Figure 1B).22 The amino terminus of cMyBP-C binds multiple components of the thick and thin filaments, interacting with actin and myosin, and the 40 kD fragment contains elements of both these binding sites as schematically shown in Figure 1B. When these two TG lines are crossed, and in the absence of tetracycline/doxycycline, the 40 kD fragment is expressed specifically in cardiomyocytes (Figure 1C). The fragment's synthesis had no visible effect on normal protein synthetic patterns (Figure 1C) and normal levels of the intact, wild type cMyBP-C protein appear to be present.
We had previously shown that cMyBP-C function is highly dependent upon its phosphorylation state.7,9,13,18 To determine if 40 kD fragment expression had any effect on the overall phosphorylation status of intact cMyBP-C, we performed Western blotting analyses with phospho-site-specific antibodies that we had developed and used previously, which are able to detect the phosphorylation status of 3 specific residues; Serines 272, 282 and 302 in the amino terminus of cMyBP-C.18 There were no significant changes in phosphorylation observed in any of the mice, ruling out post-translational modulation of the endogenous protein as playing a role in any functional deficits that might be observed (Figure 1D).
Cardiac phenotype and sarcomere architecture
The mice began displaying overt signs of cardiac dysfunction in early adulthood. Removal of hearts from NTG, single TG and the induced, DTG mice at 12 weeks revealed grossly aberrant cardiac morphology in the hearts expressing the 40 kD fragment (Figure 2). Chamber arrangement and overall geometries were grossly perturbed, with both ventricular and atrial enlargement apparent.
Figure 2. Expression of the 40 kD fragment results in altered cardiac geometry.
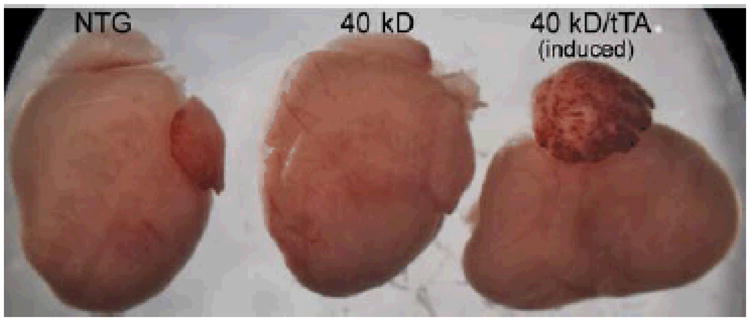
Shown are hearts derived from a 12 week old NTG, single transgenic (carrying the 40 kD transgene) and a double transgenic, induced animal. The aberrant cardiac anatomy is immediately apparent, with grossly distorted chamber geometries as a result of significant remodeling presenting.
To determine whether the 40 kD fragment incorporates normally into the sarcomere, we isolated the myofibrillar proteins from NTG, 40 kD fragment single TG and DTG mice. Western blots were performed with cMyBP-C N-terminal antibody, which detects both the full-length protein and 40 kD fragment (Figure 3A). Quantification showed no significant changes in the levels of full-length MyBP-C expression between NTG, 40 kD fragment single TG, tTA and DTG mice (Figure 3B). Significantly higher (P<0.0001) levels of the 40 kD fragment were detected only in the myofibril protein preparations from DTG hearts (Figure 3C), suggesting that the 40 kD fragment was incorporated into the sarcomere. Some “leakage” was observed in expression of the 40 kD fragment in the single TG carrying that construct (Figure 1B; “40kD”). To confirm localization of the 40 kD fragment, antibodies against cMyBP-C and c-myc were used for immunohistochemistry coupled with confocal microscopy. Results showed that the 40 kD fragment co-localized with cMyBP-C in the DTG hearts, indicating that the fragment was incorporated in a manner indistinguishable from normal, endogenous cMyBP-C at the resolution used (Figure 3D).
Figure 3. Incorporation of 40 kD protein in the sarcomere.
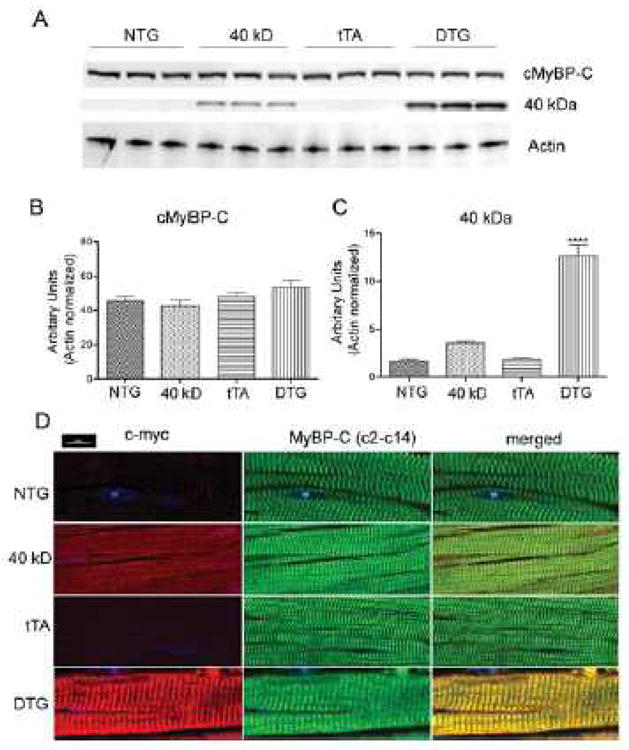
A, Myofibrils from NTG, 40 kD and tTA single TG and DTG mice expressing the 40 kD fragment were isolated, electrophoresed on acrylamide and subsequently transferred for Western blot analyses with cMyBP-C antibody. Actin was used as a loading control. B, Quantification of total, full length MyBP-C. No significant differences were found between the NTG, 40 kD, tTA and DTG groups. C, Quantification of 40 kD protein expression. Values represent mean±SE for each group (n=3). ***P<0.0001 DTG versus NTG, 40 kD, tTA controls. D, Incorporation of myc-tagged 40 kD cMyBP-C was confirmed in NTG, tTA, 40 kD and DTG mice by immunofluorescent staining of cMyBP-C with either an anti-myc (red) or anti cMyBP-C antibody (green).
Myocardial fibrosis, a hallmark of FHC,23 can contribute to sudden cardiac death, ventricular tachyarrhythmia, left ventricular dysfunction, and heart failure.24 We assessed fibrosis at 4, 12 and 16 weeks (Figure 4A, B and C, respectively). Fibrosis was seen as early as 4 weeks in the DTG mice that expressed the 40 kD fragment and progressively increased as the animals aged, with no fibrosis observed in the NTG, 40 kD and tTA TG mice (Figure 4). The heart weight to body weight ratio was also significantly increased (P<0.0001) in the DTG mice as compared to NTG, 40 kD and tTA TG mice (Figure 4D). These anatomical and histological alterations were accompanied by visible changes at the ultrastructural level as well. Disorganized sarcomeres are commonly observed in cMyBP-C-related FHC,19 thus we examined the DTG myocardial ultrastructure at 12 weeks. In the unaffected animals, the sarcomeres were in register with one another, precisely aligned and the mitochondria highly organized (Figure 5). In contrast, sarcomeres derived from the 40 kD DTG mice were consistently out of register, normal pattern organization was significantly disrupted and the overall architecture of the mitochondria severely disrupted (Figure 5). Thus, expression of the 40 kD fragment, even in the presence of wild type levels of normal protein (Figure 3B), resulted in overt cardiac pathology and sarcomeric disruption, indicating that this fragment can act in a dominant negative, “poison peptide” fashion and directly result in cardiac pathology within the whole animal context.
Figure 4. Cardiac phenotype of 40 kD mice.
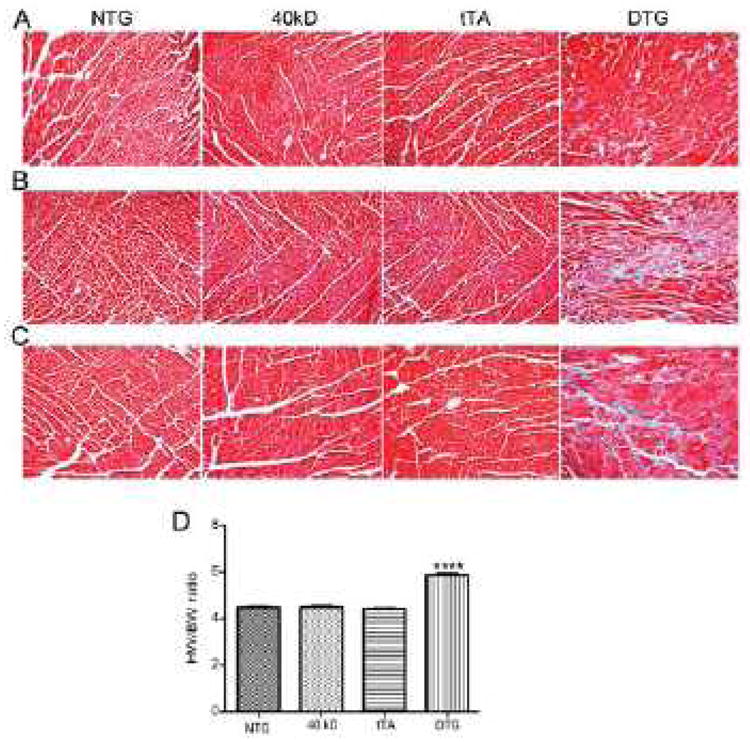
A, Representative heart sections obtained from 4 week old NTG, 40 kD, tTA and DTG mice, B, Sections from 12 week old mice or C, 16 week old mice, were stained with Masson trichrome to assess fibrosis. D, Heart weight to body weight ratios of 12 week old mice. Values represent mean±SE for each group (n=5). ***P<0.0001 DTG versus NTG, 40 kD, tTA controls.
Figure 5. Sarcomeric ultrastructural analyses.
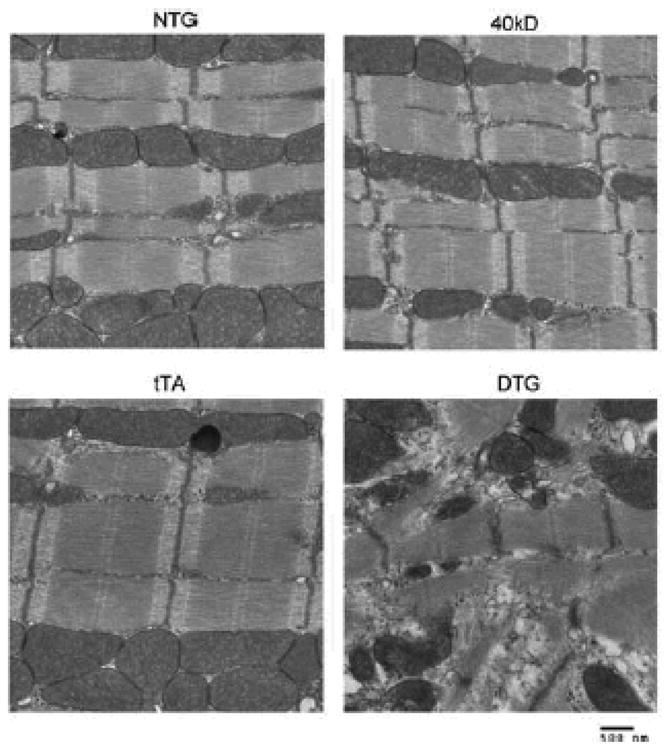
Transmission electron micrographs showing sarcomere ultrastructure of NTG, 40 kD, tTA and DTG mice (10 week old hearts).
MEK-ERK pathways are activated in hearts expressing the 40 kD fragment
Enhanced MEK and ERK activation in cardiomyocytes is strongly implicated in the pathogenesis of FHC.25 Therefore, we wished to determine if the 40 kD fragment-induced pathogenesis resulted in activation of these pathways as well. Upon phosphorylation, MEK becomes activated, and subsequently signals downstream to ERK, eventually resulting in specific transcriptional programs being upregulated.26,27 Hearts were isolated from 5 and 10 week old mice and the phosphorylation status of MEK and ERK determined by Western blot analysis using phospho-specific antibodies. MEK-ERK pathways are highly activated in the 40 kD DTG mice as compared to NTG and 40 kD single TG mice. Moreover, DTG mice displaying obvious signs of heart failure (anasarca, fluid retention, listlessness) showed the highest levels of MEK-ERK activation (Figure 6: HF).
Figure 6. Activation of MEK-ERK signaling pathways in hearts expressing the 40 kD fragment.
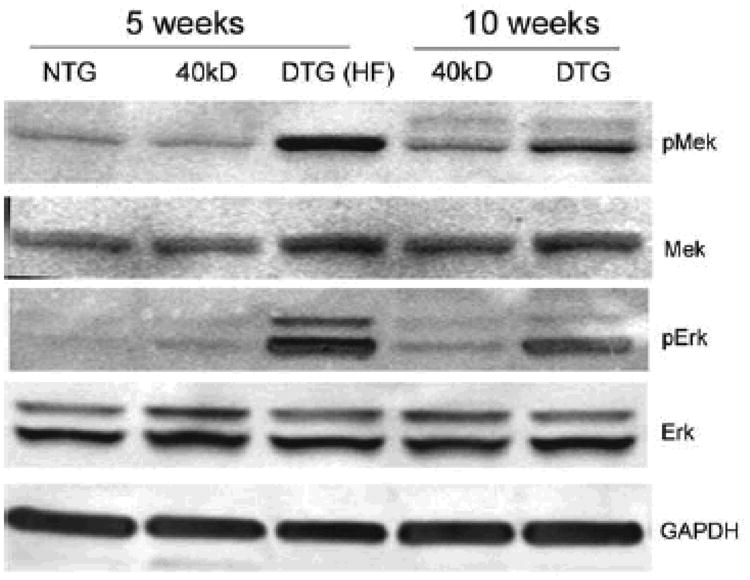
Heart lysates from 5 and 10 week old NTG, 40 kD single and DTG mice were examined for pMEK and pERK by Western blotting. The MEK-ERK pathways are highly activated in the DTG hearts, when compared with the NTG and single TG mice.
We hypothesized that aspects of the pathogenic phenotype being observed were due to upregulation of this pathway. Pathogenicity might therefore be ameliorated by MEK-ERK inhibition. We treated the 40 kD DTG mice with the MEK inhibitor U0126. The untreated 40 kD DTG mice began to die as early as 23 days after birth, with a majority of the mice deceased by 8 weeks. In contrast, only one MEK inhibitor-treated animal died within 8 weeks and there were significant increases in lifespan and probability of survival relative to the untreated cohort (Figure 7A). After 12 weeks, cardiac function was measured by M-mode echocardiography. Fractional shortening (FS) in the untreated DTG mice was significantly decreased when compared to NTG, 40 kD TG mice and tTA mice (P<0.0001). However, the U0126-treated cohort showed significantly higher values (P<0.01), relative to the untreated, induced DTG mice (Figure 7B). Left ventricular mass and conservation of normal diastolic volumes were also significantly improved (P<0.0001) (Figure 7C and D). These results suggest that activation of the MEK-ERK pathway plays a significant role in early mortality, but clearly, while MAPK inhibition can improve overall cardiac function, it cannot rescue the animal in the face of continued production of the 40 KD fragment.
Figure 7. MEK inhibition prolongs survival and improves cardiac function in 40 kD expressing mice.
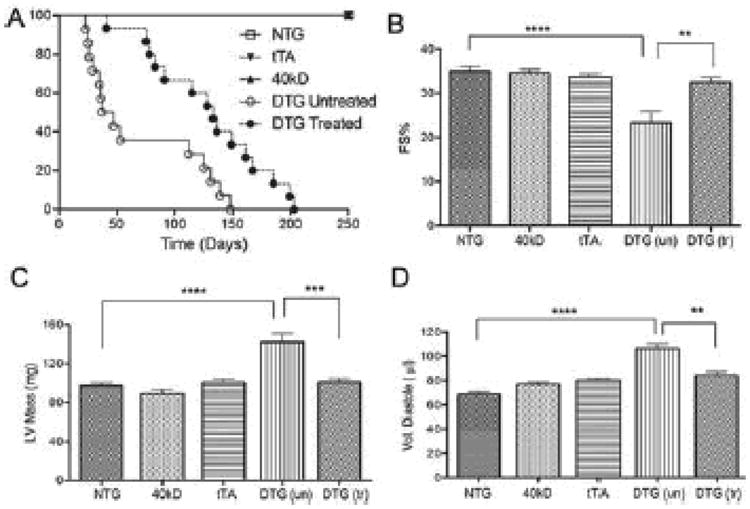
Mice were treated with U0126, a pharmacologic inhibitor of MAPK/ERK kinase 1/2 that blocks phosphorylation of ERK1/2. At 12 weeks of age, cardiac function was assayed by M-Mode echocardiography for the NTG (n=5), 40 kD single TG (n=5), tTA (n=11), untreated DTG (n=5) and treated DTG (n=9). A, Survival curves of DTG (n=16) untreated and U0126 treated (n=15) mice. B, Fractional shortening (FS). C, Left ventricular mass (LV mass) D, Volume of diastole. Values represent mean±SE for each group. ***P<0.001 DTG untreated versus NTG, 40 kD, tTA controls **P<0.01 treated DTG versus untreated DTG. ****P<0.0001.
Discussion
This report significantly extends earlier data concerning the origin and function of an endogenously produced peptide fragment derived from cMyBP-C. The 40 kD fragment, derived from the N-terminus of cMyBP-C, contains the C0 domain (99 residues), a Pro/Ala rich region (51 residues), the C1 domain (104 residues) and 17 residues of the M domain (Figure 1B). Previously we found that the 40 kD fragment is released via cleavage at a μ-calpain site and that steady state levels were increased after cardiac stress such as surgically induced pressure overload or ischemia-reperfusion injury. We also found high levels of the fragment in a number of our genetically induced models of cardiac disease (data not shown) and recently, the fragment was detected in the circulatory systems of rodents and humans after acute myocardial infarction.7,8 A recent report showed that the presence of the 40 kD fragment correlated with increased cytotoxicity, impairment of Ca2+ handling and altered contractile function of the sarcomere in vitro while, in contrast, a fragment derived from the carboxyl terminus of cMyBP-C had no detrimental effects.28 In this manuscript we detail for the first time the results of dosage-controlled expression in vivo, in the cell type in which the fragment accumulates in human disease. We show that at high doses the fragment is sufficient to cause cardiac disease, heart failure and premature death. Significantly, in some human heart failure patients, we can detect significant levels of the fragment in both ischemic and non-ischemic conditions (Figure 1A), raising the possibility that the 40 kD fragment may play a role in the natural history of human cardiac disease
It should be noted that under normal conditions in the mouse, in our hands, only about 80% of the CMyBP-C is fully phosphorylated, holding open the possibility that low levels of the fragment might accumulate at baseline with no detectable, ill effects. This appears to be the case in the human as well: the non-diseased samples contain detectable amounts of the fragment (Figure 1A). Consistent with these data is the observation that, in our mouse model in which transgene expression is not induced but there is a slight amount of “leaky” expression (Figure 3A), we detected no pathology. The data are consistent with a dose-dependent, pathogenic effect for the fragment that comes about as a consequence of its ability to interfere with normal cMyBP-C-thick/thin filament interactions.10 Consistent with the fragment's specificity for causing the observed pathogenic effects, when we turned off expression of the 40kDa fragment by giving doxycycline post-birth, we did not see any heart enlargement or sign of heart failure in those animals (data not shown).
Considering our data, and particularly elevated levels of the 40 kD fragment in cardiac samples derived from patients with heart failure, we thought it would be important to determine its potential pathogenicity. Is the 40 kD fragment sufficient to cause cardiac disease or even failure when expressed in a cardiomyocyte-specific fashion in vivo? When very low levels of the 40 kD fragment were expressed, that is, when the transgene was present but uninduced, no pathology presented. However, upon induction of 40 kD transgene expression to levels approaching those detected in stressed hearts (Figure 1A), we observed sarcomere dysgenesis, altered cardiac geometry and cardiac failure between 3 to 17 weeks after birth. The mice also developed significant cardiac hypertrophy with myofibrillar disarray and fibrosis. These symptoms developed despite the presence of normal amounts of intact cMyBP-C protein, indicating that, in vivo, accumulation of the 40 kD fragment results in a dominant, pathogenic effect. These data are consistent with more recent studies carried out in vitro by Sadayappan's group.28 In those studies, using rabbit cardiomyocytes and adenovirus infection, the pathogenic properties of the 40 kD fragment in vitro were confirmed while fragments derived from other portions of cMyBP-C stably expressed in rabbit cardiomyocytes were benign.
We next addressed the question of mechanism: where is this dominant effect concentrated? The N-terminal 40 kD region of the cMyBP-C is highly conserved across species29 and its generation is pathogenic in mammalian hearts. Reductionist approaches have confirmed its ability to impact crossbridge kinetics and effectively compete with intact cMyBP-C for actin and myosin binding,10,21 results that are consistent with our data demonstrating its correct placement in the sarcomere (Figure 3). Bacterially expressed 40 kD fragment effectively inhibited actin translocation over a surface of monomeric mouse cardiac myosin in an in vitro motility assay.21 The 40 kD fragment also stereospecifically and reversibly bound to actin,10,30 effectively serving as a brake or drag to slow myosin kinetics.10 However, we have not ruled out the possibility that the 40 kD fragment might affect thick-thin filament kinetics by also binding directly to myosin.31-33
The direct effect of the fragment's placement on sarcomere function is supported by the effect on structure (Figure 5), where altered sarcomeric architecture is readily apparent. It is worth noting that expression of the fragment had no effects on the steady state levels of full-length cMyBP-C, implying that the 40 kD fragment exerts its effect in a dominant fashion, consistent with a proposed pathogenic role in general cardiac disease, even when cMyBP-C levels are unaffected. All of the histologic and functional data are consistent with the hypothesis that the pathogenesis of the 40 kD fragment stems directly from its intercalation into the sarcomere and interference with normal thick-thin filament movement and/or kinetics. This results in altered cardiac output, cardiomyocyte remodeling and dropout, and subsequent fibrosis, all of which are observed in the 40 kD fragment model. At this point, we believe that the observed downstream effects have their primary source in altered filament mechanics. Currently, we have neither data nor evidence that the fragment can effectively signal outside of the sarcomere in terms of its ability to activate/inhibit other protective or pathogenic mechanisms.
Enhanced MEK and ERK activation in cardiomyocytes often accompanies development of cardiac pathology in FHC,25 but there is a paucity of data with respect to altered MEK-ERK pathway contribution to cMyBP-C related pathology. In the 40 kD fragment mice we defined significant activation of this pathway (Figure 5). As depletion of ERK1/2 with antisense oligonucleotides or pharmacological inhibition of MEK1/2 attenuates the hypertrophic response to agonist stimulation in cultured cardiomyocytes,34,35 we asked whether down regulation of the pathway's activity might be sufficient to ameliorate overall pathology and even prolong life. Treatment with the MEK inhibitor U0126 significantly increased life span and substantially improved heart function (Figure 7). These data provide evidence for a critical role of the MEK-ERK pathway in augmenting the development of cardiac disease in the 40 kD mice, although they clearly do not rule out the role of other pathways in the pathogenesis of the disease resulting from sarcomeric dysfunction. We hypothesize that, in the face of continued alterations of cardiac hemodynamics, multiple signaling pathways are altered that contribute to the overall pathogenicity of the secondary response. In this study, MEK inhibitor was administered during the peri- and neonatal periods before any detectable structural or functional cardiac abnormalities. A critical question to be addressed is whether MEK inhibitor would be effective in improving heart function in 40 kD fragment mice when initiated after the onset of cardiac disease, which would be more analogous to the potential for impacting the natural history of the disease in patient populations.
While this study shows that relatively high levels of the 40 kD fragment are sufficient to cause cardiac failure and death, its role in the natural history of cMyBP-C-induced human cardiomyopathy and cardiac disease is not yet defined. Clearly, in some human hearts in which the fragment is present, the levels of intact cMyBP-C are dramatically decreased (Figure 1A, ischemic hearts) and cMyBP-C haploinsufficiency could, by itself, cause increased pathology. At the same time, in the non-ischemic hearts in Figure 1A, although cMyBP-C levels are somewhat reduced, there are still appreciable amounts. In the mouse hearts, expression of the 40 kD fragment in the face of normal amounts of cMyBP-C still causes disease and premature death, leading us to hypothesize that, in the human, appreciable amounts of this fragment will impact negatively on the natural history of pre-existing cardiac disease.
Novelty and Significance.
What Is Known?
A small fragment derived from the N-terminal region of cMyBP-C is endogenously produced in stressed hearts and appears to be stable.
This fragment is capable of interacting with the contractile apparatus in isolated systems in vitro and can impact on thick-thin filament mechanics.
Levels of this fragment appear to be elevated in failing human hearts and in mouse models of cardiac disease
What New Information Does This Article Contribute?
Transgenic mice that express significant levels of the fragment specifically in cardiomyocytes develop heart disease and die prematurely, showing that, in vivo, the presence of significant levels of the fragment is sufficient to cause cardiac disease and death.
-
Expression of the fragment, even when normal amounts of wild type cMyBP-C are present, still leads to heart failure and death within 15 weeks, showing that the peptide acts in a dominant fashion.
The natural history of the disease is characterized by dramatic remodeling of the gross cardiac architecture and activation of pathogenic hypertrophic pathways.
These findings suggest that, as the fragment can be found in human hearts suffering from ischemic and non-ischemic disease, it may be a contributing factor in the development of decompensated heart failure in human cardiac disease.
This study was designed to extend data obtained in isolated systems and in cell culture to the whole heart and whole animal contexts. This N terminal fragment is stable and can accumulate to relatively high levels (20-30% of the endogenous, intact cMyBP-C). Studies using isolated thick and thin filament proteins show that the fragment influences contractile filament interactions, raising the possibility that it might impact on cardiac hemodynamics. But its role in the intact heart remained unknown. We have now defined a pathogenic role for this fragment in vivo. Using cardiomyocyte-specific transgenic expression in the mouse, we show that detectable accumulations of the fragment are sufficient to cause cardiac disease, leading to heart failure and premature death by 12-15 weeks, even when normal amounts of cMyBP-C remain. The fragment associates with the sarcomeres in a location that is consistent with it playing a dominant role in influencing normal thick and thin filament interactions. These data, along with the detection of appreciable amounts of the fragment in hearts derived from human heart failure patients, point to a potential role for this fragment in the pathogenic natural history of human heart failure.
Acknowledgments
Sources Of Funding: This work was supported by NIH grants P01HL077101, P01HL049058, R01HL105924 and The Transatlantic Network of Excellence Program grant from Le Fondation Leducq (to JR). MAR and MG are or were postdoctoral fellows of the American Heart Association (Great Rivers Affiliate).
Nonstandard Abbreviations and Acronyms
- cMyBP-C
cardiac myosin binding protein C
- TG
transgenic
- NTG
non transgenic
- tTA
tetracycline transactivator
- FHC
familial hypertrophic cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- DTG
double transgenic
- MAPK
mitogen-activated protein kinase
- MHC
α myosin heavy chain
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Offer G, Moos C, Starr R. A New Protein Of The Thick Filaments Of Vertebrate Skeletal Myofibrils. Extractions, Purification And Characterization. J Mol Biol. 1973;74:653–676. doi: 10.1016/0022-2836(73)90055-7. [DOI] [PubMed] [Google Scholar]
- 2.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg Hp, Fiszman M, Komajda M, Schwartz K. Cardiac Myosin Binding Protein-C Gene Splice Acceptor Site Mutation Is Associated With Familial Hypertrophic Cardiomyopathy. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 3.Watkins H, Conner D, Thierfelder L, Jarcho Ja, Macrae C, Mckenna Wj, Maron Bj, Seidman Jg, Seidman Ce. Mutations In The Cardiac Myosin Binding Protein-C Gene On Chromosome 11 Cause Familial Hypertrophic Cardiomyopathy. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 4.Winegrad S. Cardiac Myosin Binding Protein C: Modulator Of Contractility. Adv Exp Med Biol. 2005;565:269–281. doi: 10.1007/0-387-24990-7_20. Discussion 281-262, 405-215. [DOI] [PubMed] [Google Scholar]
- 5.Chang An, Potter Jd. Sarcomeric Protein Mutations In Dilated Cardiomyopathy. Heart Fail Rev. 2005;10:225–235. doi: 10.1007/s10741-005-5252-6. [DOI] [PubMed] [Google Scholar]
- 6.Tardiff Jc. Sarcomeric Proteins And Familial Hypertrophic Cardiomyopathy: Linking Mutations In Structural Proteins To Complex Cardiovascular Phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 7.Sadayappan S, Osinska H, Klevitsky R, Lorenz Jn, Sargent M, Molkentin Jd, Seidman Ce, Seidman Jg, Robbins J. Cardiac Myosin Binding Protein C Phosphorylation Is Cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Govindan S, Mcelligott A, Muthusamy S, Nair N, Barefield D, Martin Jl, Gongora E, Greis Kd, Luther Pk, Winegrad S, Henderson Kk, Sadayappan S. Cardiac Myosin Binding Protein-C Is A Potential Diagnostic Biomarker For Myocardial Infarction. J Mol Cell Cardiol. 2012;52:154–164. doi: 10.1016/j.yjmcc.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadayappan S, Gulick J, Klevitsky R, Lorenz Jn, Sargent M, Molkentin Jd, Robbins J. Cardiac Myosin Binding Protein-C Phosphorylation In A {Beta}-Myosin Heavy Chain Background. Circulation. 2009;119:1253–1262. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weith A, Sadayappan S, Gulick J, Previs Mj, Vanburen P, Robbins J, Warshaw Dm. Unique Single Molecule Binding Of Cardiac Myosin Binding Protein-C To Actin And Phosphorylation-Dependent Inhibition Of Actomyosin Motility Requires 17 Amino Acids Of The Motif Domain. J Mol Cell Cardiol. 2012;52:219–227. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulick J, Robbins J. Regulation Of Transgene Expression Using Tetracycline. Curr Protoc Mol Biol. 2005;Chapter 23 doi: 10.1002/0471142727.mb2312s71. Unit 23 12. [DOI] [PubMed] [Google Scholar]
- 12.Sanbe A, Gulick J, Hanks Mc, Liang Q, Osinska H, Robbins J. Reengineering Inducible Cardiac-Specific Transgenesis With An Attenuated Myosin Heavy Chain Promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 13.Sadayappan S, Gulick J, Osinska H, Martin La, Hahn Hs, Dorn GW, 2nd, Klevitsky R, Seidman Ce, Seidman Jg, Robbins J. Cardiac Myosin-Binding Protein-C Phosphorylation And Cardiac Function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Q, Sanbe A, Osinska H, Hewett Te, Klevitsky R, Robbins J. In Vivo Modeling Of Myosin Binding Protein C Familial Hypertrophic Cardiomyopathy. Circ Res. 1999;85:841–847. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
- 15.Fewell Jg, Hewett Te, Sanbe A, Klevitsky R, Hayes E, Warshaw D, Maughan D, Robbins J. Functional Significance Of Cardiac Myosin Essential Light Chain Isoform Switching In Transgenic Mice. J Clin Invest. 1998;101:2630–2639. doi: 10.1172/JCI2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decker Rs, Decker Ml, Kulikovskaya I, Nakamura S, Lee Dc, Harris K, Klocke Fj, Winegrad S. Myosin-Binding Protein C Phosphorylation, Myofibril Structure, And Contractile Function During Low-Flow Ischemia. Circulation. 2005;111:906–912. doi: 10.1161/01.CIR.0000155609.95618.75. [DOI] [PubMed] [Google Scholar]
- 17.Mcclellan G, Kulikovskaya I, Winegrad S. Changes In Cardiac Contractility Related To Calcium-Mediated Changes In Phosphorylation Of Myosin-Binding Protein C. Biophys J. 2001;81:1083–1092. doi: 10.1016/S0006-3495(01)75765-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko Vm, Lorenz Jn, Maillet M, Martin Jl, Brown Jh, Bers Dm, Molkentin Jd, James J, Robbins J. A Critical Function For Ser-282 In Cardiac Myosin Binding Protein-C Phosphorylation And Cardiac Function. Circ Res. 2011;109:141–150. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Q, Sanbe A, Osinska H, Hewett Te, Klevitsky R, Robbins J. A Mouse Model Of Myosin Binding Protein C Human Familial Hypertrophic Cardiomyopathy. J Clin Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura T, Colbert M, Krenz M, Molkentin Jd, Hahn Hs, Dorn Gw, 2nd, Robbins J. Mediating Erk 1/2 Signaling Rescues Congenital Heart Defects In A Mouse Model Of Noonan Syndrome. J Clin Invest. 2007;117:2123–2132. doi: 10.1172/JCI30756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Previs Mj, Beck Previs S, Gulick J, Robbins J, Warshaw Dm. Molecular Mechanics Of Cardiac Myosin-Binding Protein C In Native Thick Filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe Cg, Kayed R, Robbins J. Reversal Of Amyloid-Induced Heart Disease In Desmin-Related Cardiomyopathy. Proc Natl Acad Sci U S A. 2005;102:13592–13597. doi: 10.1073/pnas.0503324102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho Cy, Lopez B, Coelho-Filho Or, Lakdawala Nk, Cirino Al, Jarolim P, Kwong R, Gonzalez A, Colan Sd, Seidman Jg, Diez J, Seidman Ce. Myocardial Fibrosis As An Early Manifestation Of Hypertrophic Cardiomyopathy. N Engl J Med. 2010;363:552–563. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varnava Am, Elliott Pm, Sharma S, Mckenna Wj, Davies Mj. Hypertrophic Cardiomyopathy: The Interrelation Of Disarray, Fibrosis, And Small Vessel Disease. Heart. 2000;84:476–482. doi: 10.1136/heart.84.5.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y. Mitogen-Activated Protein Kinases In Heart Development And Diseases. Circulation. 2007;116:1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramaniam S, Unsicker K. Extracellular Signal-Regulated Kinase As An Inducer Of Non-Apoptotic Neuronal Death. Neuroscience. 2006;138:1055–1065. doi: 10.1016/j.neuroscience.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Wada T, Penninger Jm. Mitogen-Activated Protein Kinases In Apoptosis Regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 28.Govindan S, Sarkey J, Ji X, Sundaresan Nr, Gupta Mp, De Tombe Pp, Sadayappan S. Pathogenic Properties Of The N-Terminal Region Of Cardiac Myosin Binding Protein-C In Vitro. J Muscle Res Cell Motil. 2012;33:17–30. doi: 10.1007/s10974-012-9292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaffer Jf, Wong P, Bezold Kl, Harris Sp. Functional Differences Between The N-Terminal Domains Of Mouse And Human Myosin Binding Protein-C. J Biomed Biotechnol. 2010;2010:789798. doi: 10.1155/2010/789798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mun Jy, Gulick J, Robbins J, Woodhead J, Lehman W, Craig R. Electron Microscopy And 3d Reconstruction Of F-Actin Decorated With Cardiac Myosin-Binding Protein C (Cmybp-C) J Mol Biol. 2011;410:214–225. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofmann Pa, Greaser Ml, Moss Rl. C-Protein Limits Shortening Velocity Of Rabbit Skeletal Muscle Fibres At Low Levels Of Ca2+ Activation. J Physiol. 1991;439:701–715. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure And Interactions Of Myosin-Binding Protein C Domain C0: Cardiac-Specific Regulation Of Myosin At Its Neck? J Biol Chem. 2011;286:12650–12658. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Razumova Mv, Shaffer Jf, Tu Ay, Flint Gv, Regnier M, Harris Sp. Effects Of The N-Terminal Domains Of Myosin Binding Protein-C In An In Vitro Motility Assay: Evidence For Long-Lived Cross-Bridges. J Biol Chem. 2006;281:35846–35854. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- 34.Clerk A, Michael A, Sugden Ph. Stimulation Of The P38 Mitogen-Activated Protein Kinase Pathway In Neonatal Rat Ventricular Myocytes By The G Protein-Coupled Receptor Agonists, Endothelin-1 And Phenylephrine: A Role In Cardiac Myocyte Hypertrophy? J Cell Biol. 1998;142:523–535. doi: 10.1083/jcb.142.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glennon Pe, Kaddoura S, Sale Em, Sale Gj, Fuller Sj, Sugden Ph. Depletion Of Mitogen-Activated Protein Kinase Using An Antisense Oligodeoxynucleotide Approach Downregulates The Phenylephrine-Induced Hypertrophic Response In Rat Cardiac Myocytes. Circ Res. 1996;78:954–961. doi: 10.1161/01.res.78.6.954. [DOI] [PubMed] [Google Scholar]