

Abstract
Background
We utilized genetically modified mice equipped with a variable capacity to scavenge mitochondrial and cellular reactive oxygen species to investigate the pathological significance of oxidative stress in Chagas disease.
Methods and Results
C57BL/6 mice (wild type, MnSODtg, MnSOD+/−, GPx1−/−) were infected with Trypanosoma cruzi and harvested during the chronic disease phase. Chronically infected mice exhibited a substantial increase in plasma levels of inflammatory markers (nitric oxide, myeloperoxidase), lactate dehydrogenase, and myocardial levels of inflammatory infiltrate and oxidative adducts (malondialdehyde, carbonyls, 3‐nitrotyrosine) in the order of wild type=MnSOD+/−>GPx1−/−>MnSODtg. Myocardial mitochondrial damage was pronounced and associated with a >50% decline in mitochondrial DNA content in chronically infected wild‐type and GPx1−/− mice. Imaging of intact heart for cardiomyocytes and collagen by the nonlinear optical microscopy techniques of multiphoton fluorescence/second harmonic generation showed a significant increase in collagen (>10‐fold) in chronically infected wild‐type mice, whereas GPx1−/− mice exhibited a basal increase in collagen that did not change during the chronic phase. Chronically infected MnSODtg mice exhibited a marginal decline in mitochondrial DNA content and no changes in collagen signal in the myocardium. P47phox−/− mice lacking phagocyte‐generated reactive oxygen species sustained a low level of myocardial oxidative stress and mitochondrial DNA damage in response to Trypanosoma cruzi infection. Yet chronically infected p47phox−/− mice exhibited increase in myocardial inflammatory and remodeling responses, similar to that noted in chronically infected wild‐type mice.
Conclusions
Inhibition of oxidative burst of phagocytes was not sufficient to prevent pathological cardiac remodeling in Chagas disease. Instead, enhancing the mitochondrial reactive oxygen species scavenging capacity was beneficial in controlling the inflammatory and oxidative pathology and the cardiac remodeling responses that are hallmarks of chronic Chagas disease.
Keywords: cardiac remodeling; Chagas disease; mice (MnSODtg, GPx1−/−, P47phox−/−); multiphoton microscopy; oxidative stress; second harmonic generation microscopy; Trypanosoma cruzi
Introduction
Chagas disease is caused by the protozoan Trypanosoma cruzi and represents the third‐greatest tropical disease burden globally. In recent years the zoonotic presence of parasites, increased population mobility, and transmission through blood transfusion, congenital infection, and organ transplantation has increased the human cases of Chagas in the United States. Infected individuals exhibit an acute phase of peak blood and tissue parasitemia that is resolved in 2 to 3 months; however, the majority of seropositive individuals remain clinically asymptomatic throughout their lives. In ~30% to 40% of infected individuals, myocarditis evolves to cardiomyopathy with a varying extent of cardiac inflammation, tissue fibrosis, ventricular dilation, and contractile dysfunction, leading to heart failure.1–2
Several researchers have investigated the significance of myocardial inflammation in the pathogenesis of Chagas disease by using murine models in which genes or function of inflammatory mediators has been disrupted. These include mice deficient in interferon‐ γ (IFN‐γ), tumor necrosis factor–α (TNF‐α), TNF receptor, and CD4+ and/or CD8+ T cells (reviewed in references 2–3 and 2–3). Overall observation from these studies was that despite a general increase in parasite burden, the extent of cell death and tissue damage was diminished in mice deficient in inflammatory mediators compared with the wild‐type controls. These studies led to a general acceptance that persistence of inflammatory infiltrate contributes to chronic pathology of the heart, although no universal mechanism supporting activation of inflammatory responses in chronic Chagas disease has yet been proposed.
We have shown that experimental animals and humans infected by T cruzi exhibit mitochondrial dysfunction of the respiratory chain and increased formation of superoxide (O2•−) and reactive oxygen species (ROS) in the heart.4–5 Several observations that we and others have made allow us to propose that chronic persistence of inflammation and evolution of cardiomyopathy is an outcome of how the host handles oxidative stress and ROS‐induced events. First, studies in experimental models (mice, rats) and humans indicated that mitochondria damage occurred during T cruzi infection and was associated with chronic oxidative stress in the heart (reviewed in references 2,6–7 and 2,6–7). Second, glutathione, glutathione peroxidase (GPx), and Mn2+ superoxide dismutase (MnSOD), the critical antioxidants in cardiomyocytes, were either suppressed or nonresponsive in the context of chronic Chagas disease,8–10 and because of this, oxidative adducts were enhanced in cardiac biopsies of experimental animals and patients infected by T cruzi.5,11–12 Third, treatment of T cruzi–infected rodents with phenyl‐α‐tert‐butyl nitrone, a spin‐trapping antioxidant, improved cardiac hemodynamics, whereas treatment with benzonidazole (antiparasite) alone was not effective in improving cardiac function.11,13
To conclusively establish whether mitochondrial ROS plays a pathological role in Chagas disease, in this study, we have used MnSODtg mice overexpressing MnSOD in the myocardium 14 with matched wild‐type mice. MnSOD converts O2•− to H2O2 and O2 within the mitochondrial matrix and intermembrane space.15 We included GPx1−/− mice lacking GPx1, a key cellular antioxidant that uses glutathione to reduce H2O2 and lipid peroxides,16 as controls. To examine the contribution of ROS generated by phagocytic cells, we included p47phox−/− mice in the study.17 P47phox is a cytosolic subunit of the NADPH oxidase complex. The assembly and activation of NADPH oxidase result in an oxidative burst, shown to be activated in response to T cruzi infection.18 Mice were infected by T cruzi, and we investigated oxidative and inflammatory stress, mitochondrial and tissue integrity, and cardiac pathology. Further, we have used cutting‐edge 3‐dimensional imaging by multiphoton fluorescence (MPF)/second harmonic generation (SHG) to assess microarchitectural changes in chronically infected hearts. MPF/SHG can provide morphometric assessment of tissues based on intrinsic emission signals (autofluorescence from cellular and extracellular components) and SHG from noncentrosymmetric macromolecules such as fibrillar collagen19 and so far has been employed to image isolated cardiomyocytes20 and heart valve leaflets21 and recently to monitor stem cell–treated heart.22 We present data on MPF/SHG analysis of intact label‐free hearts from control and infected mice to visualize effects on heart tissue on the basis of cardiomyocyte autofluorescence and collagen SHG with quantitative analysis of SHG as an evaluation of fibrosis.
Methods
Parasites and Mice
Trypanosoma cruzi trypomastigotes (SylvioX10/4) were propagated by in vitro passage in C2C12 cells. C57BL/6 mice (wild type and p47phox−/−) were purchased from Jackson Laboratory. MnSODtg, MnSOD+/−, and GPx1−/− mice (C57BL/6 background) were kindly provided by Dr. H. Van Rammen and previously described.23–25 Mice (3 to 6 weeks old) were intraperitoneally infected (10 000 trypomastigotes/mouse), and tissues were harvested at ~120 days postinfection, corresponding to the chronic disease phase. Experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Molecular‐grade chemicals (Sigma‐Aldrich) were used.
Myeloperoxidase
Samples (10 μg of protein) were added in triplicate to 0.53 mmol/L o‐dianisidine dihydrochloride and 0.15 mmol/L H2O2 in 50 mmol/L potassium phosphate buffer (pH, 6.0). Reaction was stopped after 5 minutes, and absorbance was measured at 460 nm on a SpectraMax 190 microplate reader (Molecular Devices). One unit of myeloperoxidase (MPO) was defined as that degrading 1 nmol H2O2/min (ε=11 300M−1.cm−1).26
Nitrate/Nitrite Level
Plasma samples (10 μg of protein) were treated with 0.01 unit per 100 μL of nitrite reductase and incubated for 10 minutes with 100 μL of Griess reagent (1% sulfanilamide, 5% phosphoric acid, and 0.1% N‐[1‐napthyl] ethylenediamine dihydrochloride). Formation of diazonium salt was monitored at 545 nm (standard curve, 2 to 50 μmol/L NaNO3).27
Lactate Dehydrogenase
Plasma levels of lactate dehydrogenase (LDH) activity were measured by using a 2‐step kit (Cayman). Briefly, LDH catalyzed reduction of NAD+ to NADH by oxidation of lactate to pyruvate was coupled with reduction of tetrazolium salt to blue formazan crystals, and absorbance was monitored at 490 to 520 nm (standard curve, 0 to 1000 μU LDH).
Oxidative Stress
Tissues were homogenized in lysis buffer (50 mmol/L Tris [pH, 7.5], 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Nonidet P‐40, 2.5 mmol/L KH2PO4, and protease inhibitor cocktail, 1:10 [w/v]) and centrifuged at 3000g to remove cell debris. Tissue homogenates or plasma (5 μg of protein) was resolved on 10% acrylamide gels and transferred to polyvinylidene fluoride membrane by using a vertical Criterion Blotter (Bio‐Rad). Membranes were blocked with 5% BSA and incubated overnight at 4°C with antibodies against malondialdehyde (MDA) or 3‐nitrotyrosine (3‐NT; 1:100 dilution; Santa Cruz). Signal was developed using an HRP‐based enhanced chemiluminescence detection system (GE Healthcare) and visualized and quantitated by densitometry using a FluorChem 8800 Image Analyzer (Alpha Innotech). Membranes were stained with Sypro Ruby (Invitrogen) before blocking to confirm equal loading of samples.
For detection of protein carbonyls, samples were incubated for 25 minutes with 3% SDS and 5 mmol/L 2,4‐dinitrophenylhydrazine in 10% trifluoroacetic acid (1:1 [v/v]), neutralized with 2 mol/L Tris containing 30% glycerol, resolved on gels, and transferred to membranes. DNP‐derivatized carbonyl proteins were probed for 1 hour with rabbit anti‐DNP antibody (1:300 dilution; Invitrogen), and signal was developed as above.
Histology
Tissues were fixed in 10% formalin, dehydrated, cleared, and embedded in paraffin. Tissue sections (5 μm) were stained with hematoxylin and eosin and evaluated under light microscopy. Slides (10 sections/mouse) were scored for myocarditis as 0 (absent), 1 (focal/mild, ≤1 foci), 2 (moderate, ≥2 inflammatory foci), 3 (extensive coalescing of inflammatory foci or disseminated inflammation), or 4 (diffused inflammation, tissue necrosis, interstitial edema, and loss of integrity). Parasitic pseudocysts were scored as 0 (absent), 1 (0 to 1 foci), 2 (1 to 5 foci), or 3 (>5 foci).18 Cardiac hypertrophic morphology was determined by measuring the transnuclear width of >35 myocytes/section using a UTHSC Image Tool.28
Tissue Parasite Burden
Heart tissues (10 mg) were subjected to Proteinase‐K lysis, and total DNA was purified by the phenol/chloroform extraction/ethanol precipitation method. Total DNA (100 ng) was submitted to a real‐time PCR reaction on an iCycler thermal cycler with SYBR Green Supermix (Bio‐Rad) and Tc18SrDNA‐specific primers. Fold change was calculated by 2−ΔCt, where ΔCt represents the difference between the Ct value of the test and control samples. Data were normalized with host‐specific GAPDH sequence.29
Cytokine Gene Expression
Total RNA from heart tissue sections (10 mg/sample) was extracted and cDNA was synthesized using RNeasy (Qiagen) and iScript (Bio‐Rad) kits, respectively. First‐strand cDNA was used as a template during real‐time PCR with oligonucleotide pairs to amplify mRNA for interleukin (IL)–1β, TNF‐α, IFN‐γ, IL‐10, and TGF‐β cytokines. Relative expression of each target gene was calculated as above. All oligonucleotides used are listed in Table.
Table 1.
Oligonucleotides Used in This Study
| Gene Name | Protein Name | Genbank Accession # | Oligonucleotide Name | Oligonucleotide Sequence 5′‐3′ | Amplicon Size (bp) |
|---|---|---|---|---|---|
| mRNA amplification | |||||
| IL‐1β | Interleukin‐1β | NC_000068.7 | IL‐1β F | GAGCTTCAGGCAGGCAG | 459 |
| IL‐1β R | GGGATCCACACTCTCCAGC | ||||
| IFN‐γ | Interferon‐γ | NC_000076.6 | IFN‐γ F | CATTGAAAGCCTAGAAAGTCTG | 201 |
| IFN‐γ R | CTCATGAATGCATCCTTTTTCG | ||||
| TNF‐α | Tumor necrosis factor–α | NC_000083.6 | TNF‐α F | GTTCTATGGCCCAGACCCTCACA | 836 |
| TNF‐α R | TACCAGGGTTTGAGCTCAGC | ||||
| IL‐10 | Interleukin‐10 | NC_000067.6 | IL‐10 F | GCTCTTACTGACTGGCATGAG | 105 |
| IL‐10 R | CGCAGCTCTAGGAGCATGTG | ||||
| TGF‐β | Tumor growth factor–β | NM_011577.1 | TGF‐β F | ATACAGGGCTTTCGATTCAG | 242 |
| TGF‐β R | CAGCAGTTCTTCTCTGTGGA | ||||
| GAPDH | Glyceraldehyde 3‐phosphate | NC_000067.6 | GAPDH F | TGGCAAAGTGGAGATTGTTG | 402 |
| GAPDH R | TTCAGCTCTGGGATGACCTT | ||||
| DNA amplification | |||||
| mtCO II | Cytochrome oxidase subunit II, mitochondria | NC_012387 | CO II F | ATTGCCCTCCCCTCTCTACGCA | 100 |
| CO II R | CGTAGCTTCAGTATCATTGGTGCCC | ||||
| mtCyt b | Cytochrome b, mitochondria | NC_010339.1 | Cyt b F | GCAACCTTGACCCGATTCTTCGC | 71 |
| Cyt b R | TGAACGATTGCTAGGGCCGCG | ||||
| β‐Globin | Beta globin, nuclear | NC_000073.6 | β‐Globin F | AGCCACAGATCCTATTGCCATGC | 239 |
| β‐Globin R | TGTTGCTTGGTAAACACAGA | ||||
| Tc18SrDNA | Trypanosoma cruzi 18S ribosomal DNA | NC_018331.1 | Tc18SrDNA F | TTTT GGGC AACA GCAG GTCT | 200 |
| Tc18SrDNA R | CTGC GCCT ACGA GACA TTCC | ||||
Electron Microscopy
Heart tissue sections were fixed in Ito's fixative (1.25% formaldehyde, 2.5% glutaraldehyde, 0.03% CaCl2, and 0.03% trinitrophenol in 0.05 mol/L cacodylate buffer [pH, 7.3]) for 1 hour at room temperature and overnight at 4°C. Samples were postfixed for 1 hour in 1% osmium tetraoxide and en bloc stained with 1% uranyl acetate in 0.1 mol/L maleate buffer. After dehydration in a graded series of ethanol, tissues were embedded in Poly/Bed 812 (Polysciences). Ultrathin sections cut on a Sorvall MT‐6000 ultramicrotome were stained with uranyl acetate and lead citrate and examined on a Philips 201 transmission electron microscope at 60 kV.30
Mitochondrial DNA Content
Total DNA (20 ng) isolated from heart tissue sections was used as a template, and real‐time PCR was performed using gene‐specific primers amplifying cytochrome b and cytochrome oxidase II regions of mitochondrial DNA. Data were normalized to nuclear DNA for β‐globin.5
MPF/SHG Microscopy
Multiphoton microscopy of freshly isolated hearts was performed on a Zeiss Confocal LSM410 microscope outfitted with optics designed for ultrafast laser excitation and non‐descanned detection of fluorescence emission.31 Fluorescence excitation was provided by a femtosecond Ti:sapphire laser (Spectra‐Physics) having a 5 W frequency‐doubled Nd:YVO pump laser. The system operated with a typical pulse width of 140 fs before the objective (40 × 1.2 N.A. water), and an average incident power of 15 mW was used in all imaging trials. Epi configuration was used to collect emitted light and was detected using a cooled R6060 photomultiplier tube detector placed in a non‐descanned configuration. An illumination wavelength of 840 nm, which elicits cardiomyocytes autofluorescence based on cytosolic fluorophores and SHG, was used, and image planes were acquired from the surface at an interval of 1 μm reaching a depth of 150 μm (z‐stacks). Collection of broadband fluorescence was in the 420 to 600 nm spectral range, and SHG collection in the same region was by a 420‐nm narrow pass filter. Image reconstruction of MPF/SHG micrograph stacks from 2 to 3 regions/heart was performed using Metamorph (Molecular Devices).
To quantify fibrillar collagen, strong collagen SHG signal was separated from weak SHG signal of myosin filaments on the basis of intensity, with threshold level set above that of myosin SHG for image segmentation. A maximum intensity projection was obtained from each SHG z‐stack, threshold was applied to select for high‐intensity SHG of fibrillar collagen, and percentage of threshold region (collagen) relative to full‐image field was measured using functions in Metamorph.
Data Analysis
Data are expressed as mean±SD (n=8/group). All data were checked by histograms and Q‐Q plots to be normally distributed and analyzed using the Student t test (comparison of 2 groups) and 1‐way analysis of variance with Tukey's post hoc test (comparison of multiple groups) using SPSS software (version 14.0; SPSS Inc, Chicago, IL). Significance (*normal versus infected and #wild type versus genetically modified) is indicated by *#P<0.05, **##P<0.01, and ***###P<0.001.
Results
C57BL/6 wild‐type mice infected with 10 000 parasites exhibit peak parasitemia 14 to 45 days postinfection and develop chronic disease ~120 days postinfection.30,32 We employed this well‐established protocol of infection to wild‐type, MnSODtg, MnSOD+/−, and GPx1−/− mice in this study. All infected mice, irrespective of antioxidant status, exhibited similar levels of blood and tissue parasite burden 120 days postinfection (Figure 1). These data allowed us to assess the effects of variable mitochondrial and cellular antioxidant capacities on inflammatory and oxidative stress in chronic Chagas disease.
Figure 1.
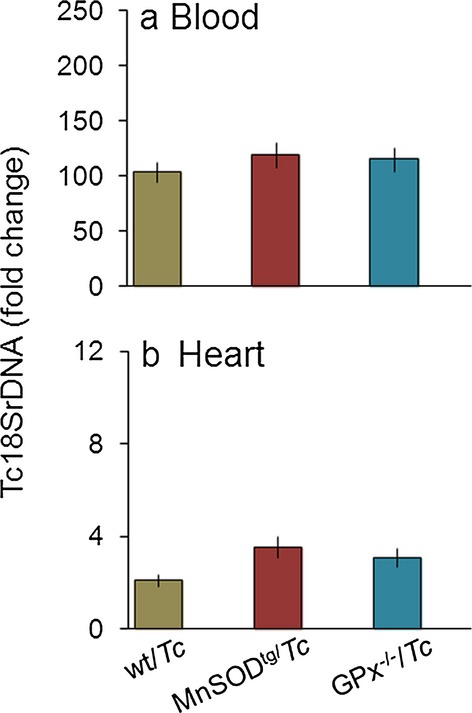
Parasite burden. C57BL/6 mice (wild type, MnSODtg, GPx−/−) were infected with Trypanosoma cruzi, and peripheral blood, and heart tissues were collected days 120 postinfection. Shown are the (A) blood and (B) tissue levels of parasite burden, determined by real‐time PCR amplification of the Tc18SrDNA sequence. Results were normalized to murine GAPDH DNA and represent fold change in TcDNA levels in chronically infected mice compared with that noted in matched normal controls. In all figures, data are presented as means±SDs (n=8/group). Significance is shown as *normal vs infected, #wild type vs genetically modified and is represented by *#P<0.05, **##P<0.01, ***###P<0.001. GPx indicates glutathione peroxidase; MnSOD, Mn2+ superoxide dismutase; PCR, polymerase chain reaction; Tc, Trypanosoma cruzi; wt, wild type.
Plasma Biomarkers of Phagocytic Activation Are Decreased in Chronically Infected MnSODtg Mice
We measured nitric oxide (•NO), LDH, and MPO levels to obtain a quantitative measure of chronic inflammation (Figure 2). Inducible nitric oxide synthase (NOS2) is a major source of •NO in activated macrophages. MPO is produced by activated neutrophils and uses H2O2 and chloride to produce reactive hypochlorous acid. LDH is a general indicator of tissue injury because of inflammatory or other cytotoxic reactions. Plasma levels of nitrite, LDH, and MPO were increased by 1.8‐fold, 1.5‐fold, and >10‐fold, respectively, in chronically infected wild‐type mice (P<0.01; Figure 2A through 2C). In comparison, chronically infected MnSODtg and GPx1−/− mice exhibited a 12% to 19% increase in plasma nitrite and LDH levels (Figure 2A and 2B). Basal plasma level of MPO was higher in MnSODtg and GPx1−/− mice compared with the levels in wild‐type controls (Figure 2C) and subsequently decreased by 48% to 68% during the chronic phase. These data suggested that neutrophil (MPO) and macrophage (NOS2/•NO) activation contributed to chronic inflammatory state in wild‐type infected mice and that these responses along with tissue injury (LDH) were minimal or absent in chronically infected MnSODtg and GPx1−/− mice.
Figure 2.
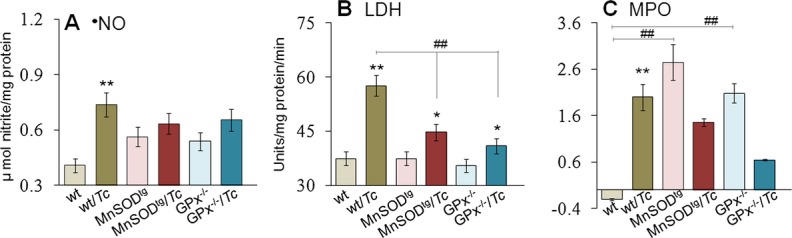
Effects of variable antioxidant status on phagocytic cell activation in response to Trypanosoma cruzi infection in mice. C57BL/6 (wild type [wt], MnSODtg, and GPx−/−) mice were infected with T cruzi (Tc) and harvested during the chronic disease phase (≥120 days postinfection). Plasma levels of (A) nitric oxide (•NO), (B) lactate dehydrogenase (LDH), and (C) myeloperoxidase (MPO) were measured by spectrophotometry. GPx indicates glutathione peroxidase; MnSODtg, Mn2+ superoxide dismutase.
Myocardial Inflammation Is Controlled in Chronically Infected MnSODtg Mice
Next, we determined the effects of MnSOD overexpression on T cruzi–induced myocarditis. Myocardial MPO activity in the chronic phase was increased by >10‐fold in wild‐type mice (P<0.01), but was barely altered in MnSODtg and GPx1−/− mice (Figure 3A). Histological studies showed the inflammatory infiltrate in heart and skeletal muscle of chronically infected mice was in the order of wild‐type ≥GPx1−/− >MnSODtg. Diffused inflammatory foci (histological score, 2 to 3) were noted in heart tissue of chronically infected wild‐type mice (Figure 3B.d). Further, chronically infected wild‐type mice exhibited a high degree of myocardial degeneration with enlarged myocytes. A notable but low level of inflammation (histological score, 1 to 2) was observed in heart and skeletal muscle of chronically infected GPx1−/− mice (Figure 3B.f). In comparison, minimal tissue inflammatory infiltrate (histological score, 0 to 1; Figure 3B.e) was detectable in chronically infected MnSODtg mice.
Figure 3.
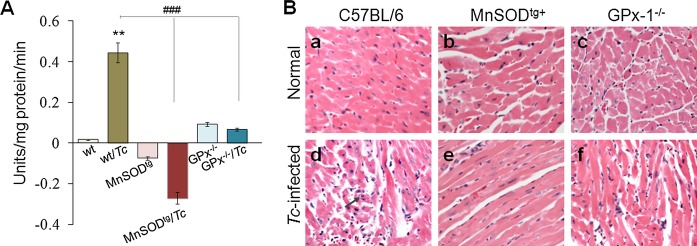
Myocardial inflammatory infiltrate is decreased in chronically infected MnSODtg mice. Mice were infected as in Figure 1. A, Myeloperoxidase activity in chronically infected murine myocardium. B, H&E staining (blue, nucleus; pink, muscle/cytoplasm/keratin) of heart tissue sections 120 days postinfection (magnification, 20×). GPx indicates glutathione peroxidase; H&E, hematoxylin and eosin; MnSOD, Mn2+ superoxide dismutase; wt, wild type.
The mRNA levels for IL‐1β, TNF‐α, IFN‐γ, IL‐10, and TGF‐β cytokines were increased by 1.7‐ to 3.4‐fold and 2.9‐ to 6.6‐fold in the myocardium of T cruzi–infected wild‐type and GPx1−/− mice, respectively, compared with the matched normal controls, and a predominance of inflammatory cytokines (IL‐1β+TNF‐α+IFN‐γ >> IL‐10+TGF‐β; P<0.05) was noted in the heart of chronically infected wild‐type and GPx1−/− mice during the disease phase (Figure 4). In MnSODtg mice, we noted no significant increase in the myocardial level of IL‐1β mRNA and a balanced type 1/type 2 cytokine response (TNF‐α+IFN‐γ=IL‐10+TGF‐β; P<0.05) during the chronic phase of disease progression (Figure 4). Together, the data presented in Figures 3 and 4 suggest that MnSODtg mice were equipped to control myocardial persistence of inflammatory infiltrate, a hallmark of Chagas disease.
Figure 4.
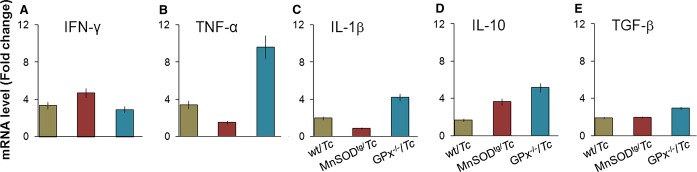
Gene expression of cytokines in myocardium of Trypanosoma cruzi–infected mice during the chronic disease phase. Wild‐type and genetically modified mice were infected with T cruzi as above. Shown are the relative changes in gene expression for IFN‐γ, TNF‐α, IL‐1β, IL‐10, and TGF‐β, determined by real‐time RT‐PCR. Results were normalized to GAPDH mRNA and represent fold change in mRNA levels in chronically infected mice compared with that noted in normal controls. GPx indicates glutathione peroxidase; IFN‐γ, interferon‐γ; IL, interleukin; MnSODtg, Mn2+ superoxide dismutase; RT‐PCR, reverse‐transcriptase polymerase chain reaction; Tc, Trypanosoma cruzi; TGF‐β, transforming growth factor–β; TNF, tumor necrosis factor; wt, wild type.
Myocardial Oxidative Stress Is Controlled in Chronically Infected MnSODtg Mice
We examined protein‐derived aldehydes and ketones produced by direct oxidation of amino acids and termed carbonyls, polypeptide‐bound 3‐NT residues formed by peroxynitrite attack and MDA that is stable breakdown products of highly reactive lipid hydroperoxides formed by the action of ROS on polyunsaturated fatty acids. Our data showed plasma carbonyls, and myocardial 3‐NT and MDA levels were increased by 2‐ to 4‐fold in chronically infected wild‐type mice (Figure 5A and 5E). The GPx1−/− and MnSOD+/− mice, like wild‐type mice, also exhibited a 2‐ to 3‐fold increase in myocardial protein carbonyl, 3‐NT, and MDA levels in response to chronic infection (Figure 5C through 5E). In comparison, chronically infected MnSODtg mice exhibited no significant increase in peripheral and myocardial carbonyls and myocardial MDA and exhibited a similar level of enhanced 3‐NT (Figure 5B), as was noted in chronically infected wild‐type mice (Figure 5B and 5E). These results suggested that the enhanced mitochondrial capacity to scavenge ROS was beneficial in preventing myocardial oxidative damage in T cruzi–infected MnSODtg mice. Enhanced mitochondrial antioxidant capacity did not alter the nitrosative stress in chronically infected MnSODtg mice.
Figure 5.
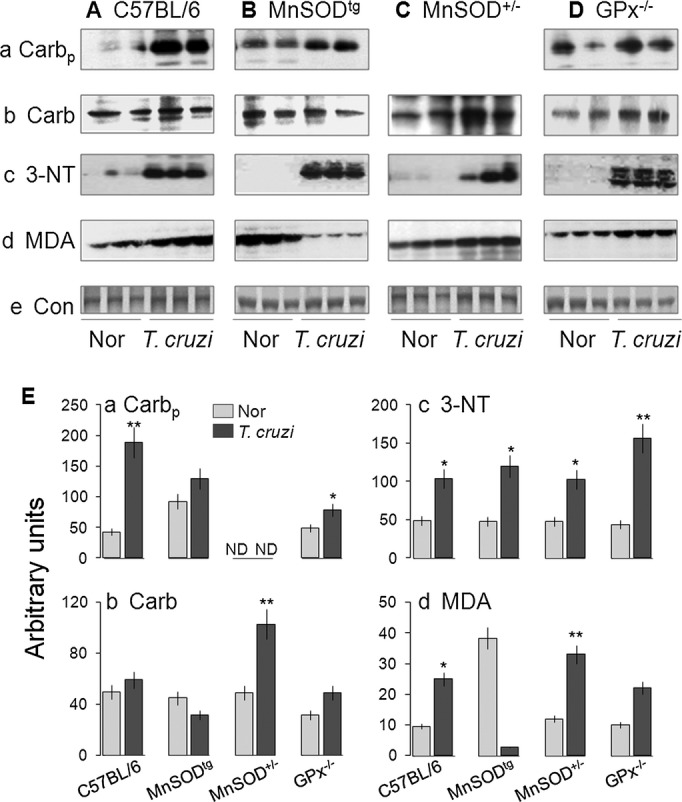
Cardiac oxidative stress is reduced in MnSODtg mice chronically infected with Trypanosoma cruzi. A through D, Cardiac tissue homogenates from normal and chronically infected C57BL/6 mice (wild type, MnSODtg, MnSOD+/−, and GPx–/–) were submitted to Western blotting for protein carbonyls (Carb), 3‐nitrotyrosine (3‐NT), and malondialdehyde (MDA). Gels were stained with Sypro Ruby to confirm equal loading of samples. Shown are representative images from 3 independent experiments (Carbp, plasma carbonyls). E, Bar graphs show the densitometric analysis of the oxidative stress markers, normalized to control (Con), in normal (Nor) and chronically infected mice. GPx indicates glutathione peroxidase; MnSODtg, Mn2+ superoxide dismutase.
Mitochondrial Abnormalities Are Controlled in Chronically Infected MnSODtg Mice
We performed electron microscopy to evaluate the extent of morphological alterations caused by chronic T cruzi infection in mice with variable antioxidant capacity. Myocardial tissue sections of chronically infected wild‐type mice exhibited evident myocardial alterations characterized by swollen or displaced mitochondria with loss of cristae and accumulation of lipid droplets alongside irregular mitochondria (Figure 6A.b). In addition, we noted large irregular nuclei, focal myofibrillar degeneration, and Z‐line distortion. In chronically infected MnSODtg mice, no significant injuries were evident: myofibrils were not damaged, mitochondria had a normal appearance, and the linear arrangement of myofibers and mitochondria was not disturbed (Figure 6A.d). The GPx1−/− mice exhibited a basal decline in mitochondrial cristae compared with the findings in wild‐type mice (Figure 6A.e), and subsequently mitochondrial mass was decreased in the chronic disease phase (Figure 6A.f). The change in mitochondrial mass was verified by quantitation of mitochondrial DNA, determined by cytochrome b and cytochrome oxidase II contents normalized to β‐globin nuclear DNA. The myocardial mitochondrial DNA level during the chronic disease phase was decreased by 50% to 57% in wild‐type and GPx1−/− mice (P<0.01) and by 30% to 32% in MnSODtg mice (Figure 6B). Together, these results suggested that the loss of myofibrils in chronically infected wild‐type and GPx1−/− mice was accompanied by compromised mitochondrial biogenesis, structure, and function, and these disease‐associated myofibrillar and mitochondrial losses were controlled, at least partially, in MnSODtg mice during the chronic disease phase.
Figure 6.
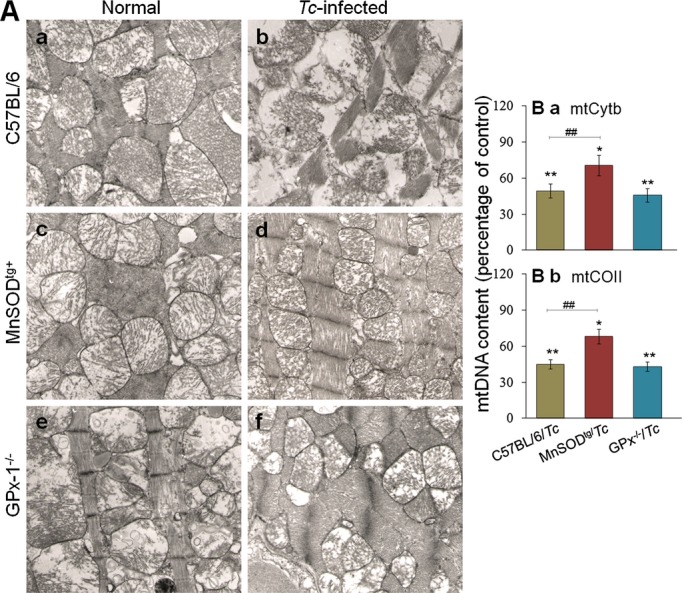
Preservation of mitochondrial structure and DNA content in chronically infected MnSODtg mice. A, Representative micrographs of heart tissue sections from chronically infected mice, submitted to transmission electron microscopy (magnification, 9400×). B, Percentage of changes in mitochondrial DNA content in cardiac biopsies of mice during the chronic disease phase was determined by real‐time PCR amplification of Cyt b (a) and CO II (b) regions of mitochondrial DNA. Data were normalized to β‐globin encoded by nuclear DNA. CO II indicates cytochrome oxidase II; Cyt b, cytochrome b; GPx, glutathione peroxidase; MnSODtg, Mn2+ superoxide dismutase; Tc, Trypanosoma cruzi.
Cardiac Remodeling by MPF/SHG Microscopy
Three‐dimensional (z‐projection) reconstructions of MPF/SHG micrograph stacks of heart are shown in Figure 5. SHG from collagen was significantly increased (>10‐fold) in chronically infected wild‐type mice (Figure 7A.b and 7B). No difference was discernible in SHG for collagen between normal and chronically infected MnSODtg mice (Figure 7A.c and 7A.d). The GPx1−/− mice exhibited a basal increase in collagen level (~5‐fold) that did not increase further in response to chronic infection (Figure 7A.e and 7A.f). Quantitative measurements of SHG area (3 regions/heart) confirmed the imaging results and demonstrated that the collagen content and distribution was increased in chronically infected hearts in the order of wild‐type >GPx1−/− >MnSODtg mice (Figure 7B).
Figure 7.
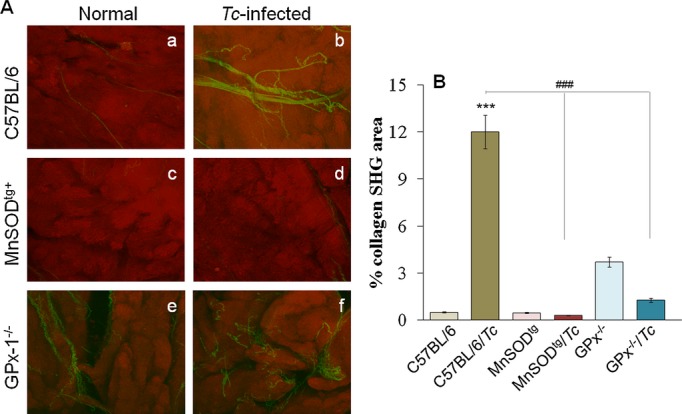
MPF/SHG microscopy of myocardial biopsies. A, Combined MPF and SHG microscopic images of normal controls and chronically infected mice. Images show separated MPF (red) from cardiomyocytes (red) and SHG (green) signal from collagen (images, 320 × 320 μm). B, Semiquantitative analysis of SHG (collagen) signal. GPx indicates glutathione peroxidase; MnSODtg, Mn2+ superoxide dismutase; MPF, multiphoton fluorescence; SHG, second harmonic generation; Tc, Trypanosoma cruzi; wt, wild type.
Cardiac Remodeling Is Not Controlled in p47phox−/− Mice Infected by T cruzi
To determine the relative contribution of phagocyte‐generated ROS in chagasic pathology, we used p47phox−/− mice. P47phox−/− mice, which lack the ability to mobilize NADPH oxidase‐mediated oxidative burst, were highly susceptible to T cruzi and therefore infected with 2000 trypomastigotes/mouse to monitor during the chronic phase. Despite the low dose, P47phox−/− mice exhibited 2‐fold and 4.5‐fold higher levels of blood and cardiac parasite burden, respectively, compared with that noted in wild‐type infected mice. Plasma nitrite and LDH levels were significantly increased (P<0.01), whereas plasma and cardiac levels of MPO were unaltered or decreased in chronically infected p47phox−/− mice (Figure 8A.a through 8A.d). Mild to moderate tissue inflammation (histological score, 1 to 2) was observed in heart and skeletal muscle of p47phox−/− mice during the chronic disease phase (Figure 8B), similar to that noted in chronically infected GPx1−/− mice (Figure 3B.c). The tissue inflammatory state was also evidenced by a 2.9‐ to 6.6‐fold increase in mRNA for IFN‐γ, IL‐1β, and TNF‐α proinflammatory cytokines in the myocardium of chronically infected p47phox−/− mice (Figure 8C). Further, p47phox−/− mice exhibited no increase in myocardial carbonyl and MDA adducts and a notable increase in 3‐NT during the chronic disease phase (Figure 8D). The mitochondrial DNA level, noted to be reduced by >50% in chronically infected wild‐type mice, was not altered in p47phox−/− mice during the chronic disease phase (Figure 8E). Yet myocardial remodeling was evidenced by a >5‐fold increase in collagen content in chronically infected p47phox−/− mice (Figure 8F). These data suggested that the phagocytes' oxidative burst and inflammatory activation are required for parasite control. Persistence of the chronic inflammatory and oxidative state in p47phox−/− mice during the disease phase, along with the results presented in Figures 2–7, suggest that mitochondria oxidative stress was the main cause of cardiac remodeling in Chagas disease.
Figure 8.
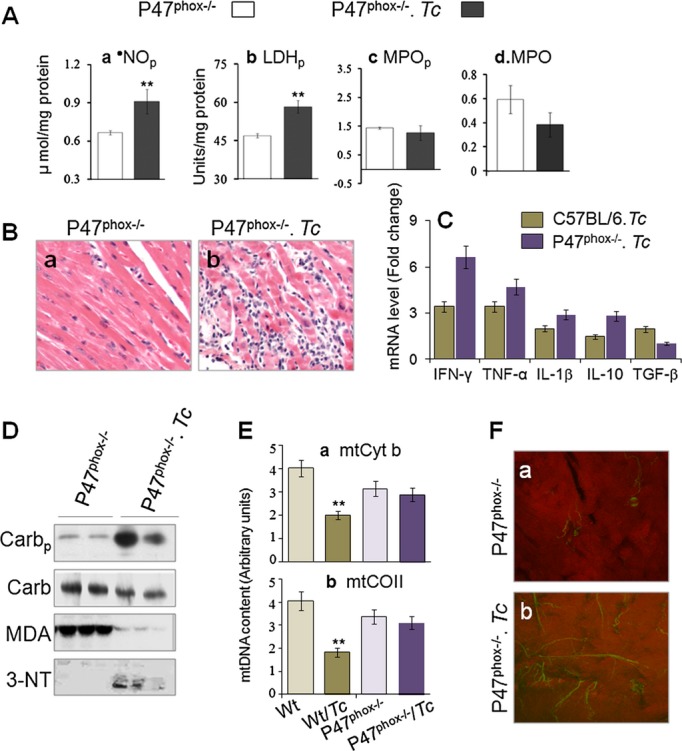
Inflammation, oxidative stress, and cardiac remodeling in p47phox−/− mice in response to chronic Trypanosoma cruzi infection. P47phox−/− mice were infected with T cruzi (2000 parasites/mouse) and euthanized 120 days postinfection. Shown are (A) plasma levels of nitrite (a), LDH (b), and MPO (c), myocardial levels of MPO (d), and (B) H&E staining of heart tissue sections from normal and chronically infected mice. C, Real‐time RT‐PCR determination of mRNA levels for cytokines as detailed in Figure 4. D, Western blotting for myocardial level of oxidative stress. E, Real‐time PCR determination of Cyt b– and CO II–encoding regions of mitochondrial DNA. F, Combined MPF and SHG microscopic images of normal and chronically infected p47phox−/− mice. 3‐NT indicates 3‐nitrotyrosine; Carb, carbonyls; Carbp, plasma carbonyls; CO II, cytochrome oxidase II; Cyt b, cytochrome b; GPx, glutathione peroxidase; H&E, hematoxylin and eosin; IFN‐γ, interferon‐γ; IL, interleukin; LDHp, plasma levels of lactate dehydrogenase; MDA, malondialdehyde; MnSODtg, Mn2+ superoxide dismutase; MPF, multiphoton fluorescence; MPO, myocardial levels of myeloperoxidase; MPOp, plasma levels of myeloperoxidase; •NOp, plasma levels of nitric oxide; RT‐PCR, reverse‐transcriptase polymerase chain reaction; SHG, second harmonic generation; Tc, Trypanosoma cruzi; TGF‐β, transforming growth factor–β; TNF, tumor necrosis factor; wt, wild type.
Discussion
Our data from this study and other reports suggest that a pro‐oxidant milieu, evidenced by increased ROS and glutathione disulfide (GSSG) levels and lipid and protein oxidation products, is present in the myocardium in chronic Chagas disease.5,8,12,33 Treatment of T cruzi–infected animals with an antioxidant resulted in a significant decline in myocardial oxidative adducts concurrent with preservation of a cardiac hemodynamic state that otherwise was compromised in the chronic disease phase in infected rats.11,13 A decline in oxidative stress in human Chagas disease patients given vitamin A has also been shown.34 These observations have supported the idea that the sustained oxidative stress is of pathological importance in chagasic cardiomyopathy, although the source of oxidative stress was unclear. In this study, we provide the first conclusive evidence that scavenging the mitochondrial ROS was beneficial in preventing oxidative damage and cardiac remodeling in Chagas disease. This was evidenced by significant control of myocardial oxidative adducts (Figure 5), preservation of mitochondrial and myofibrillar structure and arrangement (Figure 6), and distribution of myocardial collagen (Figure 7) in MnSODtg mice equipped with an extra copy of MnSOD to scavenge cardiac mitochondrial ROS.24 Importantly, MnSODtg mice exhibited a similar level of tissue parasite burden as was observed in chronically infected wild‐type mice (Figure 1), thus suggesting that the observed cardiac benefits in MnSODtg mice were not an outcome of decreased parasite persistence.
It is not known how inhibition or efficient scavenging of mitochondrial ROS is linked to preservation of cardiac structure in Chagas disease. Mice and cultured myoblasts infected with T cruzi display increased expression of ERK and cyclinD1, as well as ERK activator protein (AP‐1) and NFκB.35–36 Inhibition or scavenging of ROS has been shown to modulate ERK signaling and hypertrophic responses in neonatal and adult cardiomyocytes.37 These observations, along with the knowledge of decreased expression of hypertrophic markers in antioxidant‐treated chronically infected rats 11,13 and collagen deposition in MnSODtg mice during the chronic disease phase (Figure 7), suggest that ROS elicit pathological remodeling in chagasic myocardium through ERK/AP‐1 signaling, to be validated in future studies. The role of ROS from a mitochondrial, but not inflammatory, origin in signaling hypertrophy in chagasic hearts was substantiated by the observation that NADPH oxidase and MPO, classical mediators of inflammatory ROS, were depressed; however, the cardiac remodeling phenotype was not prevented in p47phox−/− mice infected by T cruzi (Figure 8).
Our observation of a decline in oxidative stress (protein carbonyls, MDA; Figure 5), inflammatory responses (MPO, LDH; Figure 2) and myocardial inflammatory infiltrate (Figure 3) in chronically infected MnSODtg mice provide the first indication that mitochondrial ROS also trigger chronic inflammatory responses in the heart. Others have demonstrated that trypomastigotes (or parasite proteins, eg, trans‐sialidase) activate NF‐κB that increases the resistance of myocytes and other cells to infection.35,38 We have found that ROS enhanced the nuclear translocation of RelA (p65) protein and NF‐κB‐dependent cytokine (TNF‐α, IL‐1β) gene expression in infected cardiomyocytes.39 Further, ROS caused 8‐hydroxyguanine lesions and DNA fragmentation that signaled polyadenosine ribose polymerase 1 activation and formation of poly (ADP‐ribose) polymers. Inhibition of polyadenosine ribose polymerase 1 using an RNAi or chemical inhibitors blocked poly (ADP‐ribose) modification of RelA (p65) interacting nuclear proteins and assembly of the NF‐κB transcription complex, thereby affecting cytokine gene expression.39 These studies suggested that the ROS‐PARP‐1‐RelA signaling pathway contributes to T cruzi–induced inflammatory cytokine production.40 Our data in this study validate these findings in an in vivo model and provide strong evidence that mitochondrial ROS serve as an activator of myocardial inflammatory responses in Chagas disease. It remains to be investigated how ROS may signal migration and activation of immune cells, resulting in an enhanced inflammatory infiltrate in the myocardium in chronic Chagas disease. We postulate that oxidative adducts observed in cardiac biopsies of chronically infected mice (Figure 3) and human patients5,12 serve as danger‐associated molecular patterns and thereby provide a signal for activation and migration of inflammatory phagocytes during the chronic phase. However, further studies are required to establish the interlinked effects of immune responses and antioxidant/oxidant status in the pathogenesis of Chagas disease.
In a recent study, Lim et al41 investigated the impact of homozygous deletion of GPx1 on postischemic myocardial recovery in male and female mice. The authors demonstrated that following ischemia/reperfusion, male GPx1−/− mice were highly susceptible to cardiac contractile and diastolic dysfunction and exhibited increased oxidative stress (eg, protein carbonyls). In comparison, GPx1 deficiency in female mice did not exacerbate myocardial dysfunction or oxidative stress after ischemia/reperfusion, and these benefits were found to be associated with a favorable ascorbate redox status. In this study, we observed that GPx1−/− female mice deficient in cellular glutathione antioxidant capacity were better equipped than the matched wild‐type mice to control the T cruzi–induced oxidative and inflammatory state and consequent cardiac remodeling in the heart. These data provide strong support for the notion that under stress conditions, GPx1−/− female mice can elicit a compensatory antioxidant mechanism that contribute to their ability to better control oxidative damage–associated cardiac injuries initiated by diverse etiologies. Further studies will be required to delineate the hormonal control of antioxidant/oxidant mechanisms and its significance in the chronic evolution of Chagas disease.
In vitro studies have shown that macrophages and dendritic cells play an important role in parasite control via upregulation of oxidative and nitrosative burst and inflammatory cytokines and chemokines (reviewed in reference 2). It is suggested that T cruzi initiates macrophage activation in an IFN‐γ‐dependent manner, and the resultant increase in the expression and activity of NOS2 and NADPH oxidase that produce •NO and O2•−, respectively, is essential for parasite control.42–44 TNF‐α is suggested to provide a second signal stimulating •NO and O2•− production and anti–T cruzi activity in IFN‐γ‐activated macrophages43,45–46 and thus to mediate trypanocidal function via an autocrine pathway. Our observation of a high degree of parasite burden in the myocardium of chronically infected p47phox−/− mice deficient in NADPH oxidase activity provide strong support for the notion that oxidative burst of phagocytes is required for early parasite control. Others have shown that NOS2−/− mice lacking nitric oxide production are compromised in controlling acute parasite burden.47–49 Overall, these studies provide strong evidence for the protective role of oxidative/nitrosative burst in controlling T cruzi infection and dissemination. However, note that p47phox−/− mice exhibited reduced levels of oxidative adducts and no significant changes in mitochondrial structure, but were not able to prevent cardiac inflammatory remodeling in the chronic phase of disease progression (Figure 8), likely because of parasite persistence, which can drive cellular immune responses and resultant tissue injuries. The gp91phox−/− mice that are deficient in another subunit of the NADPH oxidase and lack phagocytic oxidative burst were also found to exhibit compromised cardiac function associated with a decline in blood pressure.50 In comparison, the NOS2−/− mice, despite increased heart parasitism and no difference in cardiac inflammation compared with that noted in wild‐type controls, were better equipped to prevent cardiac injury and lesions and to preserve myocardial hemodynamics.49 Although further studies are required to delineate the mechanistic role of •NO in chronic cardiac insufficiency in Chagas disease, the observation of an increase in •NO in acutely infected gp91phox−/− mice and that T cruzi–induced cardiac insufficiency was reduced when gp91phox−/− mice were treated with an NOS2 inhibitor 50 suggest that a O2•−/•NO balance is required to preserve cardiac function, and in the absence of O2•−, overproduction of •NO contributes to disease severity by altering electrical synchrony that controls heart conduction and ventricle polarization. Another possibility is that mitochondrial ROS in chronically infected heart, along with increased •NO compensating for NADPH oxidase deficiency, produces an environment conducive to the generation of peroxynitrite, which is a powerful oxidant and cytotoxic effector molecule and can further enhance cardiac tissue injury.
A novel aspect of our studies included the use of noninvasive microscopy of fresh, label‐free heart tissue by the nonlinear optical methods of MPF/SHG. Incorporation of these methods allowed us to assess remodeling changes to the heart in chronic Chagas disease and the effects of antioxidant capacity. Tissue morphometry and inflammation revealed by autofluorescence mirrored that of hematoxylin and eosin–stained tissue sections, with the advantage that MPF/SHG was performed on the fresh intact heart and images were obtained without sectioning the tissue. SHG allowed for the quantitative assessment of collagen differences, but beyond that allowed for visualization of spatial distribution of collagen within heart tissue, again without physically sectioning the tissue. In the future, it will be of interest to conduct MPF/SHG microspectroscopy and to explore spectral windows that allow for assessment of redox potential as well.31
In summary, our data clearly demonstrate that a reduction in myocardial remodeling and inflammatory infiltrate, the hallmarks of Chagas disease, is achieved by the increased activity of MnSOD in transgenic mice. Enhancing the capacity to remove O2•− and H2O2 in double‐transgenic mice (eg, MnSODtg/GPxtg) would likely provide added protection from chronic oxidative/inflammatory stress and Chagas disease, and these factors will be tested in future studies.
Sources of Funding
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (2RO1AI054578; to N.J.G). Dr Vargas acknowledges funding from NIH/NIAID (UC7AI070083) for development of optical imaging approaches for infectious diseases.
Disclosures
None.
References
- 1.Rassi A, Jr, Rassi A, Marin‐Neto JA. Chagas disease. Lancet. 2010; 375:1388-1402 [DOI] [PubMed] [Google Scholar]
- 2.Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, Weiss LM, Nagajyothi F, Tanowitz HB, Garg NJ. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin Immunopathol. 2012; 34:753-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zacks MA, Wen JJ, Vyatkina G, Bhatia V, Garg NJ. An overview of chagasic cardiomyopathy: pathogenic importance of oxidative stress. An Acad Bras Cienc. 2005; 77:695-715 [DOI] [PubMed] [Google Scholar]
- 4.Wen JJ, Garg NJ. Mitochondrial generation of reactive oxygen species is enhanced at the Q(o) site of the complex III in the myocardium of Trypanosoma cruzi‐infected mice: beneficial effects of an antioxidant. J Bioenerg Biomembr. 2008; 40:587-598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wan X‐X, Gupta S, Zago MP, Davidson MM, Dousset P, Amoroso A, Garg NJ. Defects of mtDNA replication impaired the mitochondrial biogenesis during Trypanosoma cruzi infection in human cardiomyocytes and chagasic patients: the role of Nrf1/2 and antioxidant response. J Am Heart Assoc. 2012; 1:e003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta S, Dhiman M, Wen JJ, Garg NJ. ROS signalling of inflammatory cytokines during Trypanosoma cruzi infection. Adv Parasitol. 2011; 76:153-170 [DOI] [PubMed] [Google Scholar]
- 7.Nagajyothi F, Machado FS, Burleigh BA, Jelicks LA, Scherer PE, Mukherjee S, Lisanti MP, Weiss LM, Garg NJ, Tanowitz HB. Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell Microbiol. 2012; 14:634-643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen J‐J, Vyatkina G, Garg NJ. Oxidative damage during chagasic cardiomyopathy development: role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radic Biol Med. 2004; 37:1821-1833 [DOI] [PubMed] [Google Scholar]
- 9.Perez‐Fuentes R, Guegan JF, Barnabe C, Lopez‐Colombo A, Salgado‐Rosas H, Torres‐Rasgado E, Briones B, Romero‐Diaz M, Ramos‐Jimenez J. Sanchez‐Guillen Mdel C. Severity of chronic Chagas disease is associated with cytokine/antioxidant imbalance in chronically infected individuals. Int J Parasitol. 2003; 33:293-299 [DOI] [PubMed] [Google Scholar]
- 10.de Oliveira TB, Pedrosa RC, Filho DW. Oxidative stress in chronic cardiopathy associated with Chagas disease. Int J Cardiol. 2007; 116:357-363 [DOI] [PubMed] [Google Scholar]
- 11.Wen J‐J, Bhatia V, Popov VL, Garg NJ. Phenyl‐alpha‐tert‐butyl nitrone reverses mitochondrial decay in acute Chagas disease. Am J Pathol. 2006; 169:1953-1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhiman M, Zago MP, Nunez S, Nunez‐Burgio F, Garg NJ. Cardiac oxidized antigens are targets of immune recognition by antibodies and potential molecular determinants in Chagas disease pathogenesis. PLoS ONE. 2012; 7:e28449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen J‐J, Gupta S, Guan Z, Dhiman M, Condon D, Lui CY, Garg NJ. Phenyl‐alpha‐tert‐butyl‐nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic rats. J Am Coll Cardiol. 2010; 55:2499-2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raineri I, Carlson EJ, Gacayan R, Carra S, Oberley TD, Huang TT, Epstein CJ. Strain‐dependent high‐level expression of a transgene for manganese superoxide dismutase is associated with growth retardation and decreased fertility. Free Radic Biol Med. 2001; 31:1018-1030 [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol. 1998; 30:2281-2289 [DOI] [PubMed] [Google Scholar]
- 16.Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997; 272:16644-16651 [DOI] [PubMed] [Google Scholar]
- 17.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock‐out model of chronic granulomatous disease. J Exp Med. 1995; 182:751-758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhiman M, Garg NJ. NADPH oxidase inhibition ameliorates Trypanosoma cruzi‐induced myocarditis during Chagas disease. J Pathol. 2011; 225:583-596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton‐excited native fluorescence and second harmonic generation. Proc Natl Acad Sci USA. 2003; 100:7075-7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallace SJ, Morrison JL, Botting KJ, Kee TW. Second‐harmonic generation and two‐photon‐excited autofluorescence microscopy of cardiomyocytes: quantification of cell volume and myosin filaments. J Biomed Opt. 2008; 13:064018. [DOI] [PubMed] [Google Scholar]
- 21.Schenke‐Layland K, Stock UA, Nsair A, Xie J, Angelis E, Fonseca CG, Larbig R, Mahajan A, Shivkumar K, Fishbein MC, MacLellan WR. Cardiomyopathy is associated with structural remodelling of heart valve extracellular matrix. Eur Heart J. 2009; 30:2254-2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallenburg MA, Wu J, Li RK, Vitkin IA. Two‐photon microscopy of healthy, infarcted and stem‐cell treated regenerating heart. J Biophotonics. 2011; 4:297-304 [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995; 11:376-381 [DOI] [PubMed] [Google Scholar]
- 24.Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, Qi W, Liu Y, Liang H, Chaudhuri A, Ikeno Y, Epstein CJ, Van Remmen H, Richardson A. Overexpression of Mn Superoxide Dismutase Does Not Increase Life Span in Mice. J Gerontol A Biol Sci Med Sci. 2009; 64:1114-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001; 281:H1422-H1432 [DOI] [PubMed] [Google Scholar]
- 26.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982; 78:206-209 [DOI] [PubMed] [Google Scholar]
- 27.Kleinbongard P, Rassaf T, Dejam A, Kerber S, Kelm M. Griess method for nitrite measurement of aqueous and protein‐containing samples. Methods Enzymol. 2002; 359:158-168 [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez MN, Garg NJ, Postan M. Granulocyte colony‐stimulating factor partially repairs the damage provoked by Trypamosoma cruzi in murine myocardium. Int J Cardiol. 2013. 10.1016/j.ijcard.2013.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garg NJ, Bhatia V, Gerstner A, deFord J, Papaconstantinou J. Gene expression analysis in mitochondria from chagasic mice: alterations in specific metabolic pathways. Biochem J. 2004; 381:743-752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garg NJ, Popov VL, Papaconstantinou J. Profiling gene transcription reveals a deficiency of mitochondrial oxidative phosphorylation in Trypanosoma cruzi‐infected murine hearts: implications in chagasic myocarditis development. Biochim Biophys Acta. 2003; 1638:106-120 [DOI] [PubMed] [Google Scholar]
- 31.Edward K, Qiu S, Resto V, McCammon S, Vargas G. In vivo layer‐resolved characterization of oral dysplasia via nonlinear optical micro‐spectroscopy. Biomed Opt Express. 2012; 3:1579-1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta S, Garg NJ. Prophylactic efficacy of TcVac2R against Trypanosoma cruzi in mice. PLoS Negl Trop Dis. 2010; 4:e797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wen JJ, Dhiman M, Whorton EB, Garg NJ. Tissue‐specific oxidative imbalance and mitochondrial dysfunction during Trypanosoma cruzi infection in mice. Microbes Infect. 2008; 10:1201-1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macao LB, Filho DW, Pedrosa RC, Pereira A, Backes P, Torres MA, Frode TS. Antioxidant therapy attenuates oxidative stress in chronic cardiopathy associated with Chagas' disease. Int J Cardiol. 2007; 123:43-49 [DOI] [PubMed] [Google Scholar]
- 35.Huang H, Petkova SB, Cohen AW, Bouzahzah B, Chan J, Zhou JN, Factor SM, Weiss LM, Krishnamachary M, Mukherjee S, Wittner M, Kitsis RN, Pestell RG, Lisanti MP, Albanese C, Tanowitz HB. Activation of transcription factors AP‐1 and NF‐kappa B in murine Chagasic myocarditis. Infect Immun. 2003; 71:2859-2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouzahzah B, Yurchenko V, Nagajyothi F, Hulit J, Sadofsky M, Braunstein VL, Mukherjee S, Weiss H, Machado FS, Pestell RG, Lisanti MP, Tanowitz HB, Albanese C. Regulation of host cell cyclin D1 by Trypanosoma cruzi in myoblasts. Cell Cycle. 2008; 7:500-503 [DOI] [PubMed] [Google Scholar]
- 37.Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol. 2001; 37:676-685 [DOI] [PubMed] [Google Scholar]
- 38.Hall BS, Tam W, Sen R, Pereira ME. Cell‐specific activation of nuclear factor‐kappaB by the parasite Trypanosoma cruzi promotes resistance to intracellular infection. Mol Biol Cell. 2000; 11:153-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ba X, Gupta S, Davidson M, Garg NJ. Trypanosoma cruzi induces ROS‐PARP‐1‐RelA pathway for up regulation of cytokine expression in cardiomyocytes. J Biol Chem. 2010; 285:11596-11606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ba X, Garg NJ. Signaling Mechanism of PARP‐1 in Inflammatory Diseases. Am J Pathol. 2010; 178:946-955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim CC, Bryan NS, Jain M, Garcia‐Saura MF, Fernandez BO, Sawyer DB, Handy DE, Loscalzo J, Feelisch M, Liao R. Glutathione peroxidase deficiency exacerbates ischemia‐reperfusion injury in male but not female myocardium: insights into antioxidant compensatory mechanisms. Am J Physiol Heart Circ Physiol. 2009; 297:H2144-H2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plata F, Wietzerbin J, Pons FG, Falcoff E, Eisen H. Synergistic protection by specific antibodies and interferon against infection by Trypanosoma cruzi in vitro. Eur J Immunol. 1984; 14:930-935 [DOI] [PubMed] [Google Scholar]
- 43.Vespa GN, Cunha FQ, Silva JS. Nitric oxide is involved in control of Trypanosoma cruzi‐induced parasitemia and directly kills the parasite in vitro. Infect Immun. 1994; 62:5177-5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kayama H, Takeda K. The innate immune response to Trypanosoma cruzi infection. Microbes Infect. 2010; 12:511-517 [DOI] [PubMed] [Google Scholar]
- 45.Moncada S, Higgs EA. Endogenous nitric oxide: physiology, pathology and clinical relevance. Eur J Clin Invest. 1991; 21:361-374 [DOI] [PubMed] [Google Scholar]
- 46.Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. Trypanosoma cruzi‐infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide‐dependent trypanocidal activity. Circulation. 2000; 102:3003-3008 [DOI] [PubMed] [Google Scholar]
- 47.Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes‐Vieira J, Gazzinelli RT. Pivotal role of interleukin‐12 and interferon‐gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol. 2001; 159:1723-1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silva JS, Machado FS, Martins GA. The role of nitric oxide in the pathogenesis of Chagas disease. Front Biosci. 2003; 8:s314-s325 [DOI] [PubMed] [Google Scholar]
- 49.Carvalho CM, Silverio JC, da Silva AA, Pereira IR, Coelho JM, Britto CC, Moreira OC, Marchevsky RS, Xavier SS, Gazzinelli RT. da Gloria Bonecini‐Almeida M, Lannes‐Vieira J. Inducible nitric oxide synthase in heart tissue and nitric oxide in serum of Trypanosoma cruzi‐infected rhesus monkeys: association with heart injury. PLoS Negl Trop Dis. 2012; 6:e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santiago HC. Gonzalez Lombana CZ, Macedo JP, Utsch L, Tafuri WL, Campagnole‐Santos MJ, Alves RO, Alves‐Filho JC, Romanha AJ, Cunha FQ, Teixeira MM, Radi R, Vieira LQ. NADPH phagocyte oxidase knockout mice control Trypanosoma cruzi proliferation, but develop circulatory collapse and succumb to infection. PLoS Negl Trop Dis. 2012; 6:e1492. [DOI] [PMC free article] [PubMed] [Google Scholar]