

Abstract
Background
Cardiac c‐Kit+ cells isolated from cardiac explant‐derived cells modestly improve cardiac functions after myocardial infarction; however, their full potential has not yet been realized. For instance, the majority of potential candidates for cell therapy suffer from chronic heart failure (CHF), and it is unclear how this disease affects the explant‐derived progenitor cells. Therefore, the objective of this study was to determine the effect of CHF on the number and phenotype of cardiac explant c‐Kit+ progenitors and elucidate mechanisms of their regulation.
Methods and Results
Myocardial infarction was created by left anterior descending coronary artery occlusion. Sham‐operated animals were used as a control group. CHF‐developed infarcted animals were selected on the basis of left ventricle end‐diastolic pressure ≥20 mm Hg and scar size ≥30%. Here, we found that CHF atrial explants produced less c‐Kit+ cells than sham explants. CHF‐derived c‐Kit+ cells exhibited upregulated transforming growth factor–β (TGF‐β) signaling, increased level of epithelial to mesenchymal transition markers, and diminished expression of pluripotency markers compared with shams. We show that intervention with TGF‐β signaling by inhibiting TGF‐β receptor type I or Smad 2/3 using small‐molecule inhibitors improved c‐Kit+ cell yield, attenuated epithelial to mesenchymal transition markers, stimulated the pluripotency marker Nanog, and improved efficiency of c‐Kit+ cell differentiation toward cardiomyocyte‐like cells in vitro.
Conclusions
Taken together, our findings suggest that TGF‐β inhibition positively modulates c‐Kit+ cell phenotype and function in vitro, and this strategy may be considered in optimizing cardiac progenitor function and cell expansion protocols for clinical application.
Keywords: cell transplantation, chronic heart failure, heart, signaling pathways, stem cell
Introduction
Many adult tissues contain a rare population of multipotent progenitor cells, and mature cardiac tissues are no exception. These cardiac lineage‐restricted progenitors maintain cardiac tissue homeostasis after injury.1–5 Several potentially overlapping populations of endogenous cardiac progenitor cells have been identified to date on the basis of antigen expression (c‐Kit, Sca1, Isl1) or function (Hoechst efflux).1,3,6–7 In addition, c‐Kit+ cells have been isolated from cells spontaneously migrating from cardiac tissue cultured as “explants.”4–5,4–9 The origin of explant‐derived c‐Kit+ cells remains unclear; however, studies suggested that these cells may derive through dedifferentiation of cardiomyocytes and/or epicardial cells.10–11 If the regenerative power of these cells can be harnessed, they may serve as ideal candidates for cardiac cell therapy; however, the majority of patients who are candidates for cell therapy suffer from chronic heart failure, which may have a great impact on cardiac resident progenitor cells. Cardiac explant‐derived cells including c‐Kit+ cells are currently being used in clinical trials such as CADUCEUS12 and SCIPIO.13 For these clinical trials, autologous cardiac stem cells are used to treat heart diseases, including ischemic heart failure. Only a limited amount of heart tissue can be obtained to generate autologous explant‐derived cells, whereas current transplantation protocols require a substantial amount of cells (millions). Therefore, optimizing in vitro cell expansion to maximize the amount of the cells of interest is one of the key factors of stem cell therapies.
It was previously reported that both resident and explant‐derived c‐Kit+ cells can be expanded from pathological hearts.14–16 Studies showed that cardiovascular diseases such as ischemic cardiomyopathy and diabetes are associated with an increase in p16(INK4a)‐p53‐positive stem cell senescence, telomere shortening, and a decline in functionally competent cardiac stem cells.15,17–18 Here, we aimed to study the effects of congestive heart failure on the number and phenotype of cardiac explant‐derived c‐Kit+ progenitors and to elucidate the mechanism of their regulation.
We have previously shown that the epithelial/endothelial to mesenchymal transition (EMT) played a significant role in explant‐derived c‐Kit+ cell regulation.19 The EMT reflects epithelial cell plasticity and is defined by phenotypical and functional changes that are reminiscent of mesenchymal cells.20 Previous studies showed that the EMT program is redeployed in a heart after myocardial infarction (MI).21 Furthermore, myocardial fibrosis, a major consequence of adult heart ischemic injury, is partially mediated by the activation of epicardial and endothelial cells via EMT.22 Transforming growth factor–β (TGF‐β), a major EMT inducer, is stably upregulated within the heart after MI and plays a key role in scar formation and cardiac fibrosis.23–25 TGF‐β family proteins mainly transduce their signal to the nucleus through TGF‐β serine/threonine receptors and downstream effector Smad proteins.26–29 Both canonical (Smads‐dependent) and noncanonical (Smads‐independent) signaling pathways could be activated via TGF‐β type I receptor (TGF‐β‐R1).30
For this study, we have selected animals that developed chronic heart failure (CHF) condition 6 weeks after MI. Here, we generated explant‐derived cells (EDCs) from the atria of CHF‐developed infarcted rats and sham‐operated control rats. We tested whether the CHF milieu affects EDC composition and the c‐Kit+ cell phenotype and describe the effects of TGF‐β signaling modulations on c‐Kit+ cells generated from sham and CHF atrial explants.
Methods
Myocardial Infarction
Animal studies were performed in accordance with federal laws and regulations, international accreditation standards (AAALAC No 1115), and institutional policies including approval by the Institutional Animal Care and Use Committee of Banner Sun Health Research Institute (IACUC protocol #10‐02). Two‐month‐old Sprague Dawley rats (Harlan Laboratories) were anesthetized using a cocktail of ketamine, xylazine, and acepromazine (50, 15, and 2 mg/kg, respectively). The animals were prepared using aseptic methods, intubated (14G angiocatheter), and ventilated before a left thoracotomy exposing the heart. The heart was expressed, and the left anterior descending coronary artery was ligated using a 5‐0 TiCron suture as per standard protocols. The lungs were briefly hyperinflated, the chest was closed using 2‐0 silk, and the rodents were allowed to recover with a pain management regiment of buprenorphine. Sham‐operated animals underwent the same surgical procedure excluding left anterior descending coronary artery occlusion.
Cardiac Function Measurements
Hemodynamic statistics were collected 6 weeks post‐MI using a pressure‐volume catheter (Millar Instruments) inserted into the right carotid artery and advanced into the left ventricle. The animals were systemically anesthetized with Inactin (125 mg/kg) and intubated, and the steady‐state measurements were collected prior to ventilation. The data were analyzed using PVAN 3.6. software (Millar Instruments).
Selection of CHF Animals
CHF animals were selected on the basis of left ventricle end‐diastolic pressure measurement ≥20 mm Hg and scar size ≥30% of left ventricle.31–32 Approximately 35% of infarcted animals were classified as CHF and utilized in the following experiments; non‐CHF animals were excluded from this study. Atrial tissues were collected 6 weeks after MI from sham‐operated (n=10) and CHF (n=10) animals.
Heart Sections Processing and Staining
Heart tissue was embedded in tissue‐freezing media (Triangle Biomedical Science) snap‐frozen in liquid nitrogen, and sectioned in the coronal plane using a Leica CM1900 cryostat (Leica Microsystems, Bannockburn, IL). Coronal tissue sections (thickness of 5 to 7 μm) were mounted on positively charged glass slides and fixed/permeabilized in a 1:1 mixture of acetone/100% ethanol. For immunofluorescent staining, fixed tissue sections were blocked with 3% BSA in PBS and incubated with primary antibodies against vimentin (Abcam). Specific staining was visualized using corresponding secondary antibodies conjugated with Alexa 568 (Molecular Probes). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (Invitrogen).
Scar Size Assessment
Heart sections were prepared as described above and stained with Masson's Trichrome kit (Sigma‐Aldrich) according to the manufacturer's protocol. Transmitted light images of heart sections were processed using DP2‐BSW software (Olympus Corp). Scar percentage was calculated as a ratio of collagen‐enriched scar area (blue staining) to the whole left ventricle area.
Cell Culture
Cardiac explant outgrowth was generated as previously described.4,19,33 Briefly, tissue was cut into 1‐ to 2‐mm3 pieces and digested with 0.2% trypsin (ILife Technologies, Carlsbad, CA) and 0.1% collagenase IV (Life Technologies) for a total of 10 minutes. The remaining tissue fragments were cultured as “explants” in explant medium (CEM), which was composed of IMDM supplemented with 10% fetal bovine serum (FBS; Lonza), 100 U/mL penicillin G, 100 μg/mL streptomycin,' and 2 mmol/L l‐glutamine (Sigma‐Aldrich). After 21 days in culture, cells were collected by trypsinization. c‐Kit+ cells were separated from the cell outgrowths using magnetic beads (MACS, Miltenyi Biotec) according to the manufacturer's protocol and analyzed by flow cytometry to validate the purity.19 Freshly isolated c‐Kit+ cells were seeded on Geltrex‐coated plates (Life Technologies) at 1×105 cells/well in culture media composed of 1:1 IMDM:DMIM/F12 supplemented with 1× Insulin‐Selenium‐Transferrin (Life Technologies), 1× B‐27 serum supplement, 5% FBS, and 10 ng/mL basic fibroblast growth factor (R&D Systems). Cells were treated with 10 ng/mL of TGF‐β (R&D Systems), 10 μmol/L SB431542 (Sigma‐Aldrich), or 3 μmol/L SIS3 (Sigma‐Aldrich) for 7 days. Control cells were treated with 0.005% DMSO.
RNA Isolation and Quantitative Real‐Time RT‐PCR
Total RNA was extracted from c‐Kit+ cells using PureLink RNA Mini Kit (Life Technologies) according to the manufacturer's protocol. RNA was then quantified with a Quanti‐iT RiboGreen RNA Assay Kit and assessed using a BioTek Synergy HT Microplate Reader (excitation/emission 480/520 nm). Total RNA (200 ng) was reverse‐transcribed with a QuantiTect Reverse Transcription kit (Qiagen). Real‐time RT‐PCR was conducted using Rower SYBR Green Master Mix (Applied Biosystems) on a StepOnePlus Real‐time PCR System (Applied Biosystems). Specific primers were synthesized by Life Technologies (sequences are available on request). CYP A was used as a reference gene. Data analysis was performed on StepOne software version 2.1 (Applied Biosystems) using the comparative Ct (ΔΔCt) quantitation method.
TGF‐β1 ELISA
To assess the amount of TGF‐β1 released by explant‐derived cells, 0.2×106 cells were cultured for 4 or 10 days, and conditioned media were collected. Cell‐culture medium prior to adding cells was also collected to assess baseline levels of TGF‐β1. TGF‐β1 levels were measured using a commercially available TGF‐β1 ELISA kit (R&D Systems) according to the manufacturer's instructions. After conditioned medium was collected, total protein was extracted from cells using RIPA buffer (Thermo Scientific), and the protein amount was determined by a BCA Protein Assay kit (Thermo Scientific). TGF‐β1 amounts were normalized to total protein amount.
Western Blotting
Cells were lysed in RIPA buffer (Thermo Scientific) containing Halt Phosphatase and Proteinase inhibitor cocktail (Thermo Scientific) according to the manufacturer's protocol. Protein concentration was determined using a BCA Protein Assay kit (Thermo Scientific). An equal amount of protein (50 μg) was loaded in each well of 4% to 12% bis‐tris gels gel (Life Sciences) and subjected to electrophoresis. Proteins were transferred to a PVDF membrane (Millipore) and then blocked with 5% nonfat dry milk in Tris‐buffered saline followed by overnight incubation with primary antibodies at 4°C. Antibodies against p‐Smad2/3, Smad2/3 (Cell Signaling), and Nanog (Millipore) were used. Blots were probed with an anti‐β‐actin (Sigma Aldrich) antibody as a loading control. Membranes were washed in Tris‐buffered saline containing 0.05% Tween 20. Corresponding horseradish peroxidase–conjugated anti‐rabbit or anti‐mouse IgG (Invitrogen) was used as secondary antibodies. Immunoreactive proteins were detected by chemiluminescence (Thermo Scientific). Band intensity was determined using FluorChem 8900 software (Alpha Innotech Corp).
Flow Cytometry
Cells were fixed in 70% ethanol and labeled with the following antibodies: c‐Kit (Santa‐Cruz Biotechnology), vimentin and smooth muscle actin (Abcam), and CD90 (BD Biosciences). Cells were treated with secondary antibodies corresponding to either anti‐rabbit or anti‐mouse IgG conjugated with Alexa 488, phycoerythrin (PE), or PE‐Cy5.5 (Life Technologies). Direct labeling with FITC‐conjugated CD34 and PE‐Cy5.5 conjugated CD45 (BD Biosciences) antibodies was used to exclude bone marrow and hematopoietic cells. Freshly isolated bone marrow cells were used as positive controls for CD34 and CD45 labeling. For a negative control, cells were labeled with isotype IgG instead of primary antibody. Cell events were detected using a FACS Calibur flow cytometer equipped with an argon laser (BD Biosciences). Data were analyzed using CellQuest software (BD Biosciences).
To estimate the percentage of proliferating cells, cells were labeled with anti Ki67 antibody (Abcam) following by Alexa 488–conjugated IgG. Cells' replication state was evaluated by labeling DNA with 10 μg/mL propidium iodide (PI). G0/G1, S‐phase, and G2/M were determined by setting markers for PI fluorescence using a BD Biosciences FACSCalibur with CellQuest software.
The percentage of apoptotic cells was evaluated with direct labeling with FITC‐conjugated activated caspase 3 (BD Biosciences) antibodies as well as a Vybrant Apoptosis assay kit (Life Technologies) according to the manufacturer's instruction. Briefly, collected live cells were double‐labeled with FITC‐conjugated annexin V and PI. The percentage of apoptotic cells was calculated as a ratio of annexin Vpositive/PInegative cells to total number of cells using CellQuest software. UV‐irradiated c‐Kit+ cells were used as a positive control.
Cardiac Differentiation Potential of c‐Kit+ Cells In Vitro
To assess the differentiation potential of c‐Kit+ cells toward a cardiomyocyte lineage, cells were cultured in cardiac differentiation medium (proprietary formulation, EMD Millipore) supplemented with 2 μmol/L Mocetinostat (SelleckChem) for 7 days. To test the effect of TGF‐β inhibitors, some samples were cultured in the presence of 10 μmol/L SB431542 (Sigma‐Aldrich) or 3 μmol/L SIS3 (Sigma‐Aldrich). Control cells were treated with 0.005% DMSO. Expression of cardiac troponin T (TnT) was evaluated by immunocytochemistry. Cells were fixed/permeabilized with a 1:1 acetone:ethanol mixture, blocked with 3% BSA in PBS, and labeled with mouse anti‐TnT primary antibody (Abcam). Specific staining was visualized using anti‐mouse secondary antibodies conjugated with Alexa 568 (Molecular Probes). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (Invitrogen). TnT‐positive cells were quantified in 5 random microscopic fields. The percentage of TnT‐positive cells was calculated as the number of positively stained cells normalized to the total number of cells.
Imaging
Images were captured at room temperature using an Olympus IX‐51 epifluorescence microscope equipped with a DP72 device camera. Excitation/emission maximum filters of 490/520, 570/595, and 355/465 nm, were used for image acquisition. Images were processed using DP2‐BSW software (Olympus Corp). Acquisition settings were held constant within each experiment.
Statistics
Results are reported as mean±SE and median and range. The nonparametric Wilcoxon rank sum test was used to compare the differences among the sham and CHF groups. The impact of TGF‐β inhibitors was evaluated with the use of 1‐way ANOVA. Two‐way ANOVA was used to determine the interaction between disease (eg, CHF) and treatment (eg, TGF‐β inhibitors SB431542 [SB] or SIS3 [SIS]). Statistical analysis was conducted using SigmaStat 3.5 software. A value of P<0.05 was considered statistically significant.
Results
CHF Is Associated With Atrial Fibrosis
Myocardial infarction was created by a permanent occlusion of the left anterior descending coronary artery. Sham‐operated animals underwent the same surgical procedure excluding left anterior descending coronary artery occlusion. Six weeks after MI, rats exhibiting heart failure were selected on the basis of cardiac function measurement (Table) and histological scar evaluation (Figure 1A). Specifically, animals with left ventricle end‐diastolic pressure ≥20 mm Hg and scar size ≥30% of the left ventricle were selected for this study and designated as CHF rats.31–32 Approximately 35% of infarcted animals were classified as CHF and were used in the following experiments; non‐CHF animals were excluded from this study. Consistent with previous work,34 we showed that CHF is associated with atrial hypertrophy and fibrosis. Atrial fibrosis was indicated by an increase in both collagen deposition (Figure 1B) and number of vimentin‐positive fibroblasts35 (Figure 1C).
Table 1.
Hemodynamic Parameters
| Parameter | Sham | CHF | ||||||
|---|---|---|---|---|---|---|---|---|
| Average | Min | Med | Max | Average | Min | Med | Max | |
| HR, min−1 | 268±13 | 203 | 259 | 336 | 240±8 | 201 | 247 | 266 |
| ESP, mm Hg | 118.5±5 | 91 | 118.5 | 142 | 92±1* | 84 | 93.5 | 101 |
| EDP, mm Hg | 8.3±1 | 5 | 7 | 14 | 24±1* | 20.6 | 23.2 | 29 |
| ESV, mL | 132±18 | 62 | 114 | 218 | 240±65* | 154.0 | 234.4 | 346 |
| EDV, mL | 262±31 | 130 | 273 | 388 | 307±57 | 247.0 | 284.8 | 397 |
| SV, mL | 164±16 | 93 | 182 | 215 | 88±16* | 52 | 89 | 104 |
| CO, mL/min | 43.5±5 | 19 | 50 | 59 | 20.9±1* | 14 | 22 | 24 |
| Ea, mm Hg/μL | 0.8±0.1 | 0.5 | 1 | 0.6 | 1.1±0.1 | 0.8 | 1.1 | 2 |
| EF, % | 59±3.4 | 48 | 54.5 | 77 | 28.4±7* | 18.9 | 27.41 | 40 |
| dP/dTmax, mm Hg/s | 6823±350 | 5482 | 6729 | 8600 | 3962±200* | 3034 | 3937 | 4688 |
| SW, mm Hg×mL | 16 167±1928 | 7872 | 18940 | 23 102 | 5124±457* | 3040 | 5704 | 6696 |
| −dP/dTmin, mm Hg/s | 4664±268 | 5841 | 4684 | 3505 | 2515±162* | 3436 | 2249 | 1939 |
| Tau (W), ms | 15.2±1.2 | 9 | 14.3 | 21 | 21.9±1* | 16 | 22 | 26 |
| Tau (G), ms | 18.8±1.1 | 13 | 17.9 | 23 | 31.7±3.8* | 18 | 27.8 | 55 |
Data are presented as average±SE. n=9 per group. Min, med, and max, minimum, median, and maximum parameter values, respectively. CHF indicates congestive heart failure; CO, cardiac output; dP/dTmax, peak rate of pressure rise; −dP/dTmin, peak rate of pressure decline; Ea, arterial elastance; EDP, end‐diastolic pressure; EDV, end‐diastolic volume; EF, ejection fraction; ESP, end‐systolic pressure; ESV, end‐systolic volume; HR, heart rate; SV, stroke volume; SW, stroke work; Tau (G), relaxation time constant using the Gauss–Seidel method; Tau (W), relaxation time constant calculated by Weiss method (regression of log pressure).
P<0.05, CHF vs sham.
P<0.005, CHF vs sham.
Figure 1.

Characteristics of explant‐derived cells (EDCs) and c‐Kit+ cells generated from sham and CHF atria. A, Trichrome staining of rat heart cross‐section shows typical scar area (blue staining of collagen) 6 weeks after MI. B, Trichrome staining of sham and CHF atria shows blue collagen deposition in CHF atria. C, Sham and CHF atria fibroblasts were labeled with anti‐vimentin antibody (red). Nuclei were stained with DAPI (blue). Scale bar=2 mm (A), 200 μm (B), and 20 μm (C). D, Flow cytometry analysis of EDCs generated from sham‐operated (sham) and CHF atrial explants. *P<0.05, CHF vs sham. n=5 per group. E, Absolute number of c‐Kit+ cells isolated from sham or CHF EDCs. n=5 per group. *P<0.05, CHF vs sham. F, TGF‐β1 amount in culture medium conditioned by EDCs generated from sham and CHF explants after 4 or 10 days. TGF‐β1 was normalized to protein. *P<0.05, CHF vs sham. n=4 per group. G, qPCR analysis of c‐Kit+ cells isolated from sham and CHF explant‐derived cells. Fold changes were calculated as a ratio of average 2^(−ΔCт)±SE in the CHF group to the control group. n=6 per group. *P<0.05, CHF vs sham; #P<0.001, CHF vs sham. CHF indicates chronic heart failure; DAPI, 4′,6‐diamidino‐2‐phenylindole; E‐cadh, E‐cadherin; EDCs, explant‐derived cells; EMT, epithelial‐to‐mesenchymal transition; MI, myocardial infarction; qPCR, quantitative polymerase chain reaction; TGF‐β1, transforming growth factor–β1.
CHF Alters EDC Composition
We and others have previously demonstrated that cardiac explants generate several types of cells including fibroblasts and c‐Kit+ progenitors.8,19,33 Cardiac explants produce EDCs of 2 morphologically distinct types: attached cells and round phase‐bright cells. Attached cells were identified as differentiated fibroblasts, whereas round‐phase bright cells were shown to exhibit stem cell properties.8,36 Because CHF modified the cardiac atrial structure (Figure 1B and 1C), we speculated that CHF EDC composition varies from sham EDC composition. EDCs were generated from 6‐week CHF and sham animals and were characterized by flow cytometry. Compared with sham controls, we found that CHF EDCs comprised a higher number of vimentin‐positive fibroblasts35 and α‐smooth muscle actin (SMA)–positive myofibroblasts, and a lower number of c‐Kit‐positive cells (Figure 1D and 1E). CHF cells secreted a significantly greater amount of TFG‐β1 compared with sham EDCs (Figure 1F), possibly because of the high proportion of fibroblasts capable of TGF‐β secretion in CHF EDCs.23,25,37 TGF‐β was previously shown to trigger EMT and transformation of nonfibroblastic cells including progenitor cells into fibroblasts and myofibroblasts.38–39 Therefore, we speculated that in explant culture, a high level of TGF‐β secondary to the CHF condition might affect a phenotype of CHF‐derived c‐Kit+ progenitors.
Characterization of c‐Kit+ Cells Derived From CHF and Sham Explants
It was previously reported that cardiac diseases may negatively affect cardiac progenitor cell properties.15 Here, we compared phenotypes of c‐Kit+ progenitors isolated from sham and CHF EDCs. c‐Kit+ cells were negative for CD34 and CD45, excluding hematopoietic origin of these cells (Figure S1). Because TGF‐β is an EMT inducer, c‐Kit+ cells were analyzed for EMT‐related and pluripotency gene expression (Figure 1G). EMT is typically characterized by upregulation of the epithelial phenotype suppressor Snail and SMA and downregulation of E‐cadherin.40 Expression of Snail was 2.5‐fold higher in CHF‐derived c‐Kit+ cells compared with shams. In contrast, the expression of the epithelial marker E‐cadherin, which is negatively regulated by Snail, was 3‐fold lower in CHF‐derived c‐Kit+ cells. Also, we measured expression of Nanog and Sox2, key transcription factors regulating self‐renewal in stem cells.39–40 The expression of Sox2 was 4.5‐fold lower and that of Nanog was 4.9‐fold lower in CHF‐derived c‐Kit+ cells compared with sham. Taken together, the upregulation of Snail and downregulation of E‐cadherin and pluripotency genes suggest EMT‐like differentiation of CHF‐derived c‐Kit+ cells (Figure 1G). In addition, we observed 2.8‐fold higher expression of Pai1, a direct downstream target of TGF‐β signaling, in CHF‐derived c‐Kit+ cells (Figure 1G), indicating that TGF‐β signaling is upregulated in CHF c‐Kit+ cells compared with shams. Finally, we tested expression of Hey1 and Cyclin D2, readouts of Notch and Wnt signaling, respectively. Both Wnt and Notch pathways were shown to play a role in stem cell maintenance and c‐Kit+ cell generation.19,41 No differences were detected in cyclin D2 expression, although Hey1 expression was downregulated in CHF c‐Kit+ cells compared with shams, suggesting that Notch signaling is downregulated in CHF c‐Kit+ cells compared with shams. In summary, CHF c‐Kit+ cells are characterized by a higher level of TGF‐β signaling and EMT markers and a lower level of pluripotency markers compared with shams.
TGF‐β Treatment Augmented EMT Markers in Sham and Decreased Pluripotent Gene Expressions in c‐Kit+ Cells in Both Sham and CHF
We tested whether the high TGF‐β level in CHF EDC cultures might be responsible for the observed phenotypical differences between sham and CHF c‐Kit+ cells. Cells were treated with TGF‐β1 and assessed for EMT and pluripotency gene expression profiles. TGF‐β treatment of c‐Kit+ cells isolated from either CHF‐ or sham‐induced cell attachment and spindle‐shape morphology typical for fibroblasts (Figure 2A). In sham c‐Kit+ cells, TGF‐β induced Pai1 expression 3.4‐fold and Snail1 and SMA expression 3‐ and 6.5‐fold, respectively, and suppressed E‐cadherin and Sox2 expression 2.5‐ and 2‐fold, respectively, indicative of EMT‐like transformation (Figure 2B). Thus, the TGF‐β‐induced phenotype of sham c‐Kit+ cells is similar to the phenotype of untreated CHF c‐Kit+ cells (Figure S2). In contrast, CHF‐derived c‐Kit+ cells were less responsive to TGF‐β treatment; no significant differences in the expression of Pai1, Snail, and SMA were detected in CHF after the treatment; however, E‐cadherin, Nanog, and Sox2 expression was suppressed at least 2‐fold each (Figure 2B). Next, we tested if TGF‐β signaling in c‐Kit+ cells is transduced via canonical Smad signaling. c‐Kit+ cells were treated with TGF‐β in the presence of TGFβ‐R1 blocker SB or Smad3 phosphorylation blocker SIS. Expression of Pai1 was used as a readout for TGF‐β signaling.42 We found that either SB or SIS abolished the effect of TGF‐β on Pai1 expression (Figure S3), indicating that in c‐Kit+ cells, TGF‐β signaling is TGF‐β‐R1‐ and Smad3 dependent. These data are consistent with previous work showing Smad2/3 upregulation by TGF‐β in bone marrow–derived c‐Kit+ cells.43 Taken together, these data indicated that cardiac explant‐derived c‐Kit+ cells are susceptible to TGF‐β exposure in a TGF‐β‐R1‐ and Smad3‐dependent manner.
Figure 2.
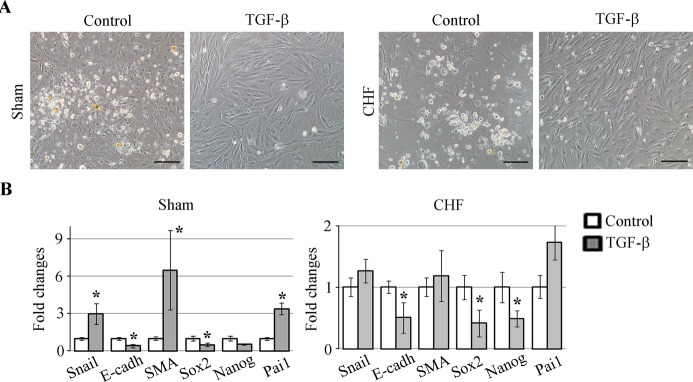
Effects of TGF‐β on sham and CHF‐derived c‐Kit+ cells. c‐Kit+ cells were isolated from sham or CHF EDCs and treated with 10 ng of TGF‐β1 for 7 days. A, TGF‐β caused morphology changes in both sham and CHF‐derived c‐Kit+ cells typical for EMT. Control cells appeared as semiadherent clusters, whereas TGF‐β‐treated cells became attached and elongated. Scale bar=100 μm. B, qRT‐PCR analyses of EMT‐ and pluripotency‐related gene expressions in TGF‐β treated c‐Kit+ cells from sham and CHF explants. Fold changes were calculated as a ratio of the expression in the TGF‐β‐treated group to the expression in the control group. n=5 per group. *P<0.05, TGF‐β treated vs untreated control. CHF indicates chronic heart failure; E‐cadh, E‐cadherin; EDC, explant‐derived cell; EMT, epithelial to mesenchymal transition; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; SMA, α‐smooth muscle actin; TGF‐β, transforming growth factor–β.
TGF‐β Inhibition in c‐Kit+ Cells Diminished EMT Markers and Enhanced Pluripotency Marker Expression
FBS is a source of TGF‐β1 in culture medium. A 10% FBS‐supplemented medium contains approximately 750 pg/mL of TGF‐β1 as measured by ELISA. Moreover, during EDC generation, c‐Kit+ cells coexist with fibroblasts, which release additional TGF‐β in culture (Figure 1F). Compared with shams, CHF c‐Kit+ cells are continuously exposed to a higher level of TGF‐β that may trigger undesirable fibroblast differentiation of CHF c‐Kit+ cells. To determine if inhibition of TGF‐β signaling in c‐Kit+ cells attenuates EMT markers and preserves pluripotency gene expression of these cells, sham or CHF c‐Kit+ cells were treated with TGF‐β inhibitors SB or SIS for 7 days and assessed for EMT and pluripotency gene expression. TGF‐β signaling suppression was confirmed by the reduction of Pai1 gene expression and the decrease in the ratio of pSmad 2/3 to total Smad 2/3 protein (Figure 3B and 3C). Both sham and CHF c‐Kit+ cells treated with SB or SIS formed clusters of small, round phase‐bright cells similar to cardiosphere‐forming cardiac stem cells8 (Figure 3A). We found that in sham c‐Kit+ cells, both SB and SIS reduced SMA gene expression 2‐ and 5‐fold, respectively, whereas in CHF c‐Kit+ cells, SMA reduction was >10‐fold for both inhibitors. Also, SB upregulated E‐cadherin expression by 6.2‐ and 3.6‐fold in sham and CHF c‐Kit+ cells, respectively. SIS upregulated E‐cadherin expression by 5‐fold in sham c‐Kit+ cells, with no effect on CHF c‐Kit+ cells (Figure 3C). In contrast, SB increased Sox2 expression by 5.2‐ and 4.1‐fold in sham and CHF cells, respectively, whereas SIS upregulated Sox2 by 4‐fold only in sham c‐Kit+ cells. Similarly, SB upregulated Nanog expression by 2‐ and 2.4‐fold in sham and CHF c‐Kit+ cells, respectively, whereas SIS upregulated Nanog by 2.2‐fold in sham cells only, with no effect in CHF cells. Concurrently, Nanog protein levels were significantly upregulated in sham c‐Kit+ cells and tended to increase in CHF c‐Kit+ cells on TGF‐β inhibitor treatment (Figure 3D and 3E); however, no difference was observed in Sox2 protein expression (data not shown). Collectively, these results indicate that although TGF‐β inhibitors normalized the majority of examined EMT markers in CHF c‐Kit+ cells toward sham controls, they only had a limited effect on stem cell markers (Figure S4).
Figure 3.

TGF‐β inhibition suppressed EMT and upregulated pluripotency gene expression in c‐Kit+ cells. TGF‐β signaling was inhibited by suppression of TGF‐β receptor type 1 (SB) or by suppression of Smad2/3 phosphorylation (SIS) for 7 days. A, SB and SIS treatments increased amounts of round phase‐bright cells compared with control. Scale bar=100 μm. B, Western blot analysis of c‐Kit+ cells treated with SB and SIS showed reductions of pSmad2/3 levels. C, qRT‐PCR analysis of EMT‐ and pluripotency‐related gene expression in SB‐ and SIS‐treated c‐Kit+ cells from sham and CHF explants. Fold changes were calculated as a ratio of the expression in the SB‐ or SIS‐treated group to the expression in the control group. n=5 per condition. *Fold changes >2. D, Western blot analysis of Nanog in SB‐ and SIS‐treated c‐Kit+ cells. β‐Actin was used as a loading control. Representative blots are shown. E, Densitometry analysis of Nanog. n=3 per condition. *P<0.05, inhibitor‐treated cells vs untreated control. CHF indicates chronic heart failure; EMT, epithelial to mesenchymal transition; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; SMA, α‐smooth muscle actin; TGF‐β, transforming growth factor–β.
TGF‐β Inhibitors Augmented In Vitro Differentiation Potential of c‐Kit+ Cells Toward Cardiomyocyte‐Like Cells
We compared sham‐ and CHF‐derived c‐Kit+ cells' capacity to differentiate toward cardiomyocyte‐like cells in vitro and determined whether TGF‐β inhibitors affect this process. Sham and CHF c‐Kit+ cells were cultured in cardiomyocyte differentiation medium in the presence or absence of TGF‐β inhibitors SB and SIS and assessed for cardiac TnT expression. Immunocytochemistry analysis showed higher differentiation potential of sham compared with CHF c‐Kit+ cells (11.7% versus 8.0% TnT+ cells in sham versus CHF c‐Kit+ cells, respectively). The presence of SB or SIS in culture medium increased the number of TnT‐expressing cells in both sham and CHF c‐Kit+ cells, but with much higher effect in sham c‐Kit+ cells (24.4% and 36.2% TnT+ cells in sham c‐Kit+ cells treated with SB and SIS, respectively; 15.4% and 19.6% in CHF c‐Kit+ cells; Figure 4A and 4B). These data demonstrated that compared with shams, CHF c‐Kit+ cells had weaker potential to differentiate toward cardiomyocyte‐like cells. Moreover, the cardiomyogenic potential of both sham and CHF c‐Kit+ cells can be augmented by TGF‐β inhibitors.
Figure 4.
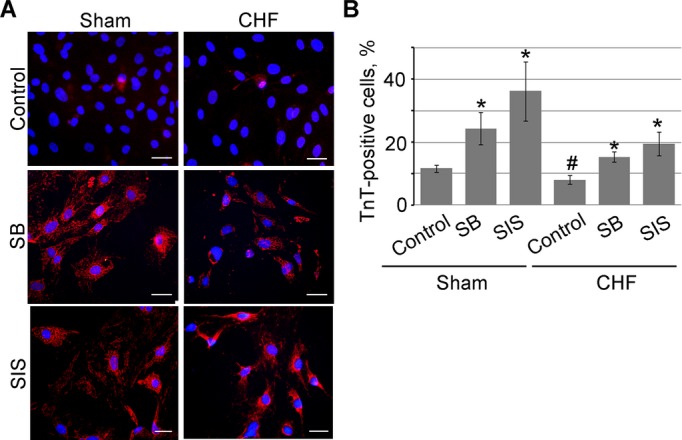
TGF‐β inhibition facilitates c‐Kit+ cell differentiation in vitro. c‐Kit+ cells were cultured in the cardiac differentiation medium in the presence of TGF‐β signaling inhibitors SB and SIS. A, Cells were labeled for cardiac troponin T (TnT, red). Nuclei were stained with DAPI (blue). Scale bar=20 μm. B, SB and SIS increased the percentage of TnT+ cells. n=3 per condition. *P<0.05 inhibitor‐treated cells vs untreated control; #P<0.05 untreated CHF vs untreated sham cells. CHF indicates chronic heart failure; DAPI, 4′,6‐diamidino‐2‐phenylindole; SB, suppression of TGF‐β receptor type 1; SIS, suppression of Smad2/3 phosphorylation; TGF‐β, transforming growth factor–β.
TGF‐β Inhibitors Increased c‐Kit+ Cell Yield From Sham and CHF Explants
We previously demonstrated that cardiac explant‐derived cells undergo EMT in culture resulting in a time‐dependent increase in fibroblast number. Therefore, in the current study we speculated that the addition of TGF‐β inhibitors in the explant culture medium can attenuate this process and improve explant‐derived progenitor cell yield. Explant outgrowths were generated in the presence of the TGF‐β inhibitors SB or SIS, and EDCs were analyzed by FACS. After 21 days in culture, no differences were detected in the apoptotic markers activated caspase 3 and annexin V (Figure S5A through S5C). Also, no differences were detected in cell proliferation and cell‐cycle status, as was demonstrated by Ki67 and propidium iodide staining (Figure S5D through S5G). Therefore, we concluded that TGF‐β inhibitors had no effect on cell growth. After 21 days in culture, we observed a higher number of small, round phase‐bright cell clusters when explants were cultured in the presence of SB and, to a lesser extent, in the presence of SIS compared with controls (Figure 5A). In addition, when generated in the presence of inhibitors, EDCs were smaller in size and appeared tightly connected, similar to cobblestone epicardial cells. Flow cytometry results demonstrated that TGF‐β inhibitors significantly decreased the percentage of vimentin‐positive fibroblasts in both sham and CHF‐derived EDCs (55% and 35% for SB and SIS, respectively in sham EDCs; 42% and 34% in CHF EDCs). In CHF EDCs, the number of SMA‐positive myofibroblasts was reduced by 63.3% and 62.8% when generated in the presence of SB and SIS, respectively. In addition, SIS reduced the number of CD90‐positive cells by 52.4% and 53%, in sham and CHF EDCs, respectively (Figure 5B). In contrast, SB treatment increased the number of c‐Kit+ cells by 36.6% and 75% in sham and CHF EDCs, respectively, whereas SIS treatment resulted in a 52% increase in the percentage of c‐Kit+ cells in sham EDCs but had no effect on CHF EDCs (Figure 5B). Our data showed no statistically significant interaction between CHF and treatments (eg, SB or SIS).
Figure 5.
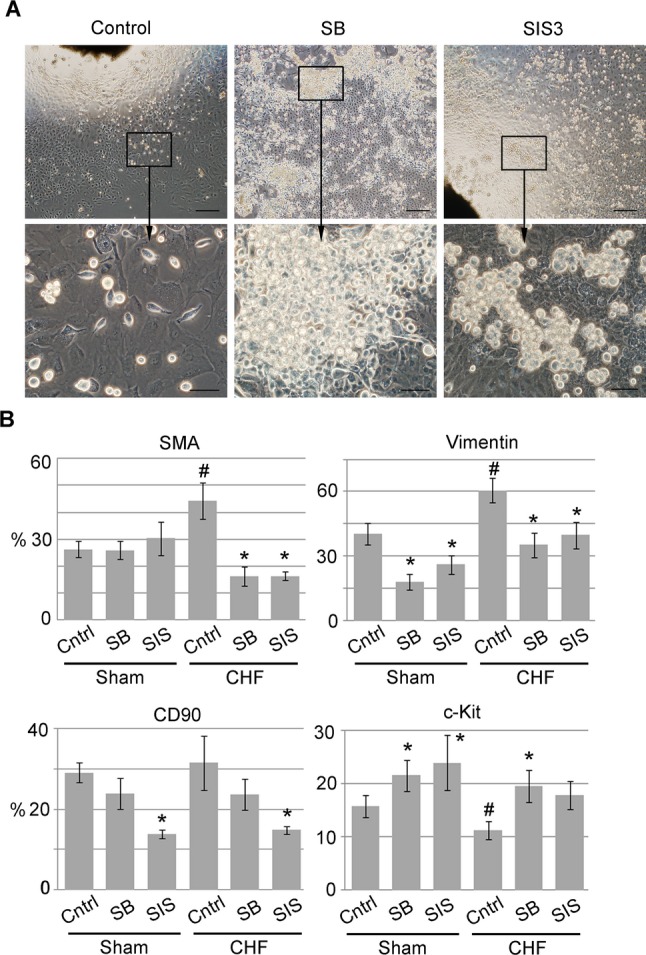
TGF‐β inhibitors increased c‐Kit+ cell yield. A, SB or SIS was added to explants culture on day 2. Vehicle solution (DMSO) was added to control explants. Scale bars=200 μm (top row) and 50 μm (bottom row). B, FACS analysis of sham or CHF EDCs generated in the presence of SB or SIS. n=5 per condition. *P<0.05, inhibitor‐treated cells vs untreated control. #P<0.05, CHF untreated vs sham untreated. CHF indicates chronic heart failure; DMSO, dimethyl sulfoxide; EDCs, explant‐derived cells; FACS, fluorescence‐activated cell sorting; SB, suppression of TGF‐β receptor type 1; SMA, α‐smooth muscle actin; SIS, suppression of Smad2/3 phosphorylation; TGF‐β, transforming growth factor–β.
In summary, our results demonstrated that TGF‐β signaling inhibition improves yield and function of c‐Kit+ cells from sham and CHF cardiac explants.
Discussion
The current study was designed to determine if heart failure affects the yield and phenotype of atrial explant‐derived c‐Kit+ progenitors and to elucidate mechanisms of their cellular regulation. We showed that CHF explants produce more fibroblasts and fewer c‐Kit+ progenitors. We also found that sham c‐Kit+ cells are more susceptible to TGF‐β signaling modulation than are CHFs; TGF‐β signaling inhibition increased c‐Kit+ cell yield, downregulated EMT markers, augmented Nanog expression, and stimulated cardiomyogenic potential in both sham and CHF c‐Kit+ cells, but with a much higher impact in shams.
The low c‐Kit+ cell yield from CHF explants could be because of several factors associated with the CHF condition. First, consistent with previous reports,34 we found that CHF causes atrial fibrosis, and therefore, CHF explants can generate more fibroblasts. Second, post‐MI cardiac resident c‐Kit+ cell depletion can contribute to the low progenitor yield. Finally, during explant culturing, other type of cells, such as cardiomyocytes and epicardial cells, can contribute to the c‐Kit+ cell pool via dedifferentiation,10–11 and therefore, a low number of CHF explant‐derived c‐Kit+ cells may reflect abnormality in the dedifferentiation mechanisms because of the CHF condition.
Previously, it was reported that EMT plays a role in the generation of stem cells.44–46 Our group have recently shown that in explant‐derived cells c‐Kit+ progenitors undergo EMT in culture.19 “Abnormal” EMT in response to injury was described in various organs including the heart.21 Generally, pathological EMT is involved in organ degeneration processes such a fibrosis.20,40 The end result of this “aggravated” form of EMT is formation of terminally differentiated fibroblasts or myofibroblasts producing extracellular matrix proteins.20,22 Recent studies demonstrated that up to 75% of myofibroblasts in fibrotic hearts are EMT derived.47 However, depending on cell type and state of differentiation, EMT does not necessarily generate fibroblasts as an end product. For example, several lines of evidence demonstrated that EMT can generate stem‐like cells.44–46 Also, during heart development, primitive cardiac progenitors undergo multiple rounds of EMT without losing the stemness.40 Explant‐derived c‐Kit+ cells exhibit many features of mesenchymal stem cells and appear to be in a fine balance between 2 fates, mesenchymal stem cells and myofibroblasts.48 It is unclear which factors regulate the molecular switch between fibroblast‐ and stem cell–producing EMTs because at the cellular level both processes are controlled by similar signaling mechanisms.40 EMT is widely regulated by signals from the extracellular environment. Thus, a sustained post‐MI TGF‐β elevation was shown to play a central role in cardiac fibrosis by inducing abnormal EMT.24–25,49 In nonfractionated EDC cultures, c‐Kit+ cells coexist with TGF‐β‐releasing fibroblasts, and compared with sham, CHF c‐Kit+ cells are continuously exposed to significantly higher TGF‐β in an amount sufficient to tilt the cell fate balance toward myofibroblasts.50 Therefore, we speculated that excessive TGF‐β exposure plays a role in the low level of stem and the high level of EMT and fibroblast markers in CHF c‐Kit+ cells. We confirmed this hypothesis by demonstrating stem cell marker loss and further upregulation of EMT markers on TGF‐β stimulation and the reverse effects of TGF‐β inhibition in c‐Kit+ cells.
Previous studies showed that small‐molecule TGF‐β inhibitors are capable of inducing stable changes in gene expression, facilitating induced pluripotent cell reprogramming by acting on a discrete cellular pathway rather than global chromatin modifications.51 Similar to cellular intermediates during reprogramming, TGF‐β inhibition stimulated Nanog expression and alleviated in vitro differentiation of c‐Kit+ cells toward a cardiomyocyte lineage, suggesting that small‐molecule TGF‐β inhibitors promote c‐Kit+ cell plasticity. Interestingly, despite a higher base level of TGF‐β signaling in CHF c‐Kit+ cells, these cells are less susceptible to TGF‐β modulations than sham cells. This sensitivity loss could be a result of downregulation of TGF‐β signaling components, signaling pathway saturation, and/or a more advanced differentiation stage of CHF c‐Kit+ cells associated with plasticity loss. This assumption is supported by a previous study showing that small‐molecule TGF‐β inhibitors effectively reactivate the pluripotency program in intermediate‐phenotype cells but not in terminally differentiated fibroblasts.51 We also found that, although both studied inhibitors downregulated EMT markers and stimulated in vitro cardiac differentiation, TGF‐β‐R1 inhibition is more efficient in improving CHF c‐Kit+ cell yield. This finding indicates that the TGF‐β signal in c‐Kit+ cells is not entirely transduced via Smad 2/3. Non‐Smad signaling including RhoA, Rac, MAPK, and PI3/Akt kinase may contribute to CHF c‐Kit+ cell regulation.52
One potential limitation of this study is a relatively small sample size. Nevertheless, we were able to detect many differences between the sham and CHF groups as well as the effects of treatments.
In conclusion, this study has described TGF‐β‐dependent phenotypical differences between sham‐ and CHF explant‐derived c‐Kit+ cells and elucidated the TGF‐β signaling role in c‐Kit+ cell regulation. Our findings are important in optimizing the scale‐up production and enhancing the efficacy of c‐Kit+ cells needed for clinical application.
Sources of Funding
This work was supported by the National Institute of Aging (NIH RO1 AG027263) and by the Sun Health Foundation.
Disclosures
None.
Acknowledgments
The authors thank Lorraine Feehery for technical support.
References
- 1.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003; 114:763-776 [DOI] [PubMed] [Google Scholar]
- 2.Johnston PV, Sasano T, Mills K, Evers R, Lee ST, Smith RR, Lardo AC, Lai S, Steenbergen C, Gerstenblith G, Lange R, Marban E. Engraftment, differentiation, and functional benefits of autologous cardiosphere‐derived cells in porcine ischemic cardiomyopathy. Circulation. 2009; 120:1075-1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003; 100:12313-12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E. Regenerative potential of cardiosphere‐derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007; 115:896-908 [DOI] [PubMed] [Google Scholar]
- 5.Carr CA, Stuckey DJ, Tan JJ, Tan SC, Gomes RS, Camelliti P, Messina E, Giacomello A, Ellison GM, Clarke K. Cardiosphere‐derived cells improve function in the infarcted rat heart for at least 16 weeks—an MRI study. PLoS ONE. 2011; 6:e25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR. Postnatal ISL1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005; 433:647-653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. CD31− but Not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ Res. 2005; 97:52-61 [DOI] [PubMed] [Google Scholar]
- 8.Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004; 95:911-921 [DOI] [PubMed] [Google Scholar]
- 9.He JQ, Vu DM, Hunt G, Chugh A, Bhatnagar A, Bolli R. Human cardiac stem cells isolated from atrial appendages stably express c‐Kit. PLoS ONE. 2011; 6:e27719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Meglio F, Castaldo C, Nurzynska D, Romano V, Miraglia R, Bancone C, Langella G, Vosa C, Montagnani S. Epithelial‐mesenchymal transition of epicardial mesothelium is a source of cardiac CD117‐positive stem cells in adult human heart. J Mol Cell Cardiol. 2010; 49:719-727 [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Li TS, Lee ST, Wawrowsky KA, Cheng K, Galang G, Malliaras K, Abraham MR, Wang C, Marban E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS ONE. 2010; 5:e12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E. Intracoronary cardiosphere‐derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012; 379:895-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, Pappas P, Tatooles A, Stoddard MF, Lima JA, Slaughter MS, Anversa P, Bolli R. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012; 126:S54-S64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan HH, Meher Homji Z, Gomes RS, Sweeney D, Thomas GN, Tan JJ, Zhang H, Perbellini F, Stuckey DJ, Watt SM, Taggart D, Clarke K, Martin‐Rendon E, Carr CA. Human cardiosphere‐derived cells from patients with chronic ischaemic heart disease can be routinely expanded from atrial but not epicardial ventricular biopsies. J Cardiovasc Transl Res. 2012; 5:678-687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cesselli D, Beltrami AP, D'Aurizio F, Marcon P, Bergamin N, Toffoletto B, Pandolfi M, Puppato E, Marino L, Signore S, Livi U, Verardo R, Piazza S, Marchionni L, Fiorini C, Schneider C, Hosoda T, Rota M, Kajstura J, Anversa P, Beltrami CA, Leri A. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011; 179:349-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D'Amario D, Fiorini C, Campbell PM, Goichberg P, Sanada F, Zheng H, Hosoda T, Rota M, Connell JM, Gallegos RP, Welt FG, Givertz MM, Mitchell RN, Leri A, Kajstura J, Pfeffer MA, Anversa P. Functionally competent cardiac stem cells can be isolated from endomyocardial biopsies of patients with advanced cardiomyopathies. Circ Res. 2011; 108:857-861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rota M, LeCapitaine N, Hosoda T, Boni A, De Angelis A, Padin‐Iruegas ME, Esposito G, Vitale S, Urbanek K, Casarsa C, Giorgio M, Luscher TF, Pelicci PG, Anversa P, Leri A, Kajstura J. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ Res. 2006; 99:42-52 [DOI] [PubMed] [Google Scholar]
- 18.Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005; 102:8692-8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zakharova L, Nural‐Guvener H, Gaballa MA. Cardiac explant‐derived cells are regulated by Notch‐modulated mesenchymal transition. PLoS ONE. 2012; 7:e37800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galichon P, Hertig A. Epithelial to mesenchymal transition as a biomarker in renal fibrosis: are we ready for the bedside? Fibrogenesis Tissue Repair. 2011; 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou B, Pu WT. Epicardial epithelial‐to‐mesenchymal transition in injured heart. J Cell Mol Med. 2011; 15:2781-2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010; 107:1304-1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Lamouille S, Derynck R. TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res. 2009; 19:156-172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bujak M, Frangogiannis NG. The role of TGF‐beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007; 74:184-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zavadil J, Bottinger EP. TGF‐beta and epithelial‐to‐mesenchymal transitions. Oncogene. 2005; 24:5764-5774 [DOI] [PubMed] [Google Scholar]
- 26.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF‐beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001; 276:17058-17062 [DOI] [PubMed] [Google Scholar]
- 27.Austin AF, Compton LA, Love JD, Brown CB, Barnett JV. Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFbeta. Dev Dyn. 2008; 237:366-376 [DOI] [PubMed] [Google Scholar]
- 28.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005; 19:2783-2810 [DOI] [PubMed] [Google Scholar]
- 29.Javelaud D, Mauviel A. Mammalian transforming growth factor‐betas: Smad signaling and physio‐pathological roles. Int J Biochem Cell Biol. 2004; 36:1161-1165 [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell. 2003; 113:685-700 [DOI] [PubMed] [Google Scholar]
- 31.Gaballa MA, Goldman S. Gene transfer of endothelial nitric oxide isoform decreases rat hindlimb vascular resistance in vivo. Hum Gene Ther. 2000; 11:1637-1646 [DOI] [PubMed] [Google Scholar]
- 32.Zakharova L, Mastroeni D, Mutlu N, Molina M, Goldman S, Diethrich E, Gaballa MA. Transplantation of cardiac progenitor cell sheet onto infarcted heart promotes cardiogenesis and improves function. Cardiovasc Res. 2010; 87:40-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis DR, Kizana E, Terrovitis J, Barth AS, Zhang Y, Smith RR, Miake J, Marban E. Isolation and expansion of functionally‐competent cardiac progenitor cells directly from heart biopsies. J Mol Cell Cardiol. 2010; 49:312-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casaclang‐Verzosa G, Gersh BJ, Tsang TS. Structural and functional remodeling of the left atrium: clinical and therapeutic implications for atrial fibrillation. J Am Coll Cardiol. 2008; 51:1-11 [DOI] [PubMed] [Google Scholar]
- 35.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005; 65:40-51 [DOI] [PubMed] [Google Scholar]
- 36.Davis DR, Zhang Y, Smith RR, Cheng K, Terrovitis J, Malliaras K, Li TS, White A, Makkar R, Marban E. Validation of the cardiosphere method to culture cardiac progenitor cells from myocardial tissue. PLoS ONE. 2009; 4:e7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taipale J, Koli K, Keski‐Oja J. Release of transforming growth factor‐beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J Biol Chem. 1992; 267:25378-25384 [PubMed] [Google Scholar]
- 38.Wang N, Ren GD, Zhou Z, Xu Y, Qin T, Yu RF, Zhang TC. Cooperation of myocardin and Smad2 in inducing differentiation of mesenchymal stem cells into smooth muscle cells. IUBMB Life. 2012; 64:331-339 [DOI] [PubMed] [Google Scholar]
- 39.Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, Liu F. Inhibition of TGF‐beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma‐associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012; 30:2810-2819 [DOI] [PubMed] [Google Scholar]
- 40.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell. 2009; 139:871-890 [DOI] [PubMed] [Google Scholar]
- 41.Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005; 6:314-322 [DOI] [PubMed] [Google Scholar]
- 42.Ghosh AK, Vaughan DE. PAI‐1 in tissue fibrosis. J Cell Physiol. 2012; 227:493-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li TS, Komota T, Ohshima M, Qin SL, Kubo M, Ueda K, Hamano K. TGF‐beta induces the differentiation of bone marrow stem cells into immature cardiomyocytes. Biochem Biophys Res Commun. 2008; 366:1074-1080 [DOI] [PubMed] [Google Scholar]
- 44.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial‐mesenchymal transition. PLoS ONE. 2008; 3:e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N, Rocco G. Epithelial to mesenchymal transition by TGFbeta‐1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS ONE. 2011; 6:e21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007; 13:952-961 [DOI] [PubMed] [Google Scholar]
- 48.Tateishi K, Ashihara E, Honsho S, Takehara N, Nomura T, Takahashi T, Ueyama T, Yamagishi M, Yaku H, Matsubara H, Oh H. Human cardiac stem cells exhibit mesenchymal features and are maintained through Akt/GSK‐3beta signaling. Biochem Biophys Res Commun. 2007; 352:635-641 [DOI] [PubMed] [Google Scholar]
- 49.Gramley F, Lorenzen J, Koellensperger E, Kettering K, Weiss C, Munzel T. Atrial fibrosis and atrial fibrillation: the role of the TGF‐beta1 signaling pathway. Int J Cardiol. 2010; 143:405-413 [DOI] [PubMed] [Google Scholar]
- 50.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta‐1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003; 278:21113-21123 [DOI] [PubMed] [Google Scholar]
- 51.Ichida JK, Blanchard J, Lam K, Son EY, Chung JE, Egli D, Loh KM, Carter AC, Di Giorgio FP, Koszka K, Huangfu D, Akutsu H, Liu DR, Rubin LL, Eggan K. A small‐molecule inhibitor of TGF‐beta signaling replaces Sox2 in reprogramming by inducing Nanog. Cell Stem Cell. 2009; 5:491-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mu Y, Gudey SK, Landstrom M. Non‐Smad signaling pathways. Cell Tissue Res. 2012; 347:11-20 [DOI] [PubMed] [Google Scholar]