

Abstract
Background
Coronary artery disease and ischemic cardiomyopathy represent the leading cause of heart failure and continue to grow at exponential rates. Despite widespread availability of coronary bypass surgery and percutaneous coronary intervention, subsequent ischemic events and progression to heart failure continue to be common occurrences. Previous studies have shown that a subgroup of patients develop collateral blood vessels that serve to connect patent and occluded arteries and restore perfusion to ischemic territories. The presence of coronary collaterals has been correlated with improved clinical outcomes; however, the molecular mechanisms governing this process remain largely unknown.
Methods and Results
To date, no mouse models of coronary arterial growth have been described. Using a closed‐chest model of myocardial ischemia, we have demonstrated that brief episodes of repetitive ischemia are sufficient to promote the growth of both large coronary arteries and the microvasculature. Induction of large coronary artery and microvascular growth resulted in improvements in myocardial perfusion after prolonged ischemia and protected from subsequent myocardial infarction. We further show that repetitive ischemia did not lead to increased expression of classic proangiogenic factors but instead resulted in activation of the innate immune system and recruitment of macrophages to growing blood vessels.
Conclusions
These studies describe a novel model of coronary angiogenesis and implicate the cardiac macrophage as a potential mediator of ischemia‐driven coronary growth.
Keywords: coronary, collateral, macrophage, repetitive ischemia, coronary angiogenesis
Introduction
Myocardial infarction (MI) and ischemic heart disease are among the leading causes of mortality in the industrial world.1 Despite efforts to minimize ischemic damage through timely revascularization and initiation of effective medical therapies, many patients go on to experience recurrent ischemic events and develop heart failure.1–2
Due to disease complexity and anatomical constraints, a significant number of patients are not candidates for either coronary bypass surgery or percutaneous coronary intervention. As a result, this population has a strikingly high annual mortality rate compared with those who undergo complete revascularization.3–4 These unfortunate results underscore the need for improved revascularization strategies.
Previously, it has been recognized that a subgroup of patients form collateral vasculature. These blood vessels connect patent and occluded arteries and restore perfusion to ischemic territories. Importantly, the presence of coronary collaterals has been correlated with improved clinical outcomes in patients with ischemic heart disease. Patients who develop angiographically visible collateral vasculature have reduced rates of cardiac mortality and major adverse cardiac events, compared with patients without collaterals. In addition, physiological assessment of collateral perfusion as measure by collateral flow index revealed improved outcomes in patients with increased collateral flow.5–9 Given these findings, there has been considerable excitement focused on identifying growth factors and signaling pathways that may promote the growth of new coronary vasculature. This approach has been termed therapeutic angiogenesis and involves administration of growth factors or gene therapy to the injured myocardium. In this context, coronary growth has been postulated to occur through a combination of angiogenesis and arteriogenesis.10–11
Little is known regarding the development and growth of coronary collaterals. In fact, much of the data available describing mechanisms of collateral growth are derived from mouse models of hindlimb ischemia. Innate immune system activation, increased classic proangiogenic growth factor expression including fibroblast growth factors and vascular endothelial growth factors (VEGF), and recruitment of endothelial progenitor cells have been implicated in collateral growth.12–13 While hindlimb ischemia represents a robust model of collateralization, it is unclear whether this system adequately depicts what occurs in the adult heart.
With respect to the heart, particular attention has been focused on understanding mechanisms of blood vessel growth in the border zone after experimental MI. In this setting, increased expression of multiple classic proangiogenic factors has been identified (fibroblast growth factors, VEGFs, and angiopoientins) with the ability to trigger coronary artery growth in animal models. However, these factors have failed to translate into the clinical arena, as multiple clinical studies revealed minimal efficacy.14–18 More recently, several other candidate molecules have been identified that can induce coronary angiogenesis after MI including sonic hedgehog, thymosin β4, PR39, and platelet‐derived growth factor β. Importantly, few of these growth factor have been demonstrated to promote large coronary artery or collateral growth.19–27 Together, these data suggest that there are critical differences between signaling pathways that promote coronary angiogenesis after MI and coronary growth in ischemic heart disease.
A key limitation toward understanding how coronary vascular growth occurs is the paucity of genetically tractable small animal models. Porcine and rat models have been described previously,28–29 but these systems are limited to the use of pharmacologic agents and gene therapy. To date, there are no published murine models. The development of a mouse model of coronary growth represents an important step in elucidating the cellular and molecular mechanisms by which coronary growth occurs in the adult heart.
Recently, a closed chest animal model has been described that allows transient and repeated occlusion of the left anterior descending coronary artery (LAD) in intact mice.30 This system has enabled investigators to study the effects of ischemia and reperfusion in both the acute and the chronic setting and avoids interpretative issues related to thoracotomy induced inflammation.31 With respect to coronary collateral development, one can modulate this technique to determine whether brief repeated ischemic events are sufficient to trigger coronary growth in intact animals. Such a scenario mirrors stuttering angina, a key trigger of coronary collateralization in patients with ischemic heart disease. Importantly, this model combines the power of mouse genetics with a clinically relevant model of ischemic injury.
Using the closed chest ischemia/reperfusion (I/R) model, we demonstrate that repetitive brief episodes of LAD ischemia are sufficient to trigger coronary growth on both the macrovascular and microvascular levels. Moreover, we demonstrate that ischemia triggered coronary growth protects from subsequent MI as a result of improved regional coronary perfusion. Last, we show minimal alterations in classic proangiogenic growth factor expression and instead implicate a role for innate immunity and cardiac macrophages in this process.
Material and Methods
Mouse Model of Ischemia‐Induced Coronary Angiogenesis
The closed‐chest model of I/R has been described previously by Entman and colleagues.30–33 Briefly, 2‐ to 4‐month‐old C57/B6 mice were anesthetized with sodium pentobarbital, intubated, and mechanically ventilated. A midline incision was made exposing the heart, and an 8‐0 Prolene suture was then placed around the proximal LAD to maximize the ischemic area. The suture was then threaded through a 1 mm piece of polyethylene tubing forming a loose snare. This served as the occluder. Each end of the suture was exteriorized through the thorax and stored in a subcutaneous pocket. The skin was then closed over the exteriorized suture ends with 5‐0 Prolene. Instrumented mice were allowed to recover for 2 weeks before induction of ischemia. A baseline echocardiogram was then performed to ensure that LAD ligation did not occur. Ischemia was induced after anesthetizing the animals with 1.5% isoflurane, opening the skin over the subcutaneous pocket, and exposing the exteriorized suture. The suture ends were dissected away from any subcutaneous tissue, and tension was exerted until ST‐segment elevation was seen on the ECG. Ischemia was confirmed by visualizing wall motion abnormality using simultaneous echocardiography. Following 15 minutes of ischemia time, tension was released and the suture ends were placed back into the subcutaneous pocket. The skin was then closed as described earlier. Sham‐operated animals underwent the identical procedure with the exception that tension was not placed on the suture ends.
Effect of I/R Injury on Coronary Growth and Infarct Size
We studied 2 groups of mice. First, to determine whether repetitive ischemia was sufficient to provoke increased coronary growth, we subjected one group of mice to 15 minutes of I/R every other day for a total of 3 sessions (brief I/R injury). The appropriate controls consisted of sham‐operated animals, as described earlier. Mice were killed 1 day after the last episode of ischemic injury. Second, to determine the functional significance of repetitive I/R injury, we subjected mice that had previously undergone 3 to 6 episodes of brief ischemic injury to 2 hours of ischemia followed by reperfusion. One hundred twenty minutes of ischemic injury has been shown to produce transmural MI in mice.34 Mice were subjected to prolonged I/R 10 days after brief repetitive ischemia, to minimize the confounding effects of ischemic preconditioning. Following reperfusion and recovery from anesthesia, echocardiography was performed to measure the baseline wall motion abnormality, left ventricular (LV) systolic function, and LV chamber size. A second echocardiogram was performed 28 days later to examine recovery of LV structure and function.35–36 Evans blue staining was performed in the mice subjected to repetitive ischemia or sham operation to examine area at risk as described previously.37 Trichrome staining was used to identify scar and measure infarct size.
Echocardiography
Mouse echocardiography was performed in the Washington University Mouse Cardiovascular Phenotyping Core facility using the VisualSonics 770 Echocardiography System. Avertin (0.005 mL/g) was used for sedation based on previously established methods of infarct quantitation.38 Both 2‐dimensional and M‐mode images were obtained in the long‐ and short‐axis views. Ejection fraction (EF) and LV dimension were calculated using standard techniques. LV dimension was normalized to body weight. The akinetic region was calculated by measuring the area of the akinetic portion of the LV myocardium and normalizing it to the area of the total LV myocardium. Measurements were performed on 3 independently acquired images per animal, by investigators who were blinded to experimental group.
Histology, Immunohistochemistry, Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Staining, and Microfil Perfusion Casting
Tissues were fixed in 2% PFA overnight at 4°C, dehydrated in 70% ethyl alcohol, and embedded in paraffin. Four micrometer sections were cut and stained with hematoxylin and eosin. For chromogenic immunohistochemical staining, 4‐μm paraffin sections were deparaffinized, rehydrated, peroxidase blocked, and antigen exposed by boiling in citric acid 10 mmol/L. Primary antibodies included rat anti‐MAC3 1:400 (BD), rabbit anti‐CD11b 1:400 (Abgent), and rat anti‐Ly6G 1:50 (BD). Biotinylated secondary antibodies were purchased from Vector Laboratories and used at 1:200. Signal was detected using the Elite ABC Kit (Vector Laboratories). For colabeling experiments and PECAM staining, tissues were fixed in 2% paraformaldehyde (PFA) overnight at 4°C, embedded in Tissue‐Tek OCT compound, (Sakura Finetek) infiltrated with sucrose, and frozen, and 10‐ to 12‐μm cryosections were cut. Primary antibodies used were rat anti‐CD31/PECAM 1:200 (BD), mouse anti‐SMA‐FITC 1:400 (Sigma‐Aldrich), and CD11b 1:400 (Abgent). Anti‐rabbit Alexa Fluor 647 was used to visualize CD11b staining in combination with SMA‐FITC. Anti‐CD31/PECAM staining was visualized using anti‐rat biotinylated secondary antibody and Elite ABC Kit (Vector Laboratories). Immunofluorescence was visualized on a Zeiss confocal microscopy system. Blood vessel density was quantified by examining at least 4 similarly oriented sections from 4 independent samples in blinded fashion. Isotype controls were used to validate all antibodies used in this study. Macrophages were considered to be associated with blood vessels if they were located within 20 μm.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed on paraffin‐embedded sections immediately after repetitive ischemia as specified by the manufacturer (In Situ Cell Death Detection Kit; R&D Systems). Picrosirius red staining was performed 10 days after repetitive ischemia. For perfusion casting, Microfil perfusion reagent (yellow, Microfil, Flow Tech, Inc) was perfused retrograde into the ascending aorta per the manufacturer's specifications. To preferentially visualize the arterial tree, microfil injection was discontinued once the coronary sinus began filling. Left coronary artery (LCA) and right coronary artery (RCA) sizes were quantified by measuring the area the vessel covered within a region of interest (ROI). Hearts were photographed at low magnification (×20), and third order branches were used to define the area covered by the LCA or RCA. Data were displayed as the percentage of the area the ROI covered. For the LCA, the ROI was the anterior wall with the LCA oriented in the center of the region. For the RCA, the ROI was the inferior wall with the RCA oriented in the center of the region.
Collaterals in Coronary Allograft Vasculopathy
To determine whether macrophages accumulate in human collateral coronary arteries, we examined endomyocardial biopsy specimens from patients with coronary allograft vasculopathy (CAV). Myocardial biopsy specimens that were previously collected for routine clinical care were obtained from the department of pathology. Before study initiation, institutional review board approval was obtained and the study was conducted in accordance to the approved protocol. Collected tissues were previously fixed in 10% formalin and stored in paraffin blocks. For patients with CAV, specimens were collected within 1 to 2 months of diagnosis and demonstrated minimal acute cellular rejection (grade O or 1A rejection). Appropriately matched control specimens were randomly selected based on the duration from transplantation, experienced fewer than 2 episodes of prior rejection and were free from major medical comorbidities such as end‐stage renal disease, malignancy, or chronic infection. Myocardial biopsy specimens were cut into 4‐μm sections using standard techniques and mounted on glass slides. Each paraffin block typically contained 3 or 4 myocardial specimens. Immunohistochemistry was performed using CD68 (KP1). The primary antibody was detected using HRP conjugated secondary antibodies and DAB substrate. CD68 immunostained sections were counterstained with hematoxylin. These assays were performed by the Anatomic and Molecular Pathology core as a component of the Institute of Clinical and Translational Science at Washington University School of Medicine. CD68‐positive staining was quantified using the following protocol. For each myocardial specimen, 2 independent fields from each of 3 stained sections were photographed at ×20 magnification. Image J software was used to quantify the percent area stained. The resulting measurements were then averaged to produce a single value to be used in the final analysis. The number of perivascular CD68+ cells was quantified by manually counting the number of CD68+ cells located within 20 μm of intramyocardial blood vessels per field. All specimens were stained, photographed, and quantified in blinded fashion.
RNA Isolation, cDNA Synthesis, and, Quantitative RT‐PCR
RNA was isolated from 4 independent biological samples using the RNeasy kit (Qiagen). cDNA was synthesized using the high‐capacity cDNA reverse transcription kit (Ambion). Quantitative RT‐PCR was performed on an ABI 7200 machine using either TaqMan‐based or SYBR green–based assays. Taqman probes for Vegf‐A, Vegf‐B, Vegf‐C, Vegf‐D, Angpt1, Angpt2, Ptch1, Fgf2, Pdgf‐β, Tgfβ1, Tgfβ2, Tgfβ3, and Hprt were purchased from Applied Biosystems. SYBR green primers were obtained from IDT. All samples were normalized to Hprt or Gapdh and then scaled relative to controls, where control samples were set at a value of 1. Thus, results for all experimental samples were graphed as relative expression compared with control.
Microarray Analysis
RNA was harvested as described and analyzed in the Genome Technology Access Center microarray core facility at Washington University. RNA from 4 independent biological samples was hybridized to the Illumina mouse Ref8 BeadDhip platform. Quality control standards for hybridization, labeling, staining, background signal, and basal level of housekeeping gene expression for each chip were performed and verified. Following hybridization, the microarray was scanned and analyzed using the GenomeStudio software (Illumina, Inc). Background subtraction, log transformation, and quantile normalization were performed using Partek GS (Partek). Hierarchical clustering and PCA analysis were used to access data quality. Differentially expressed genes were identified using a threshold change of 1.4‐fold and unadjusted P‐value <0.05 by ANOVA. We used a threshold change of 1.4‐fold to enrich for alterations in gene expression that were most likely to have a functional impact on phenotype at the expense of decreased sensitivity. Gene Ontology (GO) pathway analysis using the Database for Annotation Visualization and Integrated Discovery (DAVID) to identify signaling pathways that were significantly altered.
Statistical Analysis
Data are expressed as mean±SD. Student's t test or ANOVA was used for comparisons between groups. P‐values of <0.05 were considered significant. Bonferroni correction was performed when multiple hypotheses were tested. In these cases, the type I error (α=0.05) was split equally among each test such that P <0.025 and <0.017 indicated statistical significance after correction for testing 2 or 3 hypotheses, respectively.
Results
Mouse Model of Repetitive Ischemia
To determine whether brief episodes of repetitive ischemia constitute a sufficient stimulus to trigger coronary growth, we subjected animals to 15‐minute episodes of repetitive LAD ischemia using the closed‐chest I/R system (Figure 1A and 1B). The extent of cardiac dysfunction after a single 15‐minute episode of I/R injury was examined by serial echocardiographic analysis of LV function. At baseline, instrumented animals demonstrated a small focal wall motion abnormality located in the anterior wall adjacent to the occluder but had preserved global systolic function. Immediately after the induction of ischemia, LV systolic function decreased and a large wall motion abnormality was present affecting the anterior, apical, and distal portions of the inferior wall. At 10 minutes after reperfusion, systolic function improved; however, a large residual wall motion abnormality persisted. LV systolic function and wall motion returned to baseline 24 hours after reperfusion (Figure 1C through 1H). Reversibility of myocardial dysfunction was similarly observed after 3 serial episodes of ischemia on alternate days (data not shown).
Figure 1.

Mouse model of ischemia‐induced coronary angiogenesis. A, Diagram depicting the closed‐chest ischemia reperfusion model. An occluder is loosely placed around the LAD proximal to the major lateral branch. Ischemia is induced by placing tension on a suture threaded through the occluder. The yellow area indicates the ischemic territory and representative ECGs are shown before and during ischemia. B, Schematic describing the repetitive ischemia protocol. Mice were instrumented 2 weeks before the induction of ischemia. Ischemia was induced for 15 minutes every other day and animals were analyzed during the next 7 days. C through F, End‐systolic images after 15 minutes of ischemia. The dashed yellow line indicates the location of a wall motion abnormality, and the dashed blue line denotes the endocardial border. Prior to the inducing of ischemia (C), there is a small wall motion abnormality that corresponds to the site of occluder placement. Induction of a single episode of ischemia (D) results in a large wall motion abnormality extending from the anterior wall to the mid inferior wall. Immediately after reperfusion (E), there is a residual wall motion abnormality that resolves within 24 hours (F). G and H, Quantification of ejection fraction and akinetic region at baseline, during ischemia, 10 minutes after reperfusion, and 24 hours after reperfusion. *P<0.05 compared with baseline and 24 hours after reperfusion, **P<0.017 compared with all other time points. I, H&E–stained sections showing normal myocardial architecture after 3 episodes of repetitive ischemia. J, TUNEL staining demonstrating minimal cell death (white arrowheads) after 3 episodes of repetitive ischemia. K, Picrosirius red staining showing myocardial fibrosis in sham and mice that underwent 3 episodes of repetitive ischemia. L, Quantification of TUNEL and Picrosirius red staining. **P<0.025 compared with all other groups. Ao indicates aorta; ECG, echocardiography; H&E, hematoxylin and eosin; IR, ischemia and reperfusion; LAD, left anterior descending coronary artery; LCA, left coronary artery; LCX, left circumflex artery; LPL, lateral posterior lateral branch; LV, left ventricle; RCA, right coronary artery; RSC, right septal conal branch; RV, right ventricle; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Histological examination of cardiac tissue after 3 episodes of repetitive ischemia did not reveal evidence of gross cardiomyocyte disarray, myocardial necrosis, inflammatory cell infiltration, or scar formation as typically seen after MI (Figure 1I). TUNEL staining demonstrated rare foci of cardiomyocyte cell death and Picrosirius red staining showed minimal myocardial fibrosis within the ischemic area after 3 episodes of repetitive ischemia (Figure 1J and 1K). While quantification of TUNEL‐positive cells and fibrotic area demonstrated significant increases in myocardial cell death and fibrosis after repetitive ischemia, these events were minimal compared with mice that underwent 90 minutes of I/R injury (Figures 1L and S1). Due to the low frequency of cell death after repetitive I/R injury, we did not further delineate which cell types were most affected. These data indicate that brief episodes of I/R injury lead to reversible cardiac dysfunction with minimal myocardial fibrosis and cell death, as previously reported.30–33
Repetitive LAD Ischemia Is Sufficient to Promote Coronary Growth
To determine whether repetitive ischemia is sufficient to trigger coronary growth, we examined the coronary macrovasculature and microvasculature after 3 rounds of repetitive ischemia. One week after repetitive LAD ischemia, the coronary arterial tree was visualized using microfil perfusion casting (Figure 2A through 2H). These experiments revealed that both sham and repetitive ischemia animals had similar LCA size as expressed by area of the ROI (anterior wall) covered (65.7% versus 63.9%, P=0.61). In contrast, mice subjected to repetitive ischemia demonstrated significant growth of the RCA (Figure 2I). In sham animals, the RCA was small compared with the LCA and covered only the basal inferior wall. In mice that underwent repetitive LAD ischemia, the RCA covered the entire inferior wall and contained branches extending toward the lateral and septal walls. This represented nearly a 3‐fold increase in RCA size (32.9% versus 77.3%, P<0.05). While the lateral wall was supplied by diagonal branches of the LAD in shams, mice that underwent repetitive ischemia demonstrated contributions from both diagonal branches and the RCA (Figure 2G and 2H, arrows).
Figure 2.

Induction of coronary growth after repetitive ischemia. A–H, Microfil perfusion casts demonstrating enlargement of the RCA after repetitive LCA ischemia. Sham animals (A–D) display a large LCA (A) and rudimentary RCA (B). The lateral wall is perfused through diagonal branches of the LAD (C and D). Following repetitive ischemia, the LAD (E) appears unchanged compared with sham‐operated animals, while the RCA undergoes significant enlargement (F). In these animals, the lateral wall is perfused by both the RCA (white arrow) and diagonal branches arising from the LAD (G and H). The white rectangle denotes the area enlarged in (D and H). I, Quantification of the area covered by the LAD and RCA. *P<0.05 compared with sham. LAD indicates left anterior descending coroney artery; LCA, left coronary artery; RCA, right coronary artery.
To visualize intramyocardial arterioles and the microvasculature, we performed smooth muscle actin and CD31/PECAM immunostaining, respectively. For PECAM staining, we quantified the area stained using an automated method to estimate blood vessel density (Figure S2). Compared with sham animals, mice that were subjected to repetitive ischemia had increased capillary density in both the ischemic area and border zone (Figure 3A and 3B). No change in microvascular density was seen in the remote area (data not shown). Increased microvascular density was observed in both regions for at least 1 week after 3 episodes of repetitive ischemia and peaked at day 3 (Figure 3D and 3E). Consistent with this angiogenic response, similar increases in intramyocardial coronary arteriole number were seen in mice that underwent repetitive ischemia (Figure 3C and 3F).
Figure 3.

Microvascular expansion after repetitive ischemia. A and B, CD31/PECAM immunohistochemistry revealing expansion of the microvasculature in both the ischemic area (A) and border zone (B) after repetitive ischemia. C, Smooth muscle (SM) actin immunohistochemistry showing increased numbers of intramyocardial coronary arterioles in mice that underwent repetitive ischemia. D and E, Quantification of microvascular area stained in the ischemic area (D) and border zone (E) showing significant increases in capillary density peaking day 3 after repetitive ischemia. F, Quantification of the number of SM actin–containing blood vessels per ×10 field demonstrating increased intramyocardial arteriole number in mice that underwent repetitive ischemia at all time points examined. *P<0.05 compared with sham and day 3 after repetitive ischemia, **P<0.017 compared with all other time points. A and B, ×20 and C, ×10. PECAM indicates platelet endothelial cell adhesion molecule.
Ischemia‐Induced Coronary Growth Protects From MI by Decreasing the Area at Risk
To determine if repetitive ischemia‐driven coronary growth is physiologically relevant, we examined whether repetitive ischemia protected from prolonged I/R injury. For these experiments, mice were subjected to 3 rounds of repetitive ischemia followed by prolonged (120 minutes) I/R injury 10 days later (Figure 4A). One hundred twenty minutes of ischemia was selected because it produces a large anterior wall infarct similar to chronic LAD ligation.34 We chose to subject animals to MI 10 days after repetitive ischemia to minimize the effects of preconditioning.35–36
Figure 4.
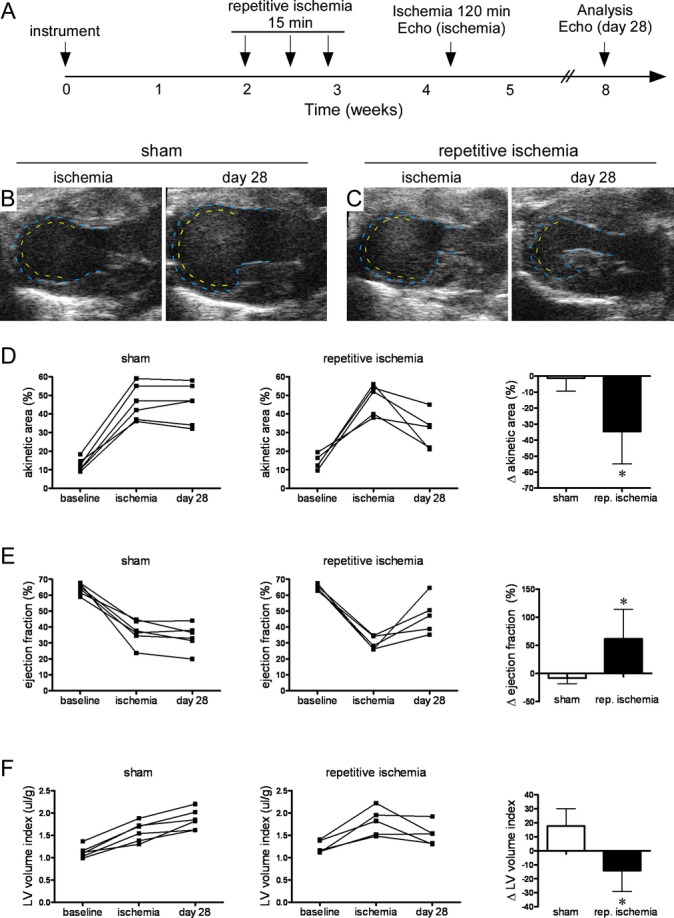
Ischemia‐induced coronary angiogenesis protects from myocardial infarction. A, Schematic depicting the experimental protocol used to test whether repetitive ischemia‐induced coronary growth protects from myocardial infarction. Echocardiograms were performed at baseline, during ischemia before myocardial infarction (ischemia) to control for variations in ischemic area, and 28 days after myocardial infarction (day 28). B and C, End‐systolic images during LAD ischemia at baseline and 28 days after myocardial infarction demonstrating that compared with sham‐operated animals (B), animals that underwent repetitive ischemia (C) have a smaller wall motion abnormality after myocardial infarction. D, Pairwise quantification of akinetic area revealing that sham animals have no change in akinetic area between ischemia and 28 days after myocardial infarction. In contrast, animals that underwent repetitive ischemia have a smaller akinetic area after myocardial infarction. E, Pairwise quantification of EF revealing that sham animals have no improvement in EF between ischemia and 28 days after myocardial infarction. In contrast, animals that underwent repetitive ischemia have improved EF after myocardial infarction. F, Pairwise quantification of LV remodeling as assessed by LV diastolic dimension indexed to body weight revealing that sham animals undergo increased LV size between ischemia and 28 days after myocardial infarction. In contrast, animals that underwent repetitive ischemia have a slight reduction in LV dimension. Also, ∆ akinetic area, EF and LV volume index indicate change between ischemia and 28 days after MI. *P<0.05 compared with sham. EF indicates ejection fraction; LAD, left anterior descending coronary artery; LV, left ventricle; MI, myocardial infarction.
Echocardiographic imaging was performed at baseline, immediately after MI, and 28 days after MI to assess the initial ischemic area, changes in LV systolic function, and LV remodeling. Immediately after MI, sham and repetitive ischemia animals demonstrated comparable wall motion abnormalities. The percentage of the ventricle that was akinetic (45.9% versus 48.1%, P=0.70) and LV EF (32.7% versus 29.8%, P=0.21) were comparable, indicating similar extents of injury between experimental groups. Sham animals displayed reduced EF immediately after MI, failed to recover LV systolic function over the next 28 days, and had increased LV chamber size consistent with the development of adverse remodeling. In contrast, mice subjected to repetitive ischemia demonstrated significant relative improvements in EF and did not undergo LV dilation 28 days after MI (Figure 4B through 4F).
Histological examination of hearts after prolonged I/R injury revealed a significant regional decrease in the area of trichrome staining in the LV inferior and lateral walls of mice subjected to repetitive ischemia compared with sham controls (11.0% versus 21.3%, P<0.01), consistent with less extensive infarction (Figure 5A through 5C). While we cannot exclude other mechanisms such as alterations in scar contraction, these findings are consistent with the pattern of RCA growth visualized by arterial casting and suggest that coronary growth and improved blood flow to the inferior and lateral walls may account for the smaller infarct sizes and improved LV function in the mice that underwent repetitive coronary ischemia.
Figure 5.

Reduced infarct size and decreased area at risk after ischemia induced angiogenesis. A and C, Compared with sham‐treated animals, repetitive ischemia resulted in decreased infarct size after myocardial infarction. Whole mount (A) and trichrome‐stained sections (B) reveal decreased scar size in animals that underwent repetitive ischemia compared with shams. The black arrow indicates that position of the left anterior descending coronary artery (LAD) occluder and the blue dashed lines mark the border between scar and viable muscle. Quantification of infarct size (C) demonstrates smaller infarcts after repetitive ischemia. *P<0.05 compared with sham. D and F, Evans blue perfusion staining (D) and respective threshold images (E) during LAD ischemia demonstrating a dose‐dependent decrease in area at risk in animals that underwent repetitive ischemia. The dashed yellow line (D) and solid blue lines (E) outline the area at risk. Quantification of area at risk (F) showing a dose dependent decrease after repetitive ischemia. **P<0.025 compared with all other time points.
To further explore this possibility, we performed Evans blue perfusion staining after either 3 or 6 rounds of repetitive ischemia (Figure 5D through 5F). Compared with sham animals that displayed a large area at risk encompassing the anterior, lateral, and inferior walls, mice that underwent repetitive ischemia had a smaller area at risk (38.4% versus 28.5%, P<0.01). Mice subjected to 3 rounds of ischemia demonstrated a localized area at risk in the anterior and lateral walls, sparing the inferior wall. Further reductions in area at risk were observed in mice that underwent 6 rounds of repetitive ischemia (19.9%, P<0.05 compared with the other groups). Together, these data indicate that repetitive ischemia promotes coronary growth and protects from MI by decreasing the area at risk, consistent with regional improvements in myocardial perfusion. Moreover, these effects are dose dependent.
Expression Profiling During Ischemia‐Induced Coronary Growth
We performed microarray‐based gene expression profiling to search for genes and pathways involved in ischemia‐induced coronary growth. RNA was isolated from the ventricular myocardium of animals that were subjected to 3 rounds of repetitive ischemia. To gain insights into temporal changes in gene expression, hearts were harvested on days 1, 3, and 5 after the third ischemic episode. Using a threshold of 1.4‐fold, we observed that compared with sham animals, the largest changes in gene expression occurred within the first few days after repetitive ischemia. Four hundred five genes (277 upregulated, 128 downregulated), 129 genes (105 upregulated, 24 downregulated), and 40 genes were altered on days 1, 3, and 5 after repetitive ischemia, respectively (Figure 6A, Tables S1 through S3). Consistent with these results, hierarchical clustering revealed that the largest alterations in global gene expression occurred at day 1 after repetitive ischemia, whereas the changes in gene expression were less marked on days 3 and 5 (Figure 6B).
Figure 6.

Microarray gene expression profiling during ischemia‐induced coronary angiogenesis. A, Venn diagram describing the distribution of upregulated and downregulated genes at day 1, 3, and 5 after repetitive ischemia compared with sham controls. B, Hierarchical cluster analysis revealing that the largest changes in gene expression occurs at days 1 and 3 after repetitive ischemia. C, Bar graphs depicting the number of genes regulated for each pathway identified through GO pathway analysis. ECM indicates extracellular matrix; GO, Gene Ontology.
GO pathway analysis uncovered enrichment of genes belonging to pathways associated with innate immunity, extracellular matrix (ECM) structure, and blood vessel development on day 1 after repetitive ischemia (Figure 6, Table). In contrast, on day 3 after repetitive ischemia, there was a shift in gene expression from genes involved in innate immunity and inflammation pathways toward pathways associated with cell adhesion, ECM organization, and vascular development. Given the smaller number of genes with altered expression on days 5 after repetitive ischemia, no pathways were significantly altered in this analysis.
Table 1.
GO Pathway Analysis of Gene Expression Following Repetitive Ischemia

Classic Proangiogenic Factor Expression During Ischemia‐Induced Coronary Growth
Despite regulation of pathways associated with blood vessel development on days 1 and 3 after repetitive ischemia, we were surprised to find that microarray profiling failed to demonstrate significant upregulation of many classically described proangiogenic factors. To confirm these findings, we performed quantitative RT‐PCR assays. Consistently, these experiments failed to show increased expression of several well‐accepted proangiogenic molecules (Figure 7A). We could not detect any increase in Vegf‐A, Vegf‐B, Vegf‐C, Vegf‐D, Pgf, Pdgf‐β, Tgfβ1‐3, Ptch1, Thymosinβ4, Mmp2, Mmp9, Nos2, or Nos3 mRNA expression. In contrast, we did observe modest increased expression of Fgf2 and Angpt2. Each of these factors has been implicated in either hindlimb ischemia or MI border zone angiogenesis models.11–12,11–21,11–27,39,22,40
Figure 7.

Classic proangiogenic growth factor expression after repetitive coronary ischemia. A, Quantitative RT‐PCR assays showing expression of classic proangiogenic growth factors in hearts 1 day after repetitive ischemia relative to sham controls. Only Angpt2 and Fgf2 were upregulated after repetitive ischemia. *P<0.05 compared with sham. B, Immunostaining revealing unchanged VEGF‐A expression (red) in mice that underwent repetitive ischemia compared with sham controls. In contrast, VEGF‐A expression is increased in the border zone after myocardial infarction. DAPI (blue) and cardiac actin (cActin, green) demarcate the border zone. C, Western blot demonstrating no change in VEGF‐A protein levels between sham control and hearts that underwent repetitive ischemia. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor.
Given the fundamental importance of VEGF signaling in vascular growth, we further examined whether VEGF‐A might be modulated on the protein level. We performed VEGF‐A immunostaining on sections obtained from sham controls, mice subjected to repetitive ischemia, or mice that underwent experimental MI. Sections were co‐stained for cardiac actin to identify cardiomyocytes and the border zone. Only low levels of VEGF‐A protein could be detected in the myocardium from sham and repetitive ischemia animals. In contrast, robust VEGF‐A protein expression was observed in the border zone after MI (Figure 7B). Consistent with these results, Western blotting demonstrated comparable VEGF‐A protein levels in sham‐operated mice and mice that underwent repetitive ischemia (Figure 7C). These data strongly suggest that classic proangiogenic growth factors previously implicated in hindlimb ischemia models and experimental MI models are not involved in ischemia‐induced coronary growth.
Involvement of Macrophages in Ischemia‐Induced Coronary Growth
As noted, GO pathway analysis revealed selective regulation of genes associated with innate immune responses on day 1 after repetitive ischemia (Table). To further explore the possibility that the innate immune system might be involved in ischemia‐induced coronary growth, we examined sections of cardiac tissue in the area at risk after repetitive ischemia. Histological analysis revealed a cellular infiltrate surrounding intramyocardial blood vessels, present at days 1 and 3 after repetitive ischemia. The cellular infiltrate largely resolved by day 5 (Figure 8A). Immunohistochemical staining demonstrated that these cells expressed MAC3 and CD11b consistent with macrophages (Figures 8B and S3). Further characterization showed that these cells were negative for Gr1, CD11c, CD3, CD19, and NK1.1 (Figure S1, data not shown). Co‐labeling with CD11b and smooth muscle actin antibodies confirmed the presence of macrophages associated with the intramyocardial coronary vasculature (Figure 8C and 8D). Quantitative analysis demonstrated significantly increased number of MAC3‐ and CD11b‐positive cells surrounding the vasculature in mice that underwent repetitive ischemia (Figure 8E and 8F). These data are consistent with prior studies demonstrating a role for macrophages in collateralization during hindlimb ischemia.41
Figure 8.

Macrophages surround the coronary vasculature during ischemia induced coronary angiogenesis. A, H&E staining (×20) showing a cellular infiltrate in the adventitia of intramyocardial coronary blood vessels (arrows) after repetitive ischemia. The infiltrate resolves by day 5 after repetitive ischemia. B, Immunostaining for MAC3 (×40) demonstrating that the cellular infiltrate is primarily composed of macrophages. C and D, Immunostaining for CD11b (magenta) and smooth muscle actin (SMA, green) demonstrating that compared with sham controls (C), there is an enrichment of macrophages surrounding intramyocardial blood vessels after repetitive ischemia (D). (Original magnification ×63, scale bar indicates 20 μm.) E and F, Quantification of MAC3‐ and CD11b‐positive cells in sham controls and mice that underwent repetitive ischemia. G, CD68 immunostaining (×40) of endomyocardial biopsy specimens from controls and patients with CAV according to the presence of collaterals showing increased numbers of perivascular macrophages in patients with CAV who have collateral vasculature. Scale bar indicates 40 μm. H, Quantification of perivascular macrophages displayed as the number of macrophages per blood vessel (BV). *P<0.05 compared with sham. **P<0.017 (E) and P<0.025 (H) compared with all other groups. CAV indicates coronary allograft vasculopathy; H&E, hematoxylin and eosin; MAC3, CD107b.
To further support a putative role for macrophages in ischemia‐driven coronary growth, we performed quantitative RT‐PCR assays to confirm changes in the expression of genes associated with macrophage activity. We included genes that were identified through our microarray experiments as well those that have been previously demonstrated to be important for macrophage recruitment, macrophage‐derived cytokine signaling, and ECM remodeling. This analysis demonstrated robust increases in the expression of numerous chemokines and cytokines including Mcp1, Mcp3, Mip1α, Mip1β, Mip1γ, Rantes, IL‐1β, IL‐6, and Socs3. In addition, we were able to detect increased expression of multiple macrophage‐associated genes previously implicated in ECM remodeling including Timp1, Ctgf, Integrin α5, and Galectin 3 (Figure S4). These data implicate potential roles for innate immunity, macrophage recruitment, and ECM remodeling in ischemia‐induced coronary growth.
To determine whether these findings in mice also pertain to growth of collateral arteries in humans, we questioned whether macrophages surround the intramyocardial vasculature in humans with coronary collaterals. For these studies, we obtained endomyocardial biopsy specimens from patients with CAV. CAV is a vascular process with similarities to native coronary artery disease that diffusely affects the coronary tree and is among the leading causes of mortality and allograft failure after cardiac transplantation.42–43 Given the diffuse nature of this vascular process and ease of access to biopsy tissue, CAV represents a potential model disease to study coronary collateral growth. In support of this notion, we have previously demonstrated that the development of angiographically visible collaterals in patients with CAV is associated with improved survival and allograft function.44
We obtained endomyocardial biopsy specimens suitable for immunohistochemical analysis from 23 control patients, 21 CAV patients without collaterals, and 30 CAV patients with angiographically visible collaterals. CD68 immunostaining demonstrated that compared with controls, CAV patients had higher numbers of macrophages surrounding the intramyocardial vasculature. CAV patients with collaterals had more perivascular macrophages than both CAV patients without collaterals and control patients (Figure 8G and 8H). Interestingly, while CAV patients lacking collaterals had the largest total number of macrophages (Figure S5), the majority of these cells were interstitial, rather than perivascular. As co‐labeling experiments were not technically feasible in these specimens and perivascular location was inferred by morphologic criteria, we were not able to precisely determine which type of vasculature was associated with CD68‐positive macrophages.
Discussion
Using repetitive I/R injury to mimic the phenomenon of stuttering angina, we demonstrate that repetitive brief episodes of LAD ischemia are sufficient to trigger expansion of coronary growth on the macrovascular and microvascular levels in a closed‐chest murine model of ischemic injury (Figures 2 and 3). We further show that ischemia‐induced coronary growth is functionally significant and leads to a decrease in the area at risk, as measured by using Evans blue staining, as well as decrease in infarct size, improved cardiac remodeling and improved LV ejection fraction after prolonged ischemic injury (Figure 4). Transcriptional profiling of hearts subjected to repetitive bouts of brief ischemia disclosed minimal alterations in the temporal expression of classic proangiogenic growth factors but did reveal a potentially important role for genes involved in innate immunity, cardiac macrophages, and ECM remodeling. Moreover, histological analysis of mouse hearts subjected to repetitive bouts of ischemia disclosed the presence of macrophages surrounding intramyocardial blood vessels, consistent with prior studies that have suggested a role for macrophages in the development of collateral vessels during hindlimb ischemia.41 Viewed together, these studies suggest that brief episodes of repetitive myocardial ischemia provoke increased coronary growth that is sufficient to protect the heart against prolonged ischemic injury and implicate a potential novel role for the cardiac macrophage as a potential mediator of ischemia‐driven coronary growth.
Expression of Classic Proangiogenic Factors During Ischemia‐Induced Coronary Growth
To our surprise, both candidate and unbiased gene expression profiling revealed minimal alterations in the expression of multiple classic proangiogenic growth factors after repetitive bouts of ischemic injury that were sufficient to provoke coronary growth. Many of these molecules are required for collateral growth during hindlimb ischemia and border zone–associated coronary angiogenesis after experimental MI. Among these, VEGF‐A is the best studied and has been implicated in most forms of blood vessel growth.34 Surprisingly, we were unable to detect increased VEGF‐A mRNA or protein on either the global or local level. Among classic proangiogenic growth factors, only Fgf2 and Angpt2 were found to be upregulated during ischemia‐induced coronary growth.
These data suggest that there are fundamental differences between the molecular mechanisms that govern ischemia‐induced coronary growth, coronary angiogenesis after experimental MI, and hindlimb ischemia. For example, robust increases in VEGF‐A expression are readily detectable after hindlimb ischemia or experimental MI.12–13,15,17 Surprisingly, we did not observe similar increases in VEGF‐A or several other classic proangiogenic growth factors during repetitive ischemia‐driven coronary growth. Although speculative, these findings may offer one potential explanation for why therapeutic angiogenesis trials using classic proangiogenic factors in patients with ischemic heart disease have failed to significantly improve clinical outcomes.
Role of the Innate Immune System and Cardiac Macrophage in Ischemia‐Induced Coronary Growth
Expression profiling and GO pathway analysis revealed a potential role for innate immunity as a potentially important driver of ischemia‐induced coronary growth. Multiple monocyte‐ and macrophage‐associated chemokines and cytokines were upregulated after repetitive ischemia and may mediate recruitment of macrophages to growing vasculature. Although this study was not intended to delineate the proximate mechanisms of innate immune activation after repetitive I/R injury, it is noteworthy that increased oxidative stress after I/R injury is sufficient to activate proinflammatory cytokines.31,33 Furthermore, we demonstrated that macrophages surround small intramyocardial arterioles during periods of coronary growth. Consistent with a role for macrophages in human coronary collateral development, immunostaining of biopsy specimens from CAV patients revealed that those with collaterals had significantly increased numbers of CD68‐positive macrophages surrounding intramyocardial blood vessels.
These findings are consistent with the known role for innate immunity and macrophages in hindlimb collateral development. Mice deficient in CCR2 display diminished numbers of macrophages surrounding collateral blood vessels and impaired ability to generate collaterals after hindlimb ischemia.45 Conversely, infusion of MCP‐1 (ligand for CCR2) improves collateral growth.46 Csfop/op mice, which are deficient in monocytes and macrophages, have impaired collateral growth, and infusion of granulocyte colony‐stimulating factor is sufficient to promote collateral growth after hindlimb ischemia.47–48 Together, these data suggest that macrophages are important mediators of collateral growth in the periphery. The molecular mechanism by which macrophages mediate collateral growth in this setting are largely unknown but are thought to involve direct contact with endothelial cells, macrophage‐derived proangiogenic growth factors, and cytokines.41
Consistent with a conserved role for macrophages in coronary collateral growth, infusion of granulocyte colony‐stimulating factor into patients with ischemic heart disease improved collateral flow as measured by collateral flow index.49 Whether cardiac macrophages are necessary for adult coronary growth and whether cardiac macrophages mediate vascular growth through mechanisms similar to those used in hindlimb ischemia models are unknown and will undoubtedly be a topic of future investigation. The development of genetically tractable models of coronary growth, such as presented here, will likely aid in the ability of investigators to answer these questions.
Limitations
While the experiments presented here implicate activation of the innate immune system and involvement of cardiac macrophages in ischemia‐driven coronary growth rather than classic proangiogenic growth factors, we have yet to rigorously prove this is the case. Further studies using genetic tools will be essential to appropriately define the requirement for macrophages in this process. In addition, identifying potential triggers of innate immune system activation such as generation reactive oxygen species and ultimately defining the molecular mechanisms by which they may govern coronary growth represent important issues that are yet to be resolved.
Although we cannot formally exclude a potential role for preconditioning as a mechanism for the smaller infarct size and improved cardiac remodeling and improved LV function in the mice subjected to repetitive ischemia, we believe that this is unlikely for 3 reasons. First, the early and late windows of preconditioning occur within 1 to 2 hours and 3 to 5 days, respectively, after ischemic injury.35–36 Thus, based on the timing of the prolonged ischemic injury (10 days after repetitive ischemia), preconditioning is unlikely to explain our findings. Further, the observation that the extent of LV dysfunction and LV dilation were not different in the sham and repetitive ischemia groups of mice immediately after prolonged I/R injury also suggests that preconditioning was not the major mechanism for the observed findings. Third, we did not observe a significant upregulation of genes implicated in late preconditioning such as iNos and Cox. Thus, our data suggest that increased coronary growth is the most likely mechanism for the decrease in infarct size and improved LV structural and functional recovery after prolonged ischemic injury.
Conclusions
Using a clinically relevant closed chest mouse model of ischemic injury, we show for the first time that brief bouts of repetitive myocardial ischemia provoke increased coronary growth that is sufficient to protect the heart against prolonged ischemic injury. These results are important for 2 reasons. First, although these studies did not identify an important role for the classic proangiogenic growth factors, they do suggest a potentially important role for innate immunity and cardiac macrophages in terms of mediating coronary growth. Second, these studies clearly demonstrate that coronary growth can be studied longitudinally using a tractable closed chest mouse model that can be genetically manipulated. Indeed, studies that have examined the molecular mechanisms for coronary angiogenesis and collateralization thus far rely on large animal models that are not amenable to genetic dissection, and/or rely on surrogate processes such as hindlimb ischemia in the mouse. While some mechanisms for angiogenesis are likely to be conserved in different experimental model systems, it is not clear to what extent signaling pathways that control collateralization in the hindlimb are also involved in coronary growth. Accordingly, subsequent studies using this closed chest murine model system may advance our understanding of the basic mechanisms that are responsible for mediating coronary growth and collateralization in the adult mammalian heart.
Sources of Funding
This project was made possible by the Washington University Institute of Clinical and Translational Sciences grant UL1 RR024992 from the National Center for Research Resources (NCRR)/National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH) and NIH R01 HL111094. KL was supported by NIH T32 HL007081 and the Physician Scientist Training Program.
Disclosures
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013; 127:e6-e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reitsma JB, Dalstra JA, Bonsel GJ, van der Meulen JH, Koster RW, Gunning‐Schepers LJ, Tijssen JG. Cardiovascular disease in the Netherlands, 1975 to 1995: decline in mortality, but increasing numbers of patients with chronic conditions. Heart. 1999; 82:52-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mukherjee D, Comella K, Bhatt DL, Roe MT, Patel V, Ellis SG. Clinical outcome of a cohort of patients eligible for therapeutic angiogenesis or transmyocardial revascularization. Am Heart J. 2001; 142:72-74 [DOI] [PubMed] [Google Scholar]
- 4.Williams B, Menon M, Satran D, Hayward D, Hodges JS, Burke MN, Johnson RK, Poulose AK, Traverse JH, Henry TD. Patients with coronary artery disease not amenable to traditional revascularization: prevalence and 3‐year mortality. Catheter Cardiovasc Interv. 2010; 75:886-891 [DOI] [PubMed] [Google Scholar]
- 5.Billinger M, Kloos P, Eberli FR, Windecker S, Meier B, Seiler C. Physiologically assessed coronary collateral flow and adverse cardiac ischemic events: a follow‐up study in 403 patients with coronary artery disease. J Am Coll Cardiol. 2002; 40:1545-1550 [DOI] [PubMed] [Google Scholar]
- 6.Nathoe HM, Koerselman J, Buskens E, van Dijk D, Stella PR, Plokker TH, Doevendans PA, Grobbee DE, de Jaegere PPOctopus Study Group Determinants and prognostic significance of collaterals in patients undergoing coronary revascularization. Am J Cardiol. 2006; 98:31-35 [DOI] [PubMed] [Google Scholar]
- 7.Steg PG, Kerner A, Mancini GB, Reynolds HR, Carvalho AC, Fridrich V, White HD, Forman SA, Lamas GA, Hochman JS, Buller CEOAT Investigators Impact of collateral flow to the occluded infarct‐related artery on clinical outcomes in patients with recent myocardial infarction: a report from the randomized occluded artery trial. Circulation. 2010; 121:2724-2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier P, Gloekler S, Zbinden R, Beckh S, de Marchi SF, Zbinden S, Wustmann K, Billinger M, Vogel R, Cook S, Wenaweser P, Togni M, Windecker S, Meier B, Seiler C. Beneficial effect of recruitable collaterals: a 10‐year follow‐up study in patients with stable coronary artery disease undergoing quantitative collateral measurements. Circulation. 2007; 116:975-983 [DOI] [PubMed] [Google Scholar]
- 9.Meier P, Hemingway H, Lansky AJ, Knapp G, Pitt B, Seiler C. The impact of the coronary collateral circulation on mortality: a meta‐analysis. Eur Heart J. 2012; 33:614-621 [DOI] [PubMed] [Google Scholar]
- 10.Mitsos S, Katsanos K, Koletsis E, Kagadis GC, Anastasiou N, Diamantopoulos A, Karnabatidis D, Dougenis D. Therapeutic angiogenesis for myocardial ischemia revisited: basic biological concepts and focus on latest clinical trials. Angiogenesis. 2012; 15:1-22 [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000; 6:389-395 [DOI] [PubMed] [Google Scholar]
- 12.Cao Y. Therapeutic angiogenesis for ischemic disorders: what is missing for clinical benefits? Discov Med. 2010; 9:179-184 [PubMed] [Google Scholar]
- 13.Schirmer SH, van Nooijen FC, Piek JJ, van Royen N. Stimulation of collateral artery growth: travelling further down the road to clinical application. Heart. 2009; 95:191-197 [DOI] [PubMed] [Google Scholar]
- 14.Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, Schatz RA, Asahara T, Isner JM, Kuntz RE. Phase 1/2 placebo‐controlled, double‐blind, dose‐escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation. 2002; 105:2012-2018 [DOI] [PubMed] [Google Scholar]
- 15.Maulik N, Thirunavukkarasu M. Growth factors and cell therapy in myocardial regeneration. J Mol Cell Cardiol. 2008; 44:219-227 [DOI] [PubMed] [Google Scholar]
- 16.Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, Udelson JE, Gervino EV, Pike M, Whitehouse MJ, Moon T, Chronos NA. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor‐2: double‐blind, randomized, controlled clinical trial. Circulation. 2002; 105:788-793 [DOI] [PubMed] [Google Scholar]
- 17.Syed IS, Sanborn TA, Rosengart TK. Therapeutic angiogenesis: a biologic bypass. Cardiology. 2004; 101:131-143 [DOI] [PubMed] [Google Scholar]
- 18.Uchida Y, Yanagisawa‐Miwa A, Nakamura F, Yamada K, Tomaru T, Kimura K, Morita T. Angiogenic therapy of acute myocardial infarction by intrapericardial injection of basic fibroblast growth factor and heparin sulfate: an experimental study. Am Heart J. 1995; 130:1182-1188 [DOI] [PubMed] [Google Scholar]
- 19.Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake PR, Shapiro R, Taylor FR, Baker DP, Asahara T, Isner JM. The morphogen sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med. 2001; 7:706-711 [DOI] [PubMed] [Google Scholar]
- 20.Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, Thorne T, Takenaka H, Aikawa R, Goukassian D, Von Samson P, Hamada H, Yoon YS, Silver M, Eaton E, Ma H, Heyd L, Kearney M, Munger W, Porter JA, Kishore R, Losordo DW. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med. 2005; 11:1197-1204 [DOI] [PubMed] [Google Scholar]
- 21.Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J Clin Invest. 2008; 118:2404-2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smart N, Risebro CA, Melville AA, Moses K, Schwartz RJ, Chien KR, Riley PR. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007; 445:177-182 [DOI] [PubMed] [Google Scholar]
- 23.Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, Roncal C, Eriksson U, Fu Q, Elfenbein A, Hall AE, Carmeliet P, Moons L, Simons M. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest. 2007; 117:3188-3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muinck ED, Nagy N, Tirziu D, Murakami M, Gurusamy N, Goswami SK, Ghatpande S, Engelman RM, Simons M, Das DK. Protection against myocardial ischemia‐reperfusion injury by the angiogenic Masterswitch protein PR 39 gene therapy: the roles of HIF1alpha stabilization and FGFR1 signaling. Antioxid Redox Signal. 2007; 9:437-445 [DOI] [PubMed] [Google Scholar]
- 25.Li J, Post M, Volk R, Gao Y, Li M, Metais C, Sato K, Tsai J, Aird W, Rosenberg RD, Hampton TG, Sellke F, Carmeliet P, Simons M. PR39, a peptide regulator of angiogenesis. Nat Med. 2000; 6:49-55 [DOI] [PubMed] [Google Scholar]
- 26.Edelberg JM, Aird WC, Wu W, Rayburn H, Mamuya WS, Mercola M, Rosenberg RD. PDGF mediates cardiac microvascular communication. J Clin Invest. 1998; 102:837-843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kupatt C, Hinkel R, Pfosser A, El‐Aouni C, Wuchrer A, Fritz A, Globisch F, Thormann M, Horstkotte J, Lebherz C, Thein E, Banfi A, Boekstegers P. Cotransfection of vascular endothelial growth factor‐A and platelet‐derived growth factor‐B via recombinant adeno‐associated virus resolves chronic ischemic malperfusion role of vessel maturation. J Am Coll Cardiol. 2010; 56:414-422 [DOI] [PubMed] [Google Scholar]
- 28.Ishikawa K, Ladage D, Takewa Y, Yaniz E, Chen J, Tilemann L, Sakata S, Badimon JJ, Hajjar RJ, Kawase Y. Development of a preclinical model of ischemic cardiomyopathy in swine. Am J Physiol Heart Circ Physiol. 2011; 301:H530-H537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O'Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation. 2005; 112:2108-2113 [DOI] [PubMed] [Google Scholar]
- 30.Dewald O, Frangogiannis NG, Zoerlein MP, Duerr GD, Taffet G, Michael LH, Welz A, Entman ML. A murine model of ischemic cardiomyopathy induced by repetitive ischemia and reperfusion. Thorac Cardiovasc Surg. 2004; 52:305-311 [DOI] [PubMed] [Google Scholar]
- 31.Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci USA. 2003; 100:2700-2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou‐Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/monocyte chemoattractant protein‐1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005; 96:881-889 [DOI] [PubMed] [Google Scholar]
- 33.Nossuli TO, Frangogiannis NG, Knuefermann P, Lakshminarayanan V, Dewald O, Evans AJ, Peschon J, Mann DL, Michael LH, Entman ML. Brief murine myocardial I/R induces chemokines in a TNF‐alpha‐independent manner: role of oxygen radicals. Am J Physiol Heart Circ Physiol. 2001; 281:H2549-H2558 [DOI] [PubMed] [Google Scholar]
- 34.Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, Hartley CJ, Pham TT, Daniel SL, Funk E, Entman ML. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol. 1999; 277:H660-H668 [DOI] [PubMed] [Google Scholar]
- 35.Bolli R, Li QH, Tang XL, Guo Y, Xuan YT, Rokosh G, Dawn B. The late phase of preconditioning and its natural clinical application—gene therapy. Heart Fail Rev. 2007; 12:189-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol. 2007; 292:H19-H27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007; 117:3930-3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanno S, Lerner DL, Schuessler RB, Betsuyaku T, Yamada KA, Saffitz JE, Kovacs A. Echocardiographic evaluation of ventricular remodeling in a mouse model of myocardial infarction. J Am Soc Echocardiogr. 2002; 15:601-609 [DOI] [PubMed] [Google Scholar]
- 39.Grundmann S, Piek JJ, Pasterkamp G, Hoefer IE. Arteriogenesis: basic mechanisms and therapeutic stimulation. Eur J Clin Invest. 2007; 37:755-766 [DOI] [PubMed] [Google Scholar]
- 40.Patel‐Hett S, D'Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011; 55:353-363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fung E, Helisch A. Macrophages in collateral arteriogenesis. Front Physiol. 2012; 3:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colvin‐Adams M, Agnihotri A. Cardiac allograft vasculopathy: current knowledge and future direction. Clin Transplant. 2011; 25:175-184 [DOI] [PubMed] [Google Scholar]
- 43.Schmauss D, Weis M. Cardiac allograft vasculopathy: recent developments. Circulation. 2008; 117:2131-2141 [DOI] [PubMed] [Google Scholar]
- 44.Lavine KJ, Sintek M, Novak E, Ewald G, Geltman E, Joseph S, Pfeifer J, Mann DL. Coronary collaterals predict improved survival and allograft function in patients with coronary allograft vasculopathy. Circ Heart Fail. 2013; 6:773-784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heil M, Ziegelhoeffer T, Wagner S, Fernandez B, Helisch A, Martin S, Tribulova S, Kuziel WA, Bachmann G, Schaper W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC‐chemokine receptor‐2. Circ Res. 2004; 94:671-677 [DOI] [PubMed] [Google Scholar]
- 46.Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W. Monocyte chemotactic protein‐1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. 1997; 80:829-837 [DOI] [PubMed] [Google Scholar]
- 47.Bergmann CE, Hoefer IE, Meder B, Roth H, van Royen N, Breit SM, Jost MM, Aharinejad S, Hartmann S, Buschmann IR. Arteriogenesis depends on circulating monocytes and macrophage accumulation and is severely depressed in op/op mice. J Leukoc Biol. 2006; 80:59-65 [DOI] [PubMed] [Google Scholar]
- 48.Lee M, Aoki M, Kondo T, Kobayashi K, Okumura K, Komori K, Murohara T. Therapeutic angiogenesis with intramuscular injection of low‐dose recombinant granulocyte‐colony stimulating factor. Arterioscler Thromb Vasc Biol. 2005; 25:2535-2541 [DOI] [PubMed] [Google Scholar]
- 49.Meier P, Gloekler S, de Marchi SF, Indermuehle A, Rutz T, Traupe T, Steck H, Vogel R, Seiler C. Myocardial salvage through coronary collateral growth by granulocyte colony‐stimulating factor in chronic coronary artery disease: a controlled randomized trial. Circulation. 2009; 120:1355-1363 [DOI] [PubMed] [Google Scholar]