

Abstract
Background
We recently reported that a bifunctional nuclear transport modifier (NTM), cSN50.1 peptide, reduced atherosclerosis, plasma cholesterol, triglycerides, and glucose along with liver fat and inflammatory markers, in a murine model of familial hypercholesterolemia. We determined that cSN50.1 improved lipid homeostasis by modulating nuclear transport of sterol regulatory element‐binding proteins through interaction with importin β. Previous studies established that cSN50.1 and related NTMs also modulate nuclear transport of proinflammatory transcription factors mediated by binding of their nuclear localization sequences (NLSs) to importins/karyopherins α. However, selectivity and specificity of NTMs for importins/karyopherins α were undetermined.
Methods and Results
We analyzed interaction of the NTM hydrophilic module, N50 peptide, derived from the NLS of NFκB1/p50, with endogenous human importins/karyopherins α to determine the mechanism of NTM modulation of importin α‐mediated nuclear transport. We show that N50 peptide forms stable complexes with multiple importins/karyopherins α. However, only interaction with importin α5 (Imp α5) displayed specific, high‐affinity binding. The 2:1 stoichiometry of the N50‐Imp α5 interaction (KD1=73 nmol/L, KD2=140 nmol/L) indicated occupancy of both major and minor NLS binding pockets. Utilizing in silico 3‐dimensional (3‐D) docking models and comparative structural analysis, we identified a structural component of the Imp α5 major NLS binding pocket that may stabilize N50 binding. Imp α5 also displayed rapid stimulus‐induced turnover, which could influence its availability for nuclear transport during the inflammatory response.
Conclusions
These results provide direct evidence that N50 peptide selectively targets Imp α5, encouraging further refinement of NLS‐derived peptides as new tools to modulate inflammatory disorders.
Keywords: cell‐penetrating peptide, importin, importin α diversity region, karyopherin, NFκB, nuclear import adaptors, nuclear transport, steatohepatitis, transcription factor, vascular inflammation
Introduction
We recently reported in this journal that treatment with a cell‐penetrating nuclear transport modifier (NTM), cSN50.1 peptide, reduced atherosclerosis as well as elevated plasma cholesterol, triglycerides, and glucose in a mouse model of familial hypercholesterolemia.1 Additionally, elevated cholesterol and triglycerides in the liver were reduced, along with liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST), markers of steatohepatitis. These signs of liver inflammation were accompanied by increased phosphorylation and nuclear translocation of nuclear factor κB (NFκB) that was attenuated in NTM‐treated mice. This outcome was consistent with the well‐established antiinflammatory function of NTMs through modulation of importin α (Imp α)‐mediated nuclear transport of proinflammatory stress‐responsive transcription factors (SRTFs), such as NFκB, activator protein 1 (AP‐1), nuclear factor of activated T cells (NFAT), and signal transducer and activator of transcription 1 (STAT1).2–6 N50‐containing NTMs (SN50, cSN50, and cSN50.1) are comprised of a hydrophilic N50 motif patterned on the nuclear localization sequence (NLS) region of the NFκB1/p50 subunit (see Table 1) fused to a motif from the signal sequence hydrophobic region (SSHR) of human fibroblast growth factor 4.1 The SSHR allows peptides to cross the plasma membrane by an ATP‐ and endosome‐independent mechanism,2,5 and the N50 motif was designed to bind to importins α during stimulus‐initiated signaling and thereby limit docking of NLS‐bearing SRTFs to their adaptor proteins and reduce nuclear import of activated STRFs. The surprising correction of dyslipidemia and its sequelae (fatty liver and atherosclerosis) was attributed to a second function of NTM, namely, nuclear transport modulation of sterol regulatory element‐binding proteins (SREBPs), master regulators of genes involved in synthesis of cholesterol, triglycerides, and fatty acids, which do not display an NLS.1,7 We determined that NTM accomplished this newly discovered function by interaction of its SSHR motif with importin β (Imp β), the sole nuclear transport shuttle for SREBPs.8 However, the mechanism of NTM interaction with importins/karyopherins α (see Table 2A for nomenclature) remained unexplained. Since multiple SRTFs modulated by NTMs display monopartite or bipartite NLSs distinct from that of NFκB1/p50, it became apparent that recognition of NTMs by importins α is more complex than initially thought. Therefore, we embarked on studying interaction of the NTM module N50 with human importins α in terms of their turnover, cell type‐specific abundance, selectivity, and accessibility of their major NLS‐binding pocket.
Table 1.
Amino Acid Sequences of Peptides Used in this Study
| Peptide | Sequence |
|---|---|
| N50 | VQRKRQKLMP |
| N50M | VQRDEQKLMP |
| cN50.1 | CVQRKRQKLMPC |
| AR1 | VELRKAKKDDQMLKRRNVSSF |
| AR3 | VELRKNKRDEHLLKRRNVPHE |
| AR4 | VELRKNKRDEHLLKKRNVPQE |
| AR5 | LQLRKQKREEQLFKRRNVATA |
| AR7 | IQLRKQKREQQLFKRRNVELI |
N50 – sequence derived from the NLS region of NFκB1/p50; N50M – sequence of control peptide with KR to DE mutation (bolded); cN50.1 – sequence of cyclized version of N50. An intra‐molecular disulfide bond is formed between the two cysteines; AR1 through AR7 – sequences derived from the auto‐inhibitory region (AR) of Imp α1 through Imp α7.
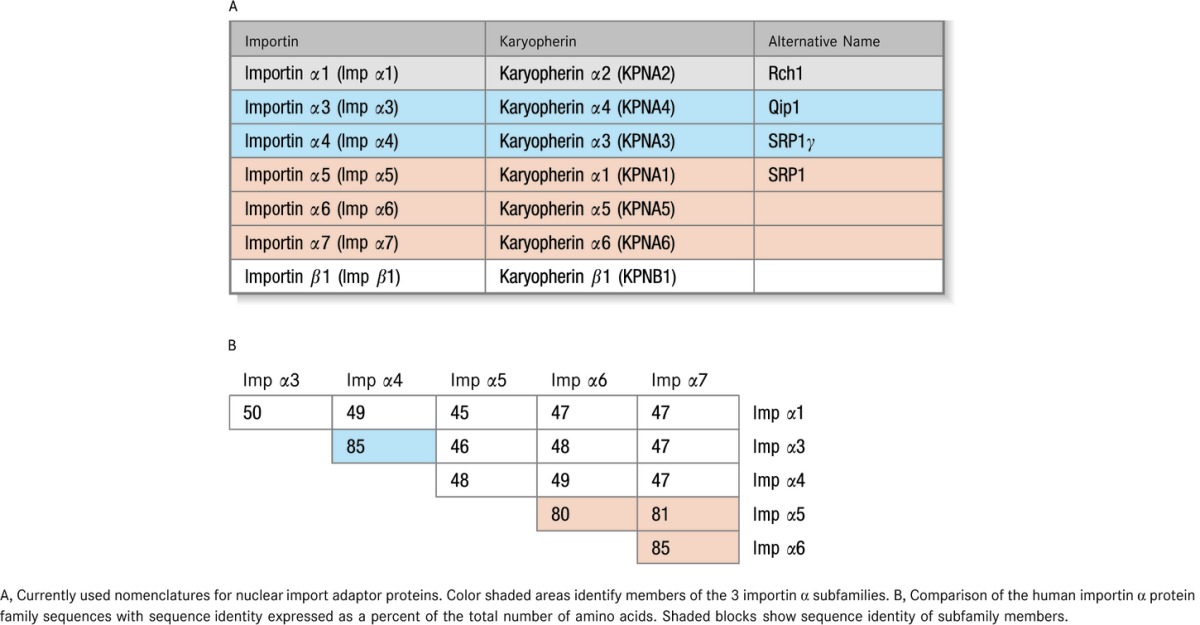
Table 2.
Nomenclature (A) and Sequence Identity (B) of Importins
Methods
Sequence Analyses of Importins/Karyopherins α
Results shown in Table 2B were determined by Align Sequences Protein BLAST (Basic Local Alignment Search Tool, National Center for Biotechnology Information, www.ncbi.nlm.nih.gov). PeptideCutter software was used to search for potential protease cleavage sites (ExPASy, Bioinformatics Resource Portal, www.expasy.org/tools), and T‐Coffee software was employed to analyze the sequences of all 6 human importins α (www.tcoffee.org). T‐Coffee uses the Pfam database to generate a comparison, allowing a combination of results obtained with several alignment methods. It produces a global alignment and a series of local alignments. The program then combines all these alignments into a multiple alignment, leading to a high degree of alignment accuracy.9
Cell Culture
Human T lymphocytes (Jurkat T cells), human epithelial cells adapted to grow in suspension (HeLa S3), and human endothelial cells (EA.hy926, human umbilical vein endothelial cells fused with A549, human lung adenocarcinoma epithelial cells) were obtained from the American Type Culture Collection and cultured according to their recommendations.
Whole Cell Lysates
Whole cell lysates were prepared in 2 steps using a modified hypotonic buffer containing NP‐40 to obtain cytosolic fractions followed by addition of salt (0.45 mol/L NaCl) to extract nuclear proteins.3
Half‐Life (t1/2) of Human Importins
To determine t1/2 of endogenously expressed importins, 10 μg/mL cycloheximide (CHX) was added to Jurkat T cells; 30 minutes later cells were either left unstimulated or stimulated with 5 nmol/L phorbol 12‐myristate 13‐acetate (PMA) and 2 μmol/L ionomycin (Iono). To ensure that protein synthesis was completely suppressed throughout the course of the experiment, extra doses of CHX were added at 8, 24, and 48 hours after stimulation. In parallel samples, 1 μmol/L of the covalent proteasome inhibitor epoxomicin was added to cells 30 minutes before the initial CHX treatment to inhibit proteasomal degradation. Approximately 107 cells were collected at each time point (1, 2, 4, 8, 24, 48, and 72 hours) after CHX treatment and whole cell lysates prepared as described above. Protein content was analyzed by quantitative immunoblotting using the Li‐COR Odyssey infrared imaging system as previously described.1,10
Synthesis and Purification of Peptides
Peptides were synthesized according to general protocols of solid phase peptide synthesis using Fmoc chemistry as described previously.1 Peptide sequences were verified by matrix‐assisted laser desorption/ionization (MALDI) mass spectroscopy in the Vanderbilt Mass Spectrometry Research Core.
Importin Binding Assays
Biotin‐labeled peptides (10 nmols) were incubated overnight with whole‐cell lysates (1.5 mg/mL total protein of Jurkat T cell) and analyzed as described previously.1 For competition binding assays, nonbiotinylated N50 peptide (at 0, 10, 30, 100, or 300 μmol/L) was incubated overnight with 1.5, 0.3 or 0.05 mg/mL total protein of Jurkat T cell lysate in equivalent volumes, followed by pull‐down of remaining unbound proteins with biotinylated N50 peptide immobilized on streptavidin (SA) beads. Proteins were analyzed by quantitative immunoblotting as described previously.1
Relative Abundance of Importins in Human Cells
An equal volume of whole‐cell lysate from each cell type, normalized by total protein concentration, was analyzed by quantitative immunoblotting with a panel of anti‐importin antibodies. Fold abundance is expressed as a ratio of corresponding band intensity normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH).
Preparation of Plasmids, Expression and Purification of Recombinant Proteins
Plasmids for expression of recombinant glutathione S‐transferase (GST) and COOH‐terminal GST‐tagged fusion proteins were prepared by standard cloning procedures using the pET‐21a (+) vector. The NH2 terminal importin β binding (IBB) domain (aa 1‐100) was deleted in constructs for human Imp α1 and Imp α5. Construct sequences were verified in the Vanderbilt Genome Sciences Research Core.
Plasmid constructs for GST‐tagged Imp α1 ΔIBB, GST‐tagged Imp α5 ΔIBB and GST alone were transformed into BL21 (DE3)‐RIL Escherichia coli for expression. Protein expression was induced at 37°C with 0.5 mmol/L isopropyl β‐D‐1‐thiogalactopyranoside for 3 hours.
GST‐tagged Imp α1 ΔIBB and Imp α5 ΔIBB proteins were expressed as inclusion bodies and isolated with Bugbuster Protein Extraction Reagent (Novagen) according to the manufacturer's recommendation, then solubilized in 6 mol/L guanidine buffer (6 mol/L GuHCl, 100 mmol/L NaH2PO4, 10 mmol/L Tris·HCl, pH 8.0) and refolded at 4°C by dialysis against refolding buffer (1 mol/L urea, 50 mmol/L glycine, 20 mmol/L HEPES [pH 8.5], 150 mmol/L NaCl, 5 mmol/L KCl, 2 mmol/L MgCl2, 1 mmol/L EDTA, 2 mmol/L GSH, and 0.2 mmol/L GSSG), followed by dialysis against FPLC Bind/Wash buffer (140 mmol/L NaCl, 2.5 mmol/L KCl, 10 mmol/L Na2HPO4, 2 mmol/L KH2PO4 [pH 7.3], 2 mmol/L DTT, 0.01% Tween 20).
The control protein, GST, was expressed as a soluble protein. The bacterial cell pellet was resuspended on ice in FPLC Bind/Wash buffer (15 mL/g) supplemented with bacterial protease inhibitors, DNase and lysozyme, then lysed by sonication and centrifuged to remove cell debris.
All proteins were purified by fast protein liquid chromatography (FPLC) on GST affinity columns according to manufacturer's protocols and dialyzed against “intracellular” buffer (140 mmol/L KCl, 10 mmol/L NaCl, 10 mmol/L Hepes [pH 7.0], 2.5 mmol/L MgCl2, 1 mmol/L EGTA, 1 mmol/L DTT, 0.02% NP‐40, and 0.01% fatty acid‐free BSA).
Determination of Binding Affinity
Peptide‐protein binding affinity was determined based on the Bio‐Layer Interferometry (BLI) technique utilized in the Octet RED96 System (ForteBio). Briefly, biotinylated N50 or N50M peptide (5 nmol/L in PBS supplemented with 0.02% NP‐40 and 0.01% fatty acid‐free BSA) was immobilized on SA biosensors (10 minutes at 37°C). Biosensors were washed in “intracellular” buffer (see Preparation of Plasmids, Expression and Purification of Recombinant Proteins for composition) then placed into target protein or GST control protein solutions at varying concentrations (50, 100, 150, and 200 nmol/L). Peptide‐protein interaction responses were recorded for 15 minutes, then protein solutions were replaced with “intracellular” buffer and dissociation responses recorded for an additional 15 minutes. Association and dissociation curves were analyzed with ForteBio Data Analysis software v. 6.3.0.40.
In Silico 3‐D Docking Models
AutoDock Vina11 was used to generate all docking models in this study. To generate 3‐D coordinates of ligands structures of QRKRQK, QRDEQK, and KKKRKVE ligands were built from l‐amino acids using the biopolymer editor in ChemBioDraw Ultra 13.0 and converted to 3‐D models using ChemBio3D Ultra 13.0 software. 3‐D models were then optimized by energy minimization and their coordinates saved as .pdb files. Receptors: 3‐D coordinates of corresponding receptors (importins/karyopherins α) were obtained from the RCSB Protein Data Bank (The Research Collaboratory for Structural Bioinformatics, http://www.rcsb.org/pdb/home/home.do) as Protein Data Bank (PDB)‐formatted files: 1q1sC.pdb (crystal structure of mouse KPNA2/Imp α1), 3feyC.pdb (crystal structure of human KPNA2/Imp α1), and 3tj3B.pdb (crystal structure of human KPNA1/Imp α5). To prepare ligand and receptor input files, input files in PDBQT format were prepared in AutoDockTools 1.5.612 (part of the MGL Tools software package) according to a general protocol.13 In general, the default values for docking parameters were used except for exhaustiveness and numbers of modes, which were set to 30 and 20, respectively. Grid spacing was set at 1 Å and the size of the docking grid was determined separately for each of the major and minor importin α NLS binding pockets: 1q1sC—major 24×32×34 Å, minor 22×34×34 Å; 3feyC—major 22×26×20 Å, minor 22×26×20 Å; 3tj3B—major 20×16×28 Å, minor 22×20×24 Å. Six independent docking models generated by AutoDock Vina were then processed, analyzed and visualized in Python Molecule Viewer 1.5.6 (PMV, part of the MGL Tools software package).
Statistical Considerations
No comparisons of treatment–induced change were implemented in any experiments included in this manuscript, therefore no formal statistical analyses were appropriate. In Figure 1 (half‐life), each corresponding data point across all panels (A through H) originates from the same cell lysate sample for each independent experiment, analyzed by immunoblotting for the different proteins. Data to generate graphs in Figure 4 (Relative Abundance, A through F) were obtained from one sample for each of the 3 cell types for each experiment, analyzed by immunoblotting for the different proteins. All data points in Figure 5B (Competition Binding Assay) were generated from the same cell lysate for each independent experiment, diluted and incubated with different concentrations of unlabeled peptide then immunoblotted for 3 different importins. No repeated measurements were conducted in these experiments. All data points in Figures 1, 4, and 5B are shown as the mean value of at least 3 independent experiments. To indicate the measure of sampling variability between experiments, errors are expressed as standard deviations.
Figure 1.
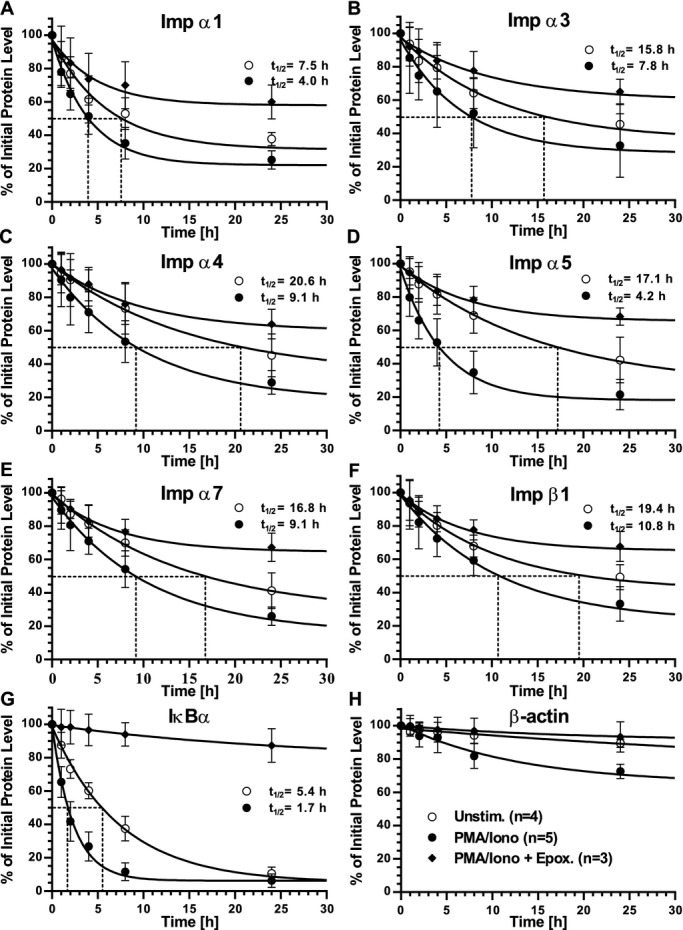
t1/2 of endogenous importins in Jurkat T cells. Graphic representation of quantitative immunoblot analyses of A, Imp α1; B, Imp α3; C, Imp α4; D, Imp α5; E, Imp α7; F, Imp β1; G, IκBα; and H, β‐actin in whole cell lysates from Jurkat T cells. Cells were left unstimulated (open circles), stimulated with PMA/Iono (solid circles), or stimulated with PMA/Iono after pretreatment with the proteasome inhibitor epoxomicin (solid diamonds). Please see (H) for symbol key. All cells were first pretreated with cyclohexamide to block de novo protein synthesis. Protein levels were quantified by immunoblotting and values at each time point normalized to β‐actin at that same time point, then calculated as a percentage of endogenous protein at t0. Dotted lines indicate t1/2. Results are shown as the mean±SD of at least 3 independent experiments. Imp α indicates importin α; Iono, ionomycin; PMA, phorbol 12‐myristate 13‐acetate; SD, standard deviation.
Figure 4.
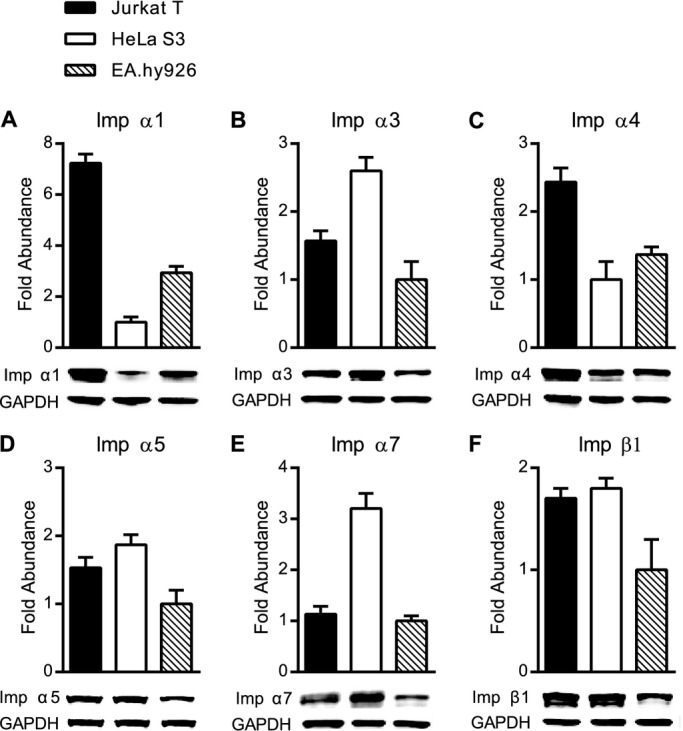
Relative abundance of endogenous importins in Jurkat T cells, HeLa S3 cells and EA.hy926 cells. A through F, Endogenous proteins in whole cell lysates from unstimulated cells were analyzed by quantitative immunoblotting and the abundance of each was calculated after normalization to GAPDH controls. For each importin, the cell type with the least abundance of that importin was set to 1.0 and the relative fold abundance of this importin in the remaining cell types is indicated. The same amount of total lysate protein from all 3 cell types was used for analysis, but was adjusted empirically for each importin based on general abundance of that importin as well as its antibody sensitivity. A, Imp α1, 6 μg; B, Imp α3, 23 μg; C, Imp α4, 17 μg; D, Imp α5, 34 μg; E, Imp α7, 23 μg; and F, Imp β1, 34 μg of total lysate protein was loaded for each cell type. Results are shown as the mean+SD from 3 independent experiments. Imp α indicates importin α; SD, standard deviation.
Figure 5.
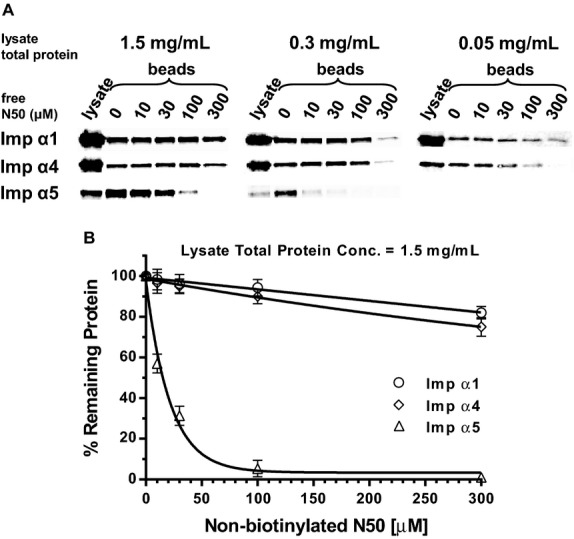
N50 peptide demonstrates specific binding to Imp α5 in a competition binding assay. Unlabeled N50 peptide (free N50) at indicated concentrations was incubated with whole cell lysate from Jurkat T cells. Biotinylated N50 peptide immobilized on agarose beads was added to pull down any importins not already complexed with free N50. Importins bound to beads were then quantified by immunoblotting. A, Immunoblots of importins pulled down from lysate total protein concentrations of 1.5, 0.3, and 0.05 mg/mL. Detection of importins in whole cell lysate is shown in the first column (lysate) for comparison to importins pulled down (beads). B, Quantitative representation of immunoblots from lysate total protein concentration of 1.5 mg/mL. The value of each importin bound to beads with no competition (0) was set to 100% and the percent of remaining importins bound after competition was calculated at each concentration of free N50 and shown as the mean±SD of 3 independent experiments. Please note that even high concentrations of free N50 peptide do not substantially inhibit binding of Imp α1 (open circles) or Imp α4 (open diamonds) to immobilized N50, but inhibition of Imp α5 (open triangles) binding to immobilized N50 is concentration dependent. Imp α indicates importin α; SD, standard deviation.
Results
Turnover of Endogenous Importins Differs in Unstimulated and Stimulated Human T Cells
The abundance of importins contributes to their function as nuclear shuttles.14 However, the intracellular turnover of endogenous importins in human cells, which contributes to their level of expression, remained unknown. Therefore, we first analyzed turnover of importins in resting and stimulated human Jurkat T cells. We employed the human T lymphocyte‐derived Jurkat T cell line for these analyses of endogenous importins. T cells are involved in vascular inflammation caused by microbial, autoimmune, and metabolic insults, and were used in our initial study of nuclear transport of proinflammatory SRTFs.3–4 Levels of endogenous importins were determined by quantitative immunoblotting with monospecific antibodies for human Imp α1, α3, α4, α5, α7, and β1. Of the 7 known importin α family members, only Imp α2 and α6 were not included in our study as Imp α2 is not expressed in mammalian cells and no monospecific antibody against Imp α6 was commercially available. As shown in Figure 1, turnover studies indicated that the t1/2 of importins ranged between 7.5 and 20.6 hours in unstimulated T cells. Upon stimulation of Jurkat T cells with PMA/Iono, standard T cell agonists that induce signal‐dependent nuclear transport of SRTFs,3 the turnover of importins was accelerated (Figure 1A through 1F). As a comparative control for our experimental system, we also determined the turnover of IκBα (Figure 1G), an inhibitor of NFκB known for its stimulus‐dependent accelerated turnover.15 IκBα sequesters NFκB1‐RelA heterodimers in the cytoplasm by shielding their NLSs.16 When inflammatory signaling is induced, IκBα is rapidly degraded by proteasomes in an ATP‐dependent manner.15,17–18 The turnover of all proteins was normalized to turnover of the cellular control protein β‐actin, which was reduced by <10% (in unstimulated cells) during the experimental time span used for these analyses (Figure 1H). In most cases, importin turnover increased ≈2‐fold in stimulated cells. However, the turnover of Imp α5 was accelerated 4‐fold (Figure 1D), notably similar to the increased rate of turnover for IκBα (Figure 1G). As IκBα turnover is regulated by phosphorylation, ubiquitination, and ATP‐dependent proteosomal degradation,15,17–18 we pretreated cells with epoxomicin, an irreversible covalent inhibitor of proteasomes.19 As anticipated, epoxomicin drastically slowed turnover of IκBα (Figure 1G). Significantly, it had a similar effect on the turnover of all importins analyzed by us, indicating their sensitivity to proteosomal degradation (Figure 1A through 1F). A search for potential protease cleavage sites in all 6 human importins using PeptideCutter software did not reveal any obvious differences between Imp α5 and other importin α isoforms. However, a multiple sequence comparison of all members of the human importin α family using T‐Coffee software identified 2 areas of significant sequence dissimilarity, which we termed importin α diversity regions (IADRs), localized adjacent to the major and minor NLS binding pockets (Figure 2).
Figure 2.
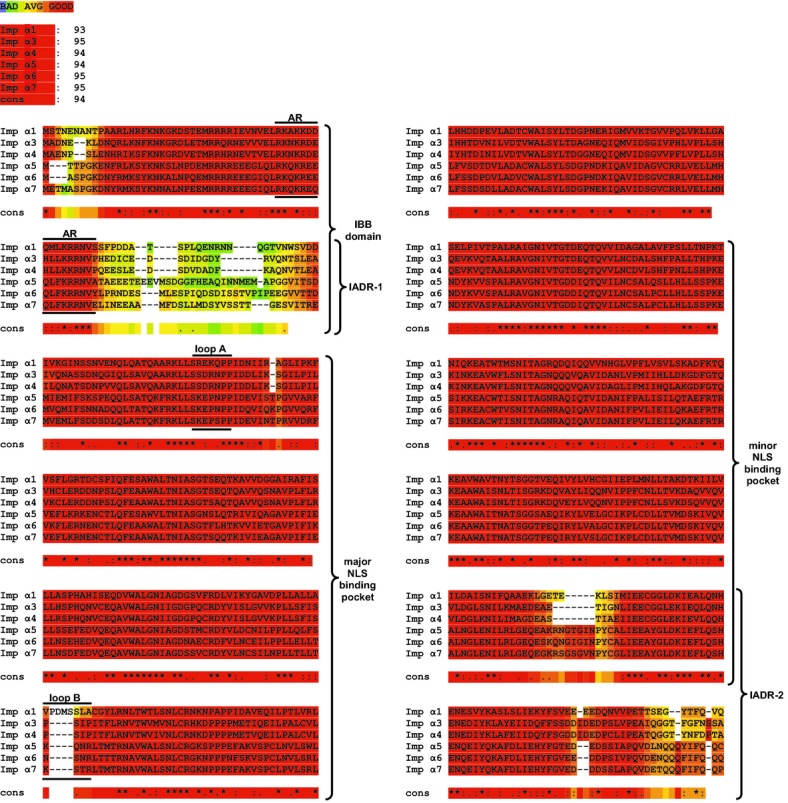
Sequence alignment. Multiple sequence alignment of the importin α family proteins by T‐Coffee software, displayed as a “heat map.” Regions with a high degree of similarity are shown in red, with decreasing similarity indicated from orange to blue (see color key at top). The overall similarity between different proteins is shown immediately below the color key, followed by the individual sequence alignments. The graphic colored output indicates the level of consistency between the final alignment and the library used by T‐Coffee. The main score is the total consistency value. A value of a 100 means full agreement between the considered alignment and its associated primary library. Please note the 2 regions of sequence divergence, denoted importin α diversity regions (IADRs), positioned near the beginning and end of the sequences. IADR‐1 and IADR‐2 are adjacent to, or overlap, the major and minor nuclear localization sequence (NLS) binding pockets, respectively. Also indicated are the positions of the auto‐inhibitory region (AR) within the importin β binding (IBB) domain, and loops A and B within the major NLS binding pocket. Symbol key: “cons” represents the consensus sequence, “*” represents the same amino acid at a given position in all proteins, “:” represents highly similar residues at a given position, and “.” represents amino acids with a similar functionality at a given position.
A Peptide Mimicking the NFκB1/p50 Nuclear Localization Sequence Interacts With Imp α1, α3, α4, and α5 in 3 Human Cell Types
To evaluate the mechanism of recognition specificity of human importins α toward NTMs, we designed studies with the knowledge that the interaction of importins α with nuclear cargo displaying an NLS is dependent on their cytoplasmic concentration as well as the binding affinity between them.14 Therefore, we analyzed lysates from untransfected human cells, retaining their proteome that comprises a physiological mix of endogenous importins and other intracellular proteins to maintain their natural relative abundance and avoid potential pitfalls related to forced expression of proteins.20
Three NLS‐based peptides mimicking the NFκB1/p50 NLS region were designed and produced for these binding studies: linear (N50), cyclized (cN50.1), and mutated (N50M) (Table 1). Peptides were labeled with biotin at the NH2 terminus and a 5 glycine connector was added between the NLS sequence and biotin to prevent steric hindrance upon binding to SA‐coated agarose beads. Interaction of these peptides with endogenous nuclear import adaptor proteins was studied in 3 human cell types: T lymphocytes (Jurkat T cells), epithelial cells (HeLa S3), and endothelial cells (EA.hy926) to determine whether cell lineage influences the combinatorial mix of importins/karyopherins α interacting with NLS peptides. Peptides were incubated with whole cell lysates from unstimulated cells and complexes formed between them and cellular proteins were then captured with SA‐coated beads followed by immunoblot analysis with antibodies to Imp α1, α3, α4, α5, α7, β1, and GAPDH. The avidity of peptide/protein interactions shows a similar pattern in all 3 cell types (Figure 3). Four out of the 6 analyzed importins, Imp α1, α3, α4, and α5, were detected in association with N50 and cN50.1 peptides. The remaining 2 importins, Imp α7 and Imp β1, although present in the lysate, were not pulled down by any of the peptides in any cell type. The mutated control peptide, N50M, was slightly reactive, as only weak interactions were observed between N50M and Imp α1 and α4, and interaction with Imp α5 was barely detectable (Figure 3).
Figure 3.
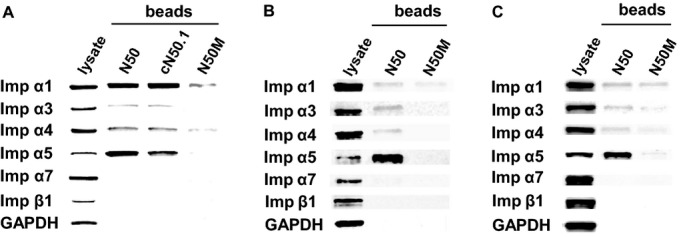
Binding of N50 peptides to endogenous importins shows variations between different human cell types. Biotinylated N50, cN50.1, and N50M peptides (A) or N50 and N50M peptides (B and C) were added to whole cell lysates from: A, Jurkat T cells; B, HeLa S3 cells; or C, EA.hy926 cells. Peptide/protein complexes were isolated with SA‐coated beads and analyzed by immunoblotting with antibodies against a panel of importins. Immunoblotting for the cellular protein GAPDH was used as a control to detect non‐specific interactions. Detection of endogenous proteins in the whole cell lysate is shown in the first column (lysate). Imp α indicates importin α; SA, streptavidin.
While differing antibody specificities preclude quantitative comparisons between different importins, using the same antibody to detect a given importin in lysates from 3 different cell types accurately reflects its relative abundance in different cell types (Figure 4), and demonstrates the variable strength of peptide/protein interactions. Likewise, the ratio between the amount of an individual importin pulled down by binding to N50 compared to the amount of that importin detected in the lysate fraction defines the relative strength of N50 binding to that importin. Thus, we determined that interaction between N50 peptide and Imp α1 is weaker in lysates from HeLa S3 cells (Figure 3B) and EA.hy926 cells (Figure 3C) as abundance of these proteins is 7‐ and 2‐fold lower, respectively, compared to Jurkat T cells (Figure 4A). A similar pattern is observed when N50 peptide interacts with Imp α4. This interaction appears stronger in the lysate obtained from Jurkat T cells (Figure 3A) compared to the same interaction in HeLa S3 cells (Figure 3B) and EA.hy926 cells (Figure 3C) as abundance of Imp α4 is 2‐fold higher in Jurkat T cells than in the other 2 cell lines (Figure 4C). In contrast, interaction of N50 peptide with Imp α3 is stronger in HeLa S3, as its abundance in this cell line is 2‐fold higher (Figure 4B). Imp α7 follows a similar pattern to Imp α3 (Figure 4E). The interaction of N50 peptide with Imp α5 is similarly strong in all 3 cell types, consistent with comparable intracellular concentrations of this importin (Figure 4D) and akin to the pattern seen with Imp β1 (Figure 4F). Nonetheless, the relative amount of Imp α5 bound to biotinylated N50 pulled down by SA‐coated beads is strikingly higher than any of the other detected proteins, indicating that the interaction between N50 and Imp α5 is stronger than the other peptide/protein interactions analyzed in this study. Based on this observation, we focused our next experiments on defining the specificity of N50 interactions with Imp α5 in competition binding assays compared to N50 interactions with Imp α1 and α4.
Specificity of Peptide Binding to Importins α
The results from the importin binding assay led us to postulate that interactions between N50 and Imp α5 were more specific than those with Imp α1 and α4. Therefore, we designed a modified competition binding assay to determine specificity of N50 binding to different importins. We added increasing concentrations of unlabeled “free” N50 peptide to different concentrations of lysate total protein from unstimulated Jurkat T cells. After overnight incubation at 4ºC, biotinylated N50 peptide, immobilized on SA beads, was added to pull down unbound proteins, which were then detected by quantitative immunoblotting (Figure 5A). Binding of Imp α5 to immobilized N50 peptide was tightly controlled by unlabeled peptide in all lysate total protein concentrations. In the most concentrated lysates (1.5 mg/mL), ≈70% inhibition was observed with 30 μmol/L of unlabeled peptide and about 95% inhibition with 100 μmol/L. In striking contrast, binding of Imp α1 and α4 to immobilized N50 peptide in the most concentrated extracts was slightly inhibited. Imp α1 and α4 band intensities were only reduced by about 20% with the highest concentration of unlabeled peptide (300 μmol/L) (Figure 5B). Their relatively unchanged binding isotherms, especially compared to the dramatic inhibition of Imp α5 in the most concentrated lysate (Figure 5B), provided additional evidence that N50 interactions with Imp α1 and Imp α4 are nonspecific.
Interaction of N50 Peptide With Imp α5 Shows Strong Binding Affinity With 2:1 Stoichiometry
Having established the specificity of N50 peptide binding to Imp α5, we set out to determine the strength of this interaction, as compared to the interaction with Imp α1, by performing binding affinity assays using GST‐tagged importin α proteins produced without their NH2‐terminal IBB domains, which contain an auto‐inhibitory region (AR) that prevents nonspecific cargos from interacting with their major and minor NLS binding pockets21 (see Figure 2). Biotinylated N50 peptide was immobilized on SA biosensors and incubated with GST‐tagged Imp α1 ΔIBB and Imp α5 ΔIBB, at 50, 100, 150, and 200 nmol/L concentrations. Biotinylated N50M peptide served as an inactive peptide control, and solutions of GST alone at corresponding concentrations were used as background control. Consistent with previous experiments showing binding specificity (see Figure 5), the maximum response recorded for interaction of N50 peptide with Imp α5 was more than 20‐fold higher than the maximum response recorded during interaction of N50 peptide with Imp α1 (Figure 6A). The responses obtained using control mutant peptide, N50M, as a ligand for interaction with Imp α5 and α1 proteins were both at the background level (Figure 6A, inset).
Figure 6.
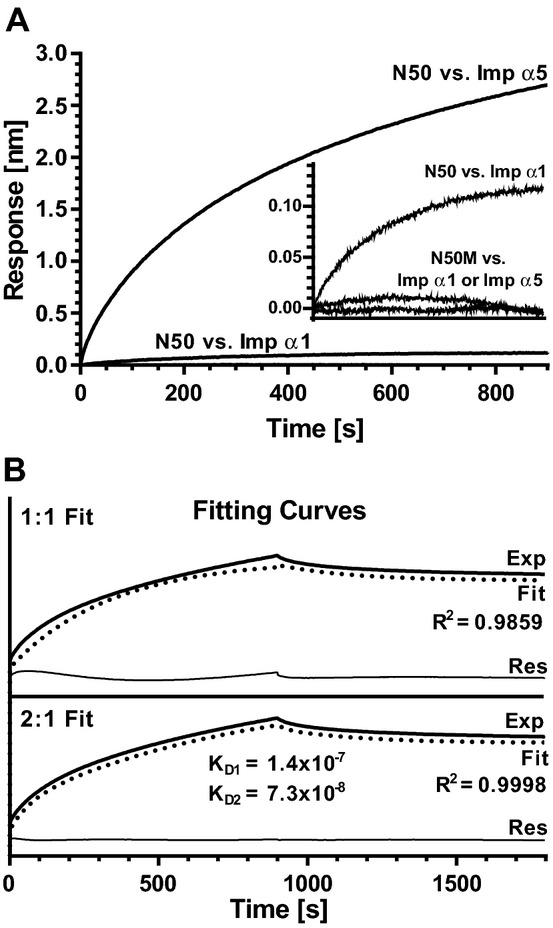
A bio‐layer interferometry (BLI) assay indicates that N50 peptide displays high binding affinity for Imp α5, but not Imp α1. Biotinylated N50 or its non‐binding mutant control N50M immobilized on SA‐coated biosensors were incubated at 30°C with 50, 100, 150 or 200 nmol/L solutions of GST‐tagged Imp α1 ΔIBB, GST‐tagged Imp α5 ΔIBB or GST alone. A, Association curves of N50 peptide with Imp α1 at 200 nmol/L and with Imp α5 at 200 nmol/L, normalized by subtracting N50 peptide binding with GST at 200 nmol/L. Association curves of N50M mutant peptide with Imp α1 and Imp α5, both at 200 nmol/L, normalized by subtracting N50M mutant peptide binding with GST at 200 nmol/L are shown in the inset panel, along with the binding isotherm for N50 peptide with Imp α1 at 200 nmol/L, for comparison. B, Fitting curves corresponding to a 1:1 (upper) or 2:1 (bottom) binding stoichiometry for interaction between N50 and Imp α5. Exp, recorded signal (heavy solid line); Fit, simulation curve (dotted line); Res, residual curve (thin solid line, Exp minus Fit). Each graph is representative of 3 independent experiments. Binding affinities were calculated using data from all concentrations of proteins in all 3 experiments. IBB indicates importin β binding; Imp α, importin α; GST, glutathione S‐transferase; SA, streptavidin.
Detailed analysis of these data indicated that interaction between N50 peptide and Imp α5 most likely proceeds according to a 2:1 kinetic model (Figure 6B). Fitting the theoretical curve with the experimental data in the 2:1 model resulted in a correlation coefficient equal to R2=0.9998 and a flat residual curve, while fitting in the 1:1 model gave R2=0.9859, resulting in an irregular‐shaped residual curve. The binding affinities in the 2:1 model were calculated as KD1=1.4×10−7 mol/L and KD2=7.3×10−8 mol/L (Figure 6B). Cumulatively, these results indicate preferential and specific binding of N50 peptide to Imp α5 whereas the observed binding to Imp α1 was proven nonspecific. The 2:1 stoichiometry of the N50 peptide binding to Imp α5 suggests occupancy of both major and minor NLS binding pockets on Imp α5.
The Auto‐Inhibitory Region of Imp α7 Impedes N50 Peptide Access to its NLS Binding Pockets
Given the high degree of homology between Imp α5 and Imp α7 (see Table 2B), we were puzzled by our inability to detect any interactions between N50 and endogenous Imp α7 in binding assays (see Figure 3). We hypothesized that the auto‐inhibitory effect of ARs differs among endogenous importins α, thereby affecting the ability of N50 peptide to access NLS binding pockets. To test this hypothesis, we designed and produced biotinylated peptides derived from the ARs of each importin α (see Table 1) and employed them in binding assays in human T cell lysate to determine their ability to compete with native AR homologs. As shown in Figure 7, the protective effect of ARs does vary among the different importins α. Imp α1 displayed the least protection by its AR, which was unable to efficiently protect its NLS binding pockets from interaction with any biotinylated ligands (row 1), or compete with native ARs to bind other importins (column 4). Conversely, Imp α7 was most protected by its AR, as most of the biotinylated peptides displayed minimal interaction with endogenous Imp α7 (row 5). Moreover, biotinylated AR7 successfully competed with native ARs of all importins α, including its own (column 8). These results indicate that the binding affinity of the Imp α7 AR to its own major and minor NLS binding pockets is stronger than the affinity of the N50‐Imp α7 interaction, thus explaining the inability of biotinylated N50 peptide to pull down Imp α7 (see Figure 3).
Figure 7.
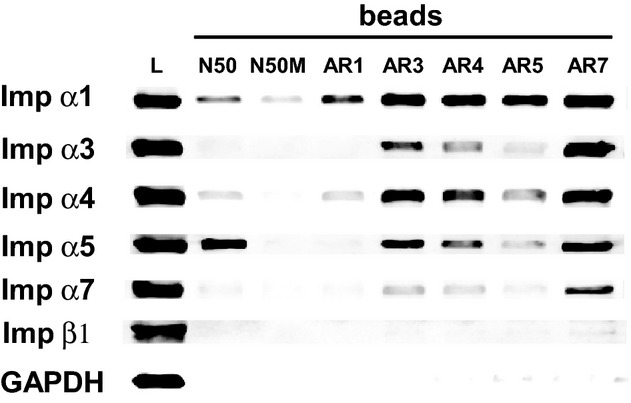
Interaction of endogenous importins in Jurkat T cells with peptides derived from their auto‐inhibitory regions. Biotinylated N50 and N50M peptides, as well as biotinylated peptides representing the auto‐inhibitory regions (AR) of Imp α1, α3, α4, α5, and α7, were added individually to whole cell lysates from Jurkat T cells. The peptide and its interacting proteins were pulled down with SA‐coated beads and analyzed by immunoblotting with antibodies against a panel of importins as in Figure 3. Immunoblotting for the cellular protein GAPDH was used as a control to detect non‐specific interactions. Detection of endogenous proteins in the whole cell lysate is shown in the first column (lysate). Imp α indicates importin α; SA, streptavidin.
Models of N50 Peptide Docking to Imp α1 and α5 Confirm Their Different Binding Characteristics
We employed 3‐D models of NLS‐Imp α docking to identify structural elements of importins α potentially responsible for stabilization/destabilization of observed interactions with N50. All docking models were generated by AutoDock Vina software11 using available crystal structures of human Imp α1 (PDB ID: 3FEY Chain C) and human Imp α5 (PDB ID: 3TJ3 Chain B). Since the crystal structure of the NFκB/p50 NLS was not available, we used ChemOffice 13.0 software to build, optimize and generate 3‐D coordinates of peptide chains to serve as ligands in our docking model. Before attempting an in silico docking analysis of p50 NLS peptide to Imp α1 and α5, we first verified the accuracy of the AutoDock Vina software by performing a control docking analysis using a binding pair that has been previously analyzed by x‐ray crystallography, and therefore has an independently verified binding conformation.
We selected a crystal structure of simian‐virus‐40 large T‐antigen (SV40) NLS bound to mouse Imp α1,22 available at RCSB Protein Data Bank (PDB ID: 1Q1S). For practical reasons, we reduced the length of the SV40 NLS sequence to 7 residues: KKKRKVE (42 rotatable bonds), then generated 3‐D coordinates and modeled its docking to the major NLS binding site of mouse Imp α1 (PDB ID: 1Q1S Chain C). The resulting ligand conformations were analyzed in PythonMoleculeViewer (PMV) and compared to the known crystal structure of this NLS (PDB ID: 1Q1S Chain B). We identified a conformation that was highly similar to its crystallographic homolog (Figure 8A), confirming the capability of the software to generate accurate docking models. The root mean square deviation (RMSD) of α carbons and all backbone atoms equaled 0.36 and 0.56 Å, respectively.
Figure 8.
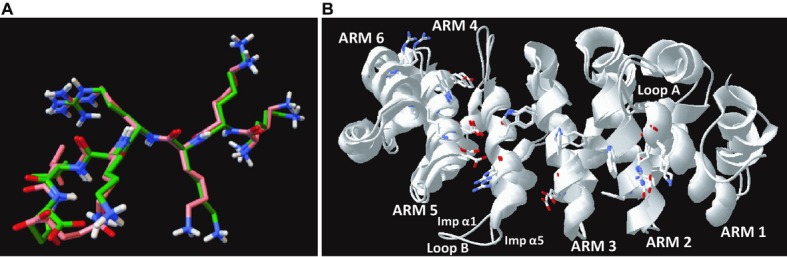
Three‐dimensional models. A, Unaligned images of the conformation of SV40 NLS obtained from the control modeling of its interaction with mouse Imp α1 (orange) and the structure of the same NLS motif obtained from x‐ray Crystallography (PDB ID: 1Q1SChain B) (green) are shown overlapped. Functional atoms are color coded: red, oxygen; blue, nitrogen; white, polar hydrogen. The RMSD of α carbons and all backbone atoms of unaligned conformations equals 0.58 and 0.77 Å, respectively. When aligned, the RMSD of α carbons and all backbone atoms equals 0.36 and 0.56 Å, respectively. Conformation of SV40 NLS were generated by AutoDock Vina 1.1.2 and visualized in PythonModelViewer 1.5.6. B, Imp α1 and α5 structures display high homology. Superimposed ribbon structures of the major NLS binding pockets from human Imp α1 (KPNA2, PDB ID: 3FEY Chain C) and human Imp α5 (KPNA1, PDB ID: 3TJ3 Chain B). The positions of structural components, including the major functional residues on the surface of NLS binding pockets, indicate a high degree of structure and sequence similarity. Loop B was identified as a region of structural diversity, in addition to IADR‐1 and IADR‐2 (see Figure 2). Structures were visualized in DeepView software (Swiss‐PdbViewer 4.1.0, Swiss Institute of Bioinformatics, http://spdbv.vital-it.ch/). ARM indicates armadillo; IADR, importin α diversity region; Imp α, importin α; NLS, nuclear localization sequence; RMSD, root mean square deviation.
After the accuracy of the software was established, we used the strict NLS sequence of N50 peptide (QRKRQK) as a ligand to model docking to the major and minor NLS binding pockets of Imp α1 and α5. This sequence corresponds to the NLS sequence of NFκB1/p50. As a control ligand, we used an inactive loss‐of‐function mutant of this NLS (QRDEQK), corresponding to the mutated sequence of N50M (see Table 1). The 3‐D coordinates of these peptide chains, each containing around 30 rotatable bonds, were generated as described in the Methods, and adapted as ligand files using AutoDockTools software. The docking models were obtained through 4 independent processes, individually covering binding to major and minor NLS binding pockets of each importin α. Conformations with the best binding affinity (lowest binding energy) were chosen for further analysis. Results showing docking of QRKRQK and its mutant QRDEQK sequences to both NLS binding pockets of Imp α1 and α5 are presented in Table 3. Even though the QRKRQK sequence occupies a similar position in the major NLS binding pocket of both importins (Figure 9C and 9D), analysis of docking affinities (KD, calculated for T=173 K with the assumption that free rotation around the majority of bonds is frozen) indicates that its binding to Imp α5 is ≈10 times stronger than to Imp α1 (0.6 and 1.9 nmol/L versus 4.5 and 19 nmol/L) (Table 3B). These calculated values are consistent with those obtained experimentally for N50 binding to Imp α5 by BLI (Table 3A). Though the high degree of similarity demonstrated in Figure 2 between the major NLS binding pockets of Imp α1 and α5 is paralleled by their structures (Figure 8B), the higher binding affinity of the QRKRQK sequence to Imp α5 suggests existence of a structural element stabilizing their interaction. Despite their high homology, a comparative structural analysis of the major NLS binding pockets of Imp α1 and α5 identified an inconsistency in the structure of loop A (the loop connecting ARM1 with ARM2 [see Figures 8B and 2]). The central position on loop A in Imp α1 is occupied by lysine (K108) (Figure 9A), while the same position on Imp α5 is occupied by proline (P114) (Figure 9B), forcing the side chain of glutamic acid (E113) to bend (Figure 9B and 9D). This structural difference may stabilize a positively charged cargo bound to the Imp α5 major NLS binding pocket. The side chain of the corresponding amino acid on Imp α1, glutamate E107, projects out from the surface and does not participate in stabilization of docked cargo (Figure 9A and 9C). When the mutated QRDEQK sequence was used as a docking ligand, binding affinity for Imp α5 to the major binding pocket decreased 25‐fold and to the minor binding pocket 8‐fold (15 nmol/L for both, see Table 3C). The ligand mutation had less effect on binding affinity for Imp α1. Mutated ligand docking affinity for the major pocket decreased 3‐fold, but conversely, binding to the minor site was almost 2‐fold stronger (11 nmol/L for both, see Table 3C). The lack of any significant loss in affinity by the mutated sequence for Imp α1 suggests that the surface of the NLS binding pocket on Imp α1 is more permissive, allowing negatively charged residues to bind with similar affinity. This might also explain the presence of a protein band corresponding to an N50M‐Imp α1 interaction in binding assays (see Figure 3). Thus molecular modeling based on energy minimization algorithms is fully consistent with experimentally obtained binding parameters of N50 peptide in regard to Imp α5 (high affinity) and Imp α1 (low affinity).
Table 3.
Characteristics of N50‐Imp α Interactions
| Imp α1 | Imp α5 | |||
|---|---|---|---|---|
| Major | Minor | Major | Minor | |
| A. VQRKRQKLMP | ||||
| KD (BLI), nmol/L | N/D | N/D | 73 | 140 |
| B. QRKRQK | ||||
| ΔG (dock), kcal/mol | −6.6 | −6.1 | −7.3 | −6.9 |
| KD (calc)*, nmol/L | 4.5 | 19 | 0.6 | 1.9 |
| C. QRDEQK | ||||
| ΔG (dock), kcal/mol | −6.3 | −6.3 | −6.2 | −6.2 |
| KD (calc)*, nmol/L | 11 | 11 | 15 | 15 |
A, N50‐Imp α binding affinity (KD) obtained from Bio‐Layer Interferometry (see Figure 6), B and C, Docking energy and calculated docking affinity obtained from modeling of B, N50 and C, N50M interactions with Imp α1 and α5. Docking energy was generated by AutoDock Vina 1.1.2. Please note that docking affinities obtained from the modeling study differ from those obtained from BLI due to different experimental conditions (temperature, ligand size, etc.). However, the binding characteristics are generally consistent between the 2 methods.
Calculated from the equation ΔG=RT·ln(KD) for T=173 K, with the assumption that rotation around the majority of bonds is frozen.
Figure 9.
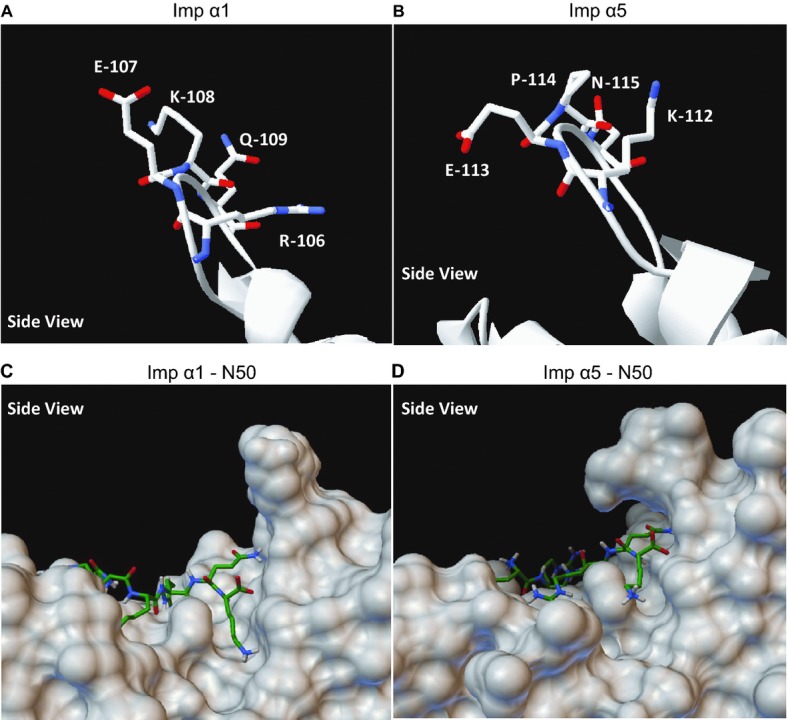
Composition of loop A in Imp α1 and α5 and its effect on docking of N50 to the major NLS binding pocket. A and B, Ribbon structure and composition of loop A of (A) human Imp α1 (KPNA2), and (B) human Imp α5 (KPNA1). Structures were visualized in DeepView. C and D, 3‐D models of p50 NLS docking to the molecular surface of (C) human Imp α1 (KPNA2), and (D) human Imp α5 (KPNA1). Please note that loop A may provide stability to the p50 NLS‐Imp α5 complex while no such role is apparent for loop A in Imp α1. Docking models were generated by AutoDock Vina 1.1.2 and visualized by PythonMoleculeViewer 1.5.6. Imp α indicates importin α; NLS, nuclear localization sequence.
Discussion
Nuclear translocation of SRTFs and other transcription factors (TFs) larger than 45 kDa is dynamically controlled by importins/karyopherins,23–26 most often through their interaction with NLSs on TFs.27–29 TFs such as NFκB, the master regulator of inflammation and immunity, contain 1 or more unique NLSs arranged as monopartite or bipartite motifs consisting of short sequences of basic residues (lysine and arginine) flanked by proline, valine, or other nonpolar amino acids.29–31 Given the limited number of importins α in the cytoplasm of human cells, it seems a combinatorial challenge for each individual TF, among the hundreds ready for delivery to the nucleus, to locate and bind its preferred importin α partner. A “Hobson's choice”, in which nuclear cargo binds to the first available importin, would be a reasonable model. Indeed, initial studies of human importins and their cargos employing a single‐cargo assay in an isolated system, found that all tested importin α family members bound cargos with similar efficiency. However, when the number of cargos was increased, a more physiological scenario, some displayed a strong preference for a single importin α,32 thereby disproving a “Hobson's choice” model. Furthermore, when the NLSs of 2 well‐characterized cargos were swapped, it was found that the NLS alone was not the only determinant of this preference.33 This implies recognition specificity of TFs for particular importins, making the turnover‐controlled availability of importins and their affinity toward NLS‐containing cargo salient features of nuclear transport.
Nuclear transport adaptors in the importin α family are highly similar (see Table 2B and Figure 2) and each comprises 10 armadillo (ARM) repeats that form major and minor NLS binding pockets located in their NH2‐ and COOH‐terminal regions, respectively.34 In this study we have determined that N50 peptide, which mimics the NLS region of NFκB1/p50, interacts in human cells with endogenous importins α and successfully competes with their auto‐inhibitory regions (ARs) for NLS binding pockets. We show that variability in the protective effect of the ARs in different importin α family members might also play a role in their binding to the NLS represented by N50, especially in the case of Imp α7, which is inaccessible to this peptide. Importantly, we demonstrate for the first time that N50 peptide preferentially binds to Imp α5 (KPNA1) with 2:1 stoichiometry. This stoichiometry is consistent with compelling evidence provided by x‐ray crystallography34–36 and Surface Plasmon Resonance data.37
This preferential, high‐affinity, targeting of Imp α5 by N50 peptide is consistent with previous studies that have reported involvement of Imp α5 in ferrying transcription factor STAT1 to the nucleus. Intracellular ablation of Imp α5, but not Imp α1, reduced nuclear import of this transcription factor.38 A homodimer of phosphorylated STAT1 is recognized by the minor NLS binding pocket of Imp α5 with a binding affinity of 191 nmol/L.39 This finding is in agreement with the inhibition of STAT1 nuclear import by SN50 peptide.3 Furthermore, nuclear import of the TF nuclear factor erythroid 2‐related factor 2, which responds to oxidative stress, depends on interaction with Imp α5 and β1 and its nuclear translocation is modulated by SN50 peptide.40 However, the exact role of Imp α5 in nuclear transport of structurally diverse SRTFs is yet to be elucidated.
Structural analysis of importins indicates a high degree of similarity (45% to 50% identity) in sequences between members of the Imp α family. Isoforms that belong to the same subfamily (eg, Imp α5, α6, and α7) display 80% to 85% sequence identity (Table 2B). Their only areas of diversity, which we term IADR (see Figure 2), are located at the beginning of the major NLS binding pocket and to a lesser extent at the end of the minor NLS binding pocket. Based on its location, IADR‐1 may influence the positioning of loop A in the major NLS binding pocket. The presence of proline (P114) in Imp α5 loop A may help stabilize docking of N50 peptide. A structural discrepancy between loop B in Imp α1 and Imp α5 may exert an opposite effect (see Figures 2 and 8B). In Imp α1, loop B is highly hydrophobic (LLAVPDMSSLA), and double the size of loop B in other importins α. This unusual connection between the H2 and H3 helices inside ARM4 introduces more flexibility to the structure, potentially destabilizing interaction of N50 peptide with the major NLS binding pocket. Conversely, loop B on Imp α5 consists of only 5 hydrophilic amino acids (SKQNR), thereby functioning as a regular turn to provide structural rigidity. The role of loop B in NLS binding requires further investigation. More detailed structure‐function analyses will also be required to definitively establish how the IADR of a given importin α contributes to preferential binding of TFs. This type of analysis would be highly valuable in the quest to develop new importin α‐specific cell‐penetrating peptide inhibitors.
Analysis of the relative abundance and turnover of endogenous importins also provides clues to their differential binding characteristics. Their abundance varies between human cell types of different lineages, with Imp α1 and α4 being highest in T lymphocytes (Jurkat T cells), while Imp α3, α5, and α7 more abundant in epithelial cells (HeLa S3). Imp β1 is least abundant in endothelial cells (EA.hy926) (Figure 4). These differences may indicate different utilization of importins among the ≈250 cell types that constitute human organ systems. Although we employed human cells representing distinct lineages, their transformed phenotypes may differ from their primary cell counterparts. Despite this caveat, we posit that differential abundance of importins in different cell types should be considered when analyzing nuclear transport mediated by a given importin.
We also found that the turnover of importins is accelerated in stimulated T cells due, at least in part, to proteasomal degradation, mostly by the β‐5 subunit and to a lesser degree by the β‐2 subunit of proteasomes inactivated by epoxomycin.19 Given the essential role of ATP‐dependent proteosomal degradation of IκBα,15,17–18 we propose that the protective effect of epoxomicin on importins turnover in stimulated cells implies a similar mechanism of proteosomal degradation. Among the 6 human importins studied here, Imp α5 has the twice the rate of turnover in stimulated human lymphocytes than any other importin we studied, suggesting that it may be more readily degraded by the epoxomicin‐targeted β‐5 subunit of the proteasome. Accelerated turnover of Imp α5 in stimulated cells indicates that Imp α5, despite its high homology with Imp α6 and Imp α7 (Table 2B), is more prone to degradation than other importins when inflammatory signaling is induced, thereby limiting availability of Imp α5 for nuclear transport of cognate TFs. This rate‐limiting mechanism may dampen excessive transport of proinflammatory transcription factors, such as STAT1, to the nucleus.
It has been reported that different members of the NFκB family such as NFκB1 (p105/p50), NFκB2 (p100/p52), RelA (p65), RelB, and c‐Rel,6 are translocated by more than 1 importin α, and that selectivity of these interactions is dependent on the varying abundance of force‐expressed proteins and transfection‐induced posttranslational modifications.41–42 A recent study of NFκB‐mediated vascular inflammation reports involvement of Imp α3 based on its expression targeted by miR‐181b, although changes in expression of Imp α5 in the vascular endothelium were noteworthy but not quantified.43
We propose that targeting Imp α5 by the N50 sequence of SN50 and its subsequent NTM congeners (cSN50 and cSN50.1) contributes to inhibition of signaling pathways evoked by metabolic, autoimmune, and microbial stimuli that cause inflammatory disorders analyzed in their preclinical models.1,44–50 It is important to note that Imp α5‐deficient mice are viable and fertile and do not show any obvious morphological or behavioral abnormalities,51 supporting the observation in these preclinical models that in vivo targeting of Imp α5 in the nuclear import machinery by NTMs is well‐tolerated.1,44–50 Further studies will be required to determine the dependency of N50 inhibitory function on Imp α5 and whether the weaker N50 interactions with other importin α family members play any significant role in mediating nuclear import of NFκB or other SRTFs in Imp α5‐deficient cells. More detailed knowledge of the interactions between nuclear transport adaptors and the TFs they ferry to the nucleus to promulgate an inflammatory response would advance our concept of targeting nuclear transport of TFs with cell‐penetrating NTM peptides.
Thus, these findings provide a structural and functional framework for further development of the next generation of NTM peptides to preferentially target nuclear transport by different members of the importin α family.
Sources of Funding
This work was supported by grants from the National Institutes of Health to Dr Hawiger (HL085833 and AA015752). Core Services performed through Vanderbilt University Medical Center's Digestive Disease Research Center supported by NIH grant P30DK058404 Core Scholarship. Additional support was provided by the Vanderbilt Clinical and Translational Science Award UL1TR000445.
Disclosures
None.
Acknowledgments
We thank Ruth Ann Veach for expert assistance in preparation of the manuscript and James E. Crowe Jr. and his colleagues for logistical help with the Octet RED96 instrument.
References
- 1.Liu Y, Major AS, Zienkiewicz J, Gabriel CL, Veach RA, Moore DJ, Collins RD, Hawiger J. Nuclear transport modulation reduces hypercholesterolemia, atherosclerosis, and fatty liver. J Am Heart Assoc. 2013; 2:e000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF‐kappa B by a synthetic peptide containing a cell membrane‐permeable motif and nuclear localization sequence. J Biol Chem. 1995; 270:14255-14258 [DOI] [PubMed] [Google Scholar]
- 3.Torgerson TR, Colosia AD, Donahue JP, Lin YZ, Hawiger J. Regulation of NF‐kappa B, AP‐1, NFAT, and STAT1 nuclear import in T lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF‐kappa B p50. J Immunol. 1998; 161:6084-6092 [PubMed] [Google Scholar]
- 4.Hawiger J. Innate immunity and inflammation: a transcriptional paradigm. Immunol Res. 2001; 23:99-109 [DOI] [PubMed] [Google Scholar]
- 5.Veach RA, Liu D, Yao S, Chen Y, Liu XY, Downs S, Hawiger J. Receptor/transporter‐independent targeting of functional peptides across the plasma membrane. J Biol Chem. 2004; 279:11425-11431 [DOI] [PubMed] [Google Scholar]
- 6.Orange JS, May MJ. Cell penetrating peptide inhibitors of nuclear factor‐kappa B. Cell Mol Life Sci. 2008; 65:3564-3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003; 100:12027-12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SJ, Sekimoto T, Yamashita E, Nagoshi E, Nakagawa A, Imamoto N, Yoshimura M, Sakai H, Chong KT, Tsukihara T, Yoneda Y. The structure of importin‐beta bound to SREBP‐2: nuclear import of a transcription factor. Science. 2003; 302:1571-1575 [DOI] [PubMed] [Google Scholar]
- 9.Notredame C, Higgins DG, Heringa J. T‐coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000; 302:205-217 [DOI] [PubMed] [Google Scholar]
- 10.Lin Q, Liu Y, Moore DJ, Elizer SK, Veach RA, Hawiger J, Ruley HE. Cutting edge: the “death” adaptor CRADD/RAIDD targets BCL10 and suppresses agonist‐induced cytokine expression in T lymphocytes. J Immunol. 2012; 188:2493-2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trott O, Olson AJ. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31:455-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999; 17:57-61 [PubMed] [Google Scholar]
- 13.Morris GM, Huey R, Olson AJ. Using autodock for ligand‐receptor docking. Curr Protoc Bioinformatics. 2008 [DOI] [PubMed] [Google Scholar]
- 14.Timney BL, Tetenbaum‐Novatt J, Agate DS, Williams R, Zhang W, Chait BT, Rout MP. Simple kinetic relationships and nonspecific competition govern nuclear import rates in vivo. J Cell Biol. 2006; 175:579-593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donald R, Ballard DW, Hawiger J. Proteolytic processing of NF‐B/IB in human monocytes: ATP‐dependent induction by pro‐inflammatory mediators. J Biol Chem. 1995; 270:9-12 [DOI] [PubMed] [Google Scholar]
- 16.Ganchi PA, Sun SC, Greene WC, Ballard DW. I kappa B/MAD‐3 masks the nuclear localization signal of NF‐kappa B p65 and requires the transactivation domain to inhibit NF‐kappa B p65 DNA binding. Mol Biol Cell. 1992; 3:1339-1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal‐induced site‐specific phosphorylation targets I kappa B alpha to the ubiquitin‐proteasome pathway. Genes Dev. 1995; 9:1586-1597 [DOI] [PubMed] [Google Scholar]
- 18.Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal‐induced degradation of I kappa B alpha requires site‐specific ubiquitination. Proc Natl Acad Sci USA. 1995; 92:11259-11263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin: 20s proteasome reveals a molecular basis for selectivity of alpha′, beta′‐epoxyketone proteasome inhibitors. J Am Chem Soc. 2000; 122:1237-1238 [Google Scholar]
- 20.Hawiger J. Cellular import of functional peptides to block intracellular signaling. Curr Opin Immunol. 1997; 9:189-194 [DOI] [PubMed] [Google Scholar]
- 21.Lott K, Cingolani G. The importin beta binding domain as a master regulator of nucleocytoplasmic transport. Biochim Biophys Acta. 2011; 1813:1578-1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontes MRM, Teh T, Toth G, John A, Pavo I, Jans DA, Kobe B. Role of flanking sequences and phosphorylation in the recognition of the simian‐virus‐40 large T‐antigen nuclear localization sequences by importin‐alpha. Biochem J. 2003; 375:339-349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newmeyer DD, Forbes DJ. Nuclear import can be separated into distinct steps in vitro: nuclear pore binding and translocation. Cell. 1988; 52:641-653 [DOI] [PubMed] [Google Scholar]
- 24.Richardson WD, Mills AD, Dilworth SM, Laskey RA, Dingwall C. Nuclear protein migration involves two steps: rapid binding at the nuclear envelope followed by slower translocation through nuclear pores. Cell. 1988; 52:655-664 [DOI] [PubMed] [Google Scholar]
- 25.Akey CW, Goldfarb DS. Protein import through the nuclear pore complex is a multistep process. J Cell Biol. 1989; 109:971-982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jans DA, Hubner S. Regulation of protein transport to the nucleus: central role of phosphorylation. Physiol Rev. 1996; 76:651-685 [DOI] [PubMed] [Google Scholar]
- 27.Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984; 39:499-509 [DOI] [PubMed] [Google Scholar]
- 28.Lanford RE, Butel JS. Construction and characterization of an SV40 mutant defective in nuclear transport of T antigen. Cell. 1984; 37:801-813 [DOI] [PubMed] [Google Scholar]
- 29.Dingwall C, Laskey RA. Nuclear targeting sequences—a consensus? Trends Biochem Sci. 1991; 16:478-481 [DOI] [PubMed] [Google Scholar]
- 30.Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large‐T antigen. Nature. 1984; 311:33-38 [DOI] [PubMed] [Google Scholar]
- 31.Robbins J, Dilwortht SM, Laskey RA, Dingwall C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991; 64:615-623 [DOI] [PubMed] [Google Scholar]
- 32.Kohler M, Speck C, Christiansen M, Bischoff FR, Prehn S, Haller H, Gorlich D, Hartmann E. Evidence for distinct substrate specificities of importin alpha family members in nuclear protein import. Mol Cell Biol. 1999; 19:7782-7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedrich B, Quensel C, Sommer T, Hartmann E, Kohler M. Nuclear localization signal and protein context both mediate importin alpha specificity of nuclear import substrates. Mol Cell Biol. 2006; 26:8697-8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell. 1998; 94:193-204 [DOI] [PubMed] [Google Scholar]
- 35.Fontes MR, Teh T, Kobe B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin‐alpha. J Mol Biol. 2000; 297:1183-1194 [DOI] [PubMed] [Google Scholar]
- 36.Fontes MR, Teh T, Jans D, Brinkworth RI, Kobe B. Structural basis for the specificity of bipartite nuclear localization sequence binding by importin‐alpha. J Biol Chem. 2003; 278:27981-27987 [DOI] [PubMed] [Google Scholar]
- 37.Catimel B, Teh T, Fontes MR, Jennings IG, Jans DA, Howlett GJ, Nice EC, Kobe B. Biophysical characterization of interactions involving importin‐alpha during nuclear import. J Biol Chem. 2001; 276:34189-34198 [DOI] [PubMed] [Google Scholar]
- 38.Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal‐dependent nuclear import of Stat1 is mediated by nuclear pore‐targeting complex formation with NPI‐1, but not Rch1. EMBO J. 1997; 16:7067-7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nardozzi J, Wenta N, Yasuhara N, Vinkemeier U, Cingolani G. Molecular basis for the recognition of phosphorylated STAT1 by importin alpha5. J Mol Biol. 2010; 402:83-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, Arinze IJ. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008; 283:8984-8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fagerlund R, Kinnunen L, Köhler M, Julkunen I, Melén K. NF‐κB is transported into the nucleus by importin α3 and importin α4. J Biol Chem. 2005; 280:15942-15951 [DOI] [PubMed] [Google Scholar]
- 42.Fagerlund R, Melén K, Cao X, Julkunen I. NF‐κB p52, RelB and c‐Rel are transported into the nucleus via a subset of importin α molecules. Cell Signal. 2008; 20:1442-1451 [DOI] [PubMed] [Google Scholar]
- 43.Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Blackwell TS, Baron RM, Feinberg MW. Microrna‐181b regulates NF‐kappaB‐mediated vascular inflammation. J Clin Invest. 2012; 122:1973-1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu XY, Robinson D, Veach RA, Liu D, Timmons S, Collins RD, Hawiger J. Peptide‐directed suppression of a pro‐inflammatory cytokine response. J Biol Chem. 2000; 275:16774-16778 [DOI] [PubMed] [Google Scholar]
- 45.Liu D, Li C, Chen Y, Burnett C, Liu XY, Downs S, Collins RD, Hawiger J. Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem. 2004; 279:48434-48442 [DOI] [PubMed] [Google Scholar]
- 46.Liu D, Zienkiewicz J, DiGiandomenico A, Hawiger J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Mol Ther. 2009; 17:796-802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duffy JY, McLean KM, Lyons JM, Czaikowski AJ, Wagner CJ, Pearl JM. Modulation of nuclear factor‐kappaB improves cardiac dysfunction associated with cardiopulmonary bypass and deep hypothermic circulatory arrest. Crit Care Med. 2009; 37:577-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Sullivan AW, Wang JH, Redmond HP. NF‐κB and p38 MAPK inhibition improve survival in endotoxin shock and in a cecal ligation and puncture model of sepsis in combination with antibiotic therapy. J Surg Res. 2009; 152:46-53 [DOI] [PubMed] [Google Scholar]
- 49.Moore DJ, Zienkiewicz J, Kendall PL, Liu D, Liu X, Veach RA, Collins RD, Hawiger J. In vivo islet protection by a nuclear import inhibitor in a mouse model of type 1 diabetes. PLoS One. 2010; 5:e13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veach RA, Zienkiewicz J, Collins RD, Hawiger J. Lethality in a murine model of pulmonary anthrax is reduced by combining nuclear transport modifier with antimicrobial therapy. PLoS One. 2012; 7:e30527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shmidt T, Hampich F, Ridders M, Schultrich S, Hans VH, Tenner K, Vilianovich L, Qadri F, Alenina N, Hartmann E, Kohler M, Bader M. Normal brain development in importin‐alpha5 deficient‐mice. Nat Cell Biol. 2007; 9:1337-1338 [DOI] [PubMed] [Google Scholar]