

Abstract
Background
Our laboratory has previously demonstrated the importance of a cytoskeletal‐based survival signaling pathway using in vitro models of ischemia/reperfusion (IR). However, the importance of this pathway in mediating stress‐elicited survival signaling in vivo is unknown.
Methods and Results
The essential cytoskeletal signaling pathway member focal adhesion kinase (FAK) was selectively deleted in adult cardiac myocytes using a tamoxifen‐inducible Cre‐Lox system (α‐MHC‐MerCreMer). Polymerase chain reaction (PCR) and Western blot were performed to confirm FAK knockout (KO). All mice were subjected to a 40‐minute coronary occlusion followed by 24 hours of reperfusion. Ischemic preconditioning (IP) was performed using a standard protocol. Control groups included wild‐type (WT) and tamoxifen‐treated α‐MHC‐MerCreMer+/−/FAKWT/WT (experimental control) mice. Infarct size was expressed as a percentage of the risk region. In WT mice IP significantly enhanced the expression of activated/phosphorylated FAK by 36.3% compared to WT mice subjected to a sham experimental protocol (P≤0.05; n=6 hearts [sham], n=4 hearts [IP]). IP significantly reduced infarct size in both WT and experimental control mice (43.7% versus 19.8%; P≤0.001; 44.7% versus 17.5%; P≤0.001, respectively). No difference in infarct size was observed between preconditioned FAK KO and nonpreconditioned controls (37.1% versus 43.7% versus 44.7%; FAK KO versus WT versus experimental control; P=NS). IP elicited a 67.2%/88.8% increase in activated phosphatidylinositol‐3‐kinase (PI3K) p85/activated Akt expression in WT mice, but failed to enhance the expression of either in preconditioned FAK KO mice.
Conclusions
Our results indicate that FAK is an essential mediator of IP‐elicited cardioprotection and provide further support for the hypothesis that cytoskeletal‐based signaling is an important component of stress‐elicited survival signaling.
Keywords: cytoskeleton, ischemia, ischemic preconditioning
Introduction
Heart disease is the leading cause of death in the United States, and ischemia/reperfusion (IR)‐induced cell death, such as seen during myocardial infarction (MI), is a major cause of morbidity and mortality among those suffering from heart disease.1–2 The best strategy to improve both survival and quality of life in patients suffering from MI is to minimize myocardial death that occurs due to IR. In the laboratory setting, certain interventions are capable of mitigating the lethal cellular injury associated with IR including hypothermia,3 heat stress,4 and ischemic preconditioning.5 However, the mechanisms responsible for this cytoprotection have yet to be completely elucidated.
Ischemic preconditioning (IP), defined as a (series of) brief episode(s) of reversible ischemia preceding a lethal period of ischemia, elicits the most robust and consistent protection from IR.6 Since the discovery of IP, there has been a substantial amount of research directed at understanding the mechanism of IP‐elicited cardioprotection. A wide range of effector molecules and signaling proteins have been implicated in the IP response including phosphatidylinositol‐3‐kinase (PI3K), Akt (protein kinase B), protein kinase C‐epsilon (PKC‐ε), and extracellular signal‐regulated kinase (ERK).7–8 Furthermore, many studies have investigated how IP may modulate cellular metabolism,9 and particular emphasis has been paid to the effect of IP on delaying the formation of the mitochondrial permeability transition pore.10 However, despite the volume of research aimed at understanding the downstream mediators of the cardioprotective effect of IP, the mechanism by which IP‐elicited cardioprotective signaling is initiated has received less attention and is consequently less completely understood. Better understanding of the mechanism by which cardioprotective signaling is initiated by IP and/or other known modulators of IR injury may shed light on novel targets for ischemic heart disease therapy.
Focal adhesions are specialized cell‐to‐matrix junctions that link the extracellular matrix to the actin‐based cytoskeleton. While cardiac myocytes are not thought to contain focal adhesions proper, they do contain analogous specialized structures known as costameres.11–12 Costameres are vinculin‐containing protein complexes which encircle cardiac myocytes perpendicular to their long axis. In addition to vinculin, costameres contain other proteins characteristic of focal adhesions including talin, α‐actinin, and β1 integrins.12 Overlying the Z disc of the sarcomere, costameres are positioned to form a physical link between the contractile apparatus and the extracellular matrix, allowing externally applied and intrinsically generated mechanical force to be transmitted bidirectionally.12 This ability to transduce mechanical force into biological activity may enable costameres to play an important role in initiating signaling cascades in response to ischemic stress.
Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase and an essential mediator of cytoskeletal and integrin signaling.13 In response to clustering of integrins, FAK becomes localized within costameres and focal adhesions at the cytoplasmic tails of integrins where it is auto‐phosphorylated and thereby activated.13–14 FAK activation is also dependent on cytoskeletal integrity, as disruption of the cytoskeleton with cytochalasin D, an inhibitor of actin polymerization, has been demonstrated to prevent stretch‐induced FAK activation in neonatal rat ventricular myocytes (NRVM).15 Once activated, FAK may interact with several other signaling and scaffolding proteins. Of particular importance is paxillin, an adaptor protein which interacts directly with integrins and FAK, helping to concentrate other proteins within the focal adhesion signaling complex.16 FAK‐mediated tyrosine phosphorylation of these proteins creates high affinity binding sites for several SH2‐domain–containing proteins found within focal adhesions, notably PI3K and Src kinases, thereby facilitating downstream signaling.13,17 This cytoskeletal signaling complex (integrin/FAK/paxillin/PI3K) has been shown to be important for maintaining cell viability in both nonmuscle cells and in cardiac myocytes.18–20
Our laboratory has previously demonstrated that heat stress in cultured neonatal rat ventricular myocytes (NRVMs) is associated with the assembly of an integrin‐FAK‐paxillin signaling complex and a reduction in cell death in response to metabolic inhibition (simulated IR).4,11 Moreover, our laboratory has demonstrated that both IP in the isolated adult rat heart and whole body heat stress in adult rats lead to the activation of FAK and protection from ischemic cell death.7 Cardioprotection in these models was associated with the activation of Akt, a well‐established cardioprotective and antiapoptotic protein.21 The ability of both IP and heat stress to activate FAK and Akt raises the possibility that IP‐elicited signaling may be initiated as part of a general cytoprotective cellular stress response. Importantly, our previous studies showed that disruption of integrin‐FAK interaction in NRVM worsened myocyte survival following simulated IR, supporting the concept that FAK plays a central role in cell survival signaling.4 Furthermore, NRVMs induced to overexpress FRNK (FAK‐related non‐kinase), an endogenous competitive inhibitor of FAK, were found to have reduced expression of activated Akt following heat stress, implicating Akt as an important mediator of cardioprotection downstream of FAK.11
Whereas our previous studies utilized in vitro and ex vivo model systems to demonstrate activation and protection resulting from a cytoskeletal‐based cardioprotective signaling pathway, this study was designed to determine whether this same pathway is activated in a well‐characterized in vivo model of myocardial infarction. To specifically address the role of FAK in the proximal activation of the signaling pathway, a mouse line was created in which FAK was inducibly knocked‐out in a myocyte‐selective fashion using the well‐established α‐MHC‐MerCreMer system.22 If cytoskeletal signaling is indeed an important component of the stress‐elicited survival signaling characteristic of IP, then loss of FAK, an essential mediator of cytoskeletal signaling, should reduce/abolish the cardioprotective effect afforded by IP as well as prevent the activation of Akt, a known downstream mediator of IP‐elicited cardioprotection.
Methods
Animals
The use of animals in this study was approved by the Animal Care and Use Committee of Louisiana State University Health Sciences Center. All reported experiments conformed to the standards in the National Institutes of Health's Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85‐23, Revised 1996). Experiments were performed on adult male 8‐ to 12‐week‐old mice. FAKflox/WT mice were received as a generous gift from Dr Jun‐Lin Guan at the University of Michigan. α‐MHC‐MerCreMer+/− mice were purchased from The Jackson Laboratory (stock # 005657). Both the floxed FAK and α‐MHC‐MerCreMer+/− mice were received on a mixed C57BL/6 and 129/SV genetic background. All mice were bred and housed in the Louisiana State University Health Sciences Center Animal Care Facility in specific pathogen‐free chambers. α‐MHC‐MerCreMer+/− mice and FAKflox/WT mice were bred together for 4 generations to generate wild‐type (WT) and transgenic animals on a uniform genetic background. This breeding strategy entailed initially crossing α‐MHC‐MerCreMer+/− and FAKflox/WT mice to produce Cre+/−/FAKflox/WT mice. These Cre+/−/FAKflox/WT mice were subsequently bred together for 3 generations, producing Cre−/−/FAKWT/WT (WT), Cre+/+/FAKWT/WT, Cre−/−/FAKflox/flox and Cre+/−/FAKflox/flox mice. WT×WT, Cre+/+/FAKWT/WT×WT, and Cre−/−/FAKflox/flox×Cre+/−/FAKflox/flox mice were then crossed to generate all animals used in the study.
Conditional FAK KO Mice
We used Cre‐Lox technology to generate an inducible, cardiac myocyte‐specific FAK knockout (KO) mouse line. FAKflox/flox mice were crossed with FAKflox/flox mice heterozygous for the α‐MHC‐MerCreMer recombinase, which has been shown to efficiently mediate genetic recombination in cardiac myocytes following tamoxifen administration.22 The reason for employing an inducible recombination strategy was that it allowed for the determination of the consequences of acute FAK loss in the adult mouse heart and it circumvented the embryonic lethal phenotype associated with total FAK deletion during development.23 Mice tail snips were sent off for genotyping to Transnetyx. The floxed FAK mice contain the third exon of the FAK gene flanked by loxP sites, and Cre‐mediated excision of the floxed FAK exon yields a truncated, nonfunctional gene product.24
Tamoxifen Administration
The tamoxifen solution was generated fresh before each use by dissolving tamoxifen powder (Sigma‐Aldrich) in corn oil to a final concentration of 20 mg/mL. To induce FAK KO, α‐MHC‐MerCreMer+/−/FAKflox/flox mice (hereafter referred to as FAK KO mice) were treated with tamoxifen via intraperitoneal injections once a day for 2 days at a calculated dose of 40 mg/kg. Mice were allowed to recover for 5 days following the tamoxifen administration regimen prior to any subsequent experimental manipulation.
PCR Analysis
Genomic DNA was extracted from cardiac and extra‐cardiac tissues using the REDExtract‐N‐Amp Tissue PCR Kit (Sigma‐Aldrich) and subjected to polymerase chain reaction (PCR) using the forward (GCTGATGTCCCAAGCTATTCC) and reverse (AGGGCTGGTCTGCGCTGACAGG) primers. Anticipated band sizes for the WT product, floxed product, and postrecombination product are 1.4 kb, 1.6 kb, and 550 bp, respectively. The cycling parameters for the PCR were as follows: 3 cycles at 94°C for 3 minutes, 67°C for 2 minutes, and 72°C for 2 minutes; 30 cycles at 94°C for 1 minute, 63°C for 1 minute, and 72°C for 2 minutes; 1 hold at 72°C for 10 minutes.
Western Blot Analysis
For analysis and quantification of tissue‐specific FAK protein reduction, adult cardiac myocytes were isolated from FAK KO mice, and protein was harvested as described below. For analysis of activated FAK (FAKpTyr397), PI3K (pPI3K p85), and Akt (AktpSer473) expression, protein was harvested via whole heart tissue homogenization in T‐PER Protein Extraction Reagent (Thermo Scientific) with protease and phosphatase inhibitor cocktails added (Roche Applied Science). Sixty micrograms of isolated myocyte protein and either 40 μg (for analysis of FAKpTyr397 and AktpSer473 expression) or 80 μg (for analysis of pPI3K p85 expression) of whole heart protein (as determined by bicinchoninic acid [BCA] protein assays) were subjected to sodium dodecyl sulphate (SDS)‐protein electrophoresis. Following electrophoresis, the samples were transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes were blocked for 1 hour at room temperature with Odyssey blocking buffer (LI‐COR Biosciences) and subsequently incubated overnight at 4°C with one of the following primary antibodies: (1) rabbit anti‐FAK (Millipore); (2) rabbit anti‐phospho FAK (Tyr397; Cell Signaling); (3) rabbit anti‐phospho PI3K p85/p55 (Tyr458/Tyr199; Cell Signaling); (4) rabbit anti‐phospho‐Akt (Ser473; Cell Signaling). Membranes were then incubated with the fluorophore‐labeled goat antirabbit secondary antibody (LI‐COR Biosciences) for 1 hour at room temperature, and final protein expression was detected and quantified using the Odyssey infrared imaging system (LI‐COR Biosciences). Quantified protein expression was reported in arbitrary units as indicated.
Isolation of Adult Cardiac Myocytes
The isolation of adult cardiac myocytes was performed using a Langendorff perfusion apparatus as previously described.25 Briefly, mice were anesthetized via an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) and anticoagulated with 200 units of heparin. The heart was rapidly excised from the thoracic cavity, cannulated via the aorta, and perfused in the Langendorff mode with calcium‐free perfusion buffer (120 mmol/L NaCl, 5.4 mmol/L KCl, 1.2 mmol/L MgSO4·7H2O, 1.2 mmol/L NaH2PO4, 20 mmol/L NaHCO3, 5.6 mmol/L glucose, 10 mmol/L butanedione monoxime [BDM; Sigma‐Aldrich], 5 mmol/L taurine, pH 7.3, gassed with 95% O2/5% CO2) for 4 minutes at a rate of 3 mL/min, followed by digestion buffer (perfusion buffer plus 25 μmol/L CaCl2 and Liberase TH [Roche Applied Science] at a concentration of 0.26 units/mL) for 10 to 16 minutes. Following digestion, the left and right ventricles were cut into small pieces with curved fine tip forceps in 2 mL of digestion buffer in a 60 mm Petri dish. The supernatant was transferred to a 15 mL conical test tube containing Stop 1 buffer (10% fetal bovine serum [FBS] in perfusion buffer plus 25 μmol/L CaCl2). The remaining pellet was triturated with 2 mL of digestion buffer with plastic transfer pipettes. On the completion of the trituration, the suspension was transferred and pooled in Stop 1 buffer and filtered utilizing a 100 μm nylon mesh filter. The filtered suspension was transferred to a 15 mL conical test tube (Pellet 1) and allowed to settle by gravity for 15 min at room temperature. The supernatant was aspirated and placed into another 15 mL conical test tube (Pellet 2) and centrifuged for 2 min at 500 rpms. Both pellets were pooled in 12 mL of Stop 2 buffer (5% FBS in perfusion buffer plus 50 μmol/L CaCl2) and transferred to a 100 mm Petri dish. A final calcium level of 1 mmol/L was reintroduced through a series of resuspensions containing increasing concentrations of CaCl2. Myocytes were subsequently plated on laminin‐coated (10 μg/mL) 60 mm Petri dishes in plating media (minimum essential media [MEM] plus 5% FBS, 2 mmol/L l‐glutamine, 1% penicillin/streptomycin, and 10 mmol/L BDM) for 1 hour at 37°C in a humidified atmosphere of 98% air, 2% CO2. The plated myocytes were then washed with 1× phosphate‐buffered saline (PBS) pH 7.4 and protein was harvested for downstream Western blot analysis through lysis with a RIPA buffer kit (Santa Cruz Biotechnology).
Echocardiographical Assessment of Cardiac Function
In preparation for cardiac functional analysis, mice were sedated with 1.5% isoflurane. The VisualSonics VEVO 770 Echocardiogram was used to obtain B‐mode 2 dimensional (2D) images and M‐mode tracings of the anterior and posterior left ventricular wall using a 2D reference sector. Cardiac function parameters examined included fractional shortening, ejection fraction, and heart rate. Left ventricular dimensions were evaluated by measuring interventricular septum end‐systolic/diastolic dimension and left ventricular end‐systolic/diastolic internal diameter. Percent ejection fraction was calculated using the following equation: (stroke volume/end diastolic volume)×100. Percent fractional shortening was calculated using the following equation: ([LVEDD−LVESD]/LVEDD)×100, where LVEDD is the left ventricular end diastolic diameter and LVESD is the left ventricular end systolic diameter.
Myocardial Ischemia/Reperfusion Protocol and Determination of Infarct Size
Figure 1 summarizes the myocardial IR surgical protocols and experimental design used in the study. Mice were anesthetized with an intraperitoneal injection of pentobarbital (60 mg/kg) and an intramuscular injection of ketamine (50 mg/kg) prior to being intubated and connected to a rodent ventilator (MiniVent Type 845; Harvard Apparatus). Ventilation was performed at a rate of 140 breaths/min with a tidal volume of 140 μL and was supplemented with 100% oxygen. Body temperature was monitored with a rectal thermometer and maintained at 37°C throughout the surgical procedure with a heating pad. Depth of anesthesia was assessed using the toe‐pinch reflex, which is established as a useful indicator of depth of anesthesia.26 Following anesthetization, a left thoracotomy was performed, and the left coronary artery (LCA) was isolated and ligated near its origin with 7‐0 cardiovascular silk suture. A short segment of PE10 polyethylene tubing was inserted between the ligation and the surface of the heart in order to protect the underlying LCA from undue trauma. Coronary occlusion was confirmed by examining the surface of the heart for tissue‐blanching in the ischemic zone. The IP protocol consisted of a standard 3 cycles of 5 minutes of ischemia followed by 5 minutes of reperfusion. A sustained 40‐minute ligation of the LCA was used to induce myocardial infarction. Following the 40‐minute coronary occlusion, the ligation was released to allow for arterial reperfusion which was confirmed by hyperemic blushing on the surface of the heart. Buprenorphine (0.03 mg/kg) was administered subcutaneously for postoperative analgesia, and mice were allowed to recover overnight. To perform the sham IP protocol, mice were anesthetized as described above. The suture was passed underneath the LCA, but remained untied. Following 30 minutes, the heart was immediately excised and homogenized for protein harvest as described above.
Figure 1.
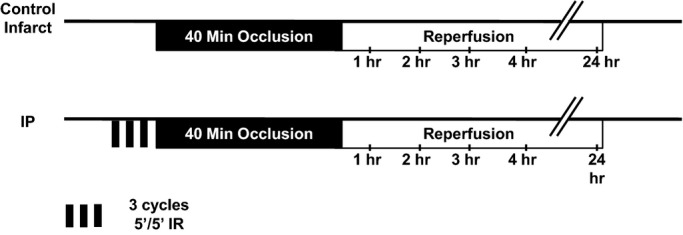
Experimental design. Mice were subjected to a 40‐minute sustained coronary occlusion followed by 24 hours of reperfusion either in the presence (IP) or absence (control infarct) of IP. IP indicates ischemic preconditioning; IR, ischemia/reperfusion.
Following 24 hours of reperfusion, mice were reanesthetized and placed on the ventilator. The right carotid artery was cannulated with a tapered PE 50 catheter, the LCA ligation was replaced, and the heart was injected with 1.5 mL of 15% Phthalo blue dye (diluted in phosphate‐buffered saline; Quantum Ink) via the carotid artery catheter to allow for the delineation between the nonischemic area and the area at risk (AAR). Following the injection of the dye, the heart was rapidly excised, fixed in a 2% SeaPlaque agarose gel solution (Lonza Rockland), and cross‐sectioned into 1 mm thick slices using a McIlwain tissue slicer (Mickle Laboratory Engineering). The slices were incubated at 37°C for 2 minutes in 1% 2,3,5‐triphenyltetrazolium chloride (Sigma‐Aldrich) solution to demarcate the infarcted tissue from the ischemic but viable tissue within the AAR. Each slice was weighed then photographed using an Olympus Q‐Color 5 digital camera mounted to an Olympus SZ61 dissecting microscope. Planimetry was performed by a blinded observer using the National Institutes of Health Image J software to measure the left ventricular area, AAR, and area of infarction. Infarct size was determined using the previously reported equation27: weight of infarct size=(A1×Wt1)+(A2×Wt2)+…+(A6×Wt6) where A is the percent area of infarction calculated by planimetry from corresponding slices 1 to 6 represented by subscripted numbers and Wt is the weight of the same numbered slices. Infarct size was ultimately expressed as a percentage of the AAR using the following equation: weight of infarct size/weight of AAR, where the AAR weight was calculated in a similar manner as the infarct weight by summing the products of the percent AAR and the corresponding slice weight. AAR as a percentage of the total left ventricular area was calculated by (weight of AAR/weight of left ventricle), where the weight of the left ventricle was calculated by summing the weights of all slices.
Statistics
All Western blot data are expressed as means±SE (in arbitrary units). All echocardiography and infarct data are expressed as means±SE. The assumption of normality was investigated in all data prior to performing statistical tests. Echocardiography data were analyzed using an unpaired t‐test analysis. Statistical analysis of FAK expression was performed with a 1‐way analysis of variance (ANOVA) with Tukey's posttest. Statistical analysis of FAKpTyr397 expression was performed using an unpaired t‐test. Statistical analysis of pPI3K p85 and AktpSer473 expression was performed with a 2‐way ANOVA with Bonferroni's posttest. Statistical analysis of infarct data was performed with a 2‐way ANOVA with Bonferroni's posttest. Prism 5 (GraphPad software) was utilized to compute statistics. Significance level was set at P≤0.05.
Results
Generation of Conditional FAK KO Mice
To generate the inducible, cardiac myocyte‐specific FAK KO transgenic mice, we crossed α‐MHC‐MerCreMer+/−/FAKflox/flox mice with FAKflox/flox mice. A total of 131 animals were bred using this breeding pair. Of these 131 mice, 65 were α‐MHC‐MerCreMer+/−/FAKflox/flox and 66 were FAKflox/flox, in accordance with the expected 1:1 Mendelian ratio.
Tamoxifen Induces Genetic Recombination and Reduces FAK Protein Expression in Cre+/−/FAKflox/flox Mice
To determine if genetic recombination had occurred in cardiac tissue harvested from FAK KO mice, cardiac and extra‐cardiac genomic DNA from control and FAK KO mice was subjected to PCR analysis. The 550 bp recombined FAK allele was detected in cardiac genomic DNA of FAK KO mice, but not in cardiac genomic DNA of α‐MHC‐MerCreMer+/−/FAKflox/flox mice not treated with tamoxifen (FAK C, Figure 2A), demonstrating that excision of the floxed FAK gene segment is dependent on the administration of tamoxifen. The recombination allele was not detected in lung, liver, skeletal muscle, or aortic tissue of FAK KO mice confirming the tissue specificity of the recombination event and consistent with the expression of the α‐MHC gene (Figure 2A).
Figure 2.
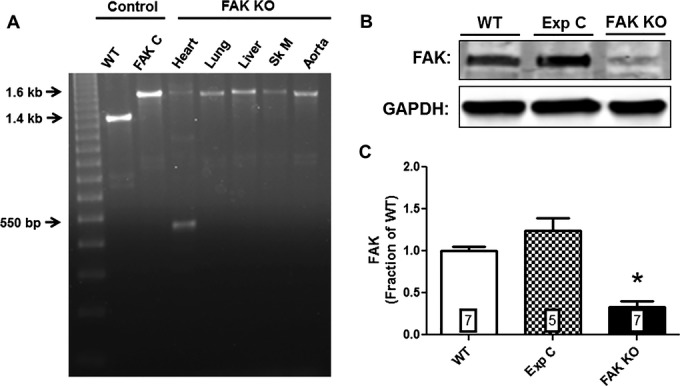
Tamoxifen induced deletion of FAK. A, Genomic DNA of wild‐type (WT) and Cre‐positive floxed FAK mice were amplified by PCR. The WT band is 1.4 kb, the floxed FAK product is 1.6 kb and the postrecombination product is 550 bp. DNA was extracted from cardiac tissue unless otherwise noted. FAK C, Cre‐positive floxed‐FAK mouse not treated with tamoxifen; Sk M, skeletal muscle. B, Representative Western blot showing FAK protein expression in lysates of isolated adult cardiac ventricular myocytes from wild‐type (WT), experimental control (Exp C), and FAK KO mice. C, Normalized integrated intensity data for myocyte FAK expression in WT, Exp C and FAK KO mice. FAK expression in FAK KO mice was reduced by 66.5% compared to WT mice (P≤0.001; n=7 hearts, both groups) and by 73% compared to Exp C mice (P≤0.001; n=5 hearts [Exp C], n=7 hearts [FAK KO]). Numbers within bars indicate sample size of each group. Exp C indicates experimental control; FAK, focal adhesion kinase; KO, knock out; PCR, polymerase chain reaction.
To evaluate if Cre‐mediated recombination of the floxed FAK allele leads to reduced FAK protein expression specifically at the myocyte level, isolated adult cardiac myocyte protein lysates were prepared and subjected to Western blot analysis. Figure 2B and 2C demonstrate that FAK protein was significantly reduced by 66.5% compared to WT mice (P≤0.001; n=7 hearts, both groups) and by 73% compared to tamoxifen‐treated α‐MHC‐MerCreMer+/−/FAKWT/WT mice (P≤0.001; n=5 hearts [Exp C], n=7 hearts [FAK KO]).
FAK Knockout Does Not Affect Baseline Cardiac Function
It has been reported that embryonic FAK KO is associated with lethal cardiac and other developmental defects.23 In addition, use of the tamoxifen/α‐MHC‐MerCreMer expression system has been previously reported to result in a dilated cardiomyopathy.28 Therefore, in developing this model system, it was important to demonstrate that neither conditional reduction of FAK expression nor tamoxifen administration adversely impact baseline cardiac function in our FAK KO mouse model.
To evaluate baseline cardiac function in FAK KO mice, we performed echocardiography at a time when FAK was confirmed to be significantly reduced (ie, 5 days after tamoxifen administration). No difference in ejection fraction, fractional shortening, or heart rate was observed in FAK KO mice compared to WT mice (see Table). Moreover, interventricular septum end‐systolic/diastolic dimension and left ventricular end‐systolic/diastolic internal diameter were unchanged between both groups (see Table). Finally, no differences in gross heart morphology were apparent in FAK KO mice compared to WT mice (Figure 3). Therefore, neither reduction of FAK protein expression nor our tamoxifen administration regimen affected baseline cardiac function of FAK KO mice.
Table 1.
Echocardiographical Analysis of Cardiac Function
| WT (n=8) | FAK KO (n=18) | |
|---|---|---|
| EF, % | 57.3±3.4 | 55.7±1.7 |
| FS, % | 29.9±2.5 | 28.5±1.1 |
| IVSd, mm | 0.72±0.08 | 0.70±0.03 |
| IVSs, mm | 0.92±0.11 | 0.94±0.06 |
| LVIDd, mm | 3.65±0.10 | 3.76±0.09 |
| LVIDs, mm | 2.55±0.13 | 2.69±0.08 |
| HR, bpm | 448.6±5.9 | 463.3±13.3 |
Values were calculated from at least 2 separate M‐mode measurements. Values are means±SE. EF indicates ejection fraction; FAK, focal adhesion kinase; FS, fractional shortening; HR, heart rate; IVSd, interventricular septum end diastolic dimension; IVSs, interventricular septum end systolic dimension; KO, knock out; LVIDd; left ventricular internal diameter at end diastole; LVIDs, left ventricular internal diameter at end systole; WT, wild‐type.
Figure 3.
Photograph comparing gross morphology of FAK KO and WT hearts at baseline. FAK KO heart was photographed 5 days after conclusion of tamoxifen administration. FAK indicates focal adhesion kinase; KO, knock out; WT, wild‐type.
IP Induces FAK Activation
To demonstrate that in vivo IP stimulates FAK activation and signaling in our model system, activated FAK expression was examined in WT mice subjected either to the IP protocol or a sham experimental protocol. To assess activated FAK expression, heart lysates from IP and non‐IP WT mice were analyzed for expression of tyrosine‐phosphorylated FAK as a surrogate for FAK activation.13 Figure 4 shows that FAKpTyr397 expression in WT mice subjected to IP was significantly increased by 36.3% compared to mice subjected to the sham procedure (P≤0.05; n=6 hearts [sham], n=4 hearts [IP]).
Figure 4.
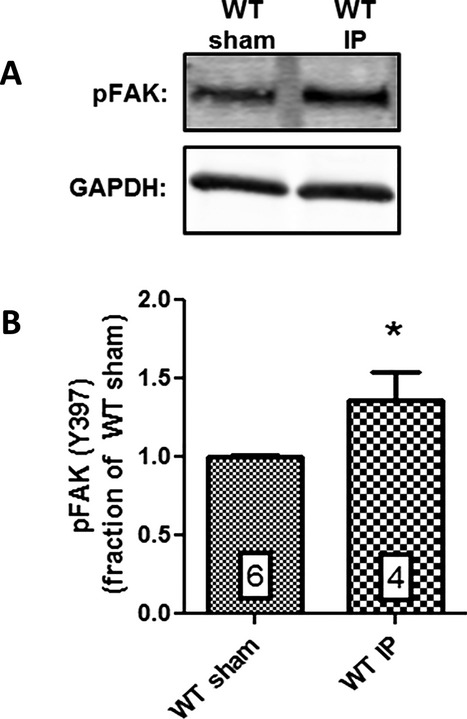
IP induces FAK activation in WT mice. A, Representative Western blot showing pFAK (Y397) expression in lysates from hearts of WT mice subjected to either the IP or sham protocol. B, Normalized integrated intensity data for cardiac pFAK expression. pFAK expression in WT mice subjected to IP was increased by 36.3% compared to WT mice subjected to the sham procedure (P≤0.05; n=6 hearts [sham], n=4 hearts [IP]). Numbers within bars indicate sample size of each group. IP indicates ischemic preconditioning; pFAK, activated/phosphorylated focal adhesion kinase; WT, wild‐type.
Effect of FAK KO on Infarct Size
If cytoskeletal signaling is an important component of stress‐elicited survival signaling hypothesized to underlie the cardioprotective effect of IP, then FAK KO should greatly impair/eliminate IP‐elicited cardioprotection. To assess the role of FAK in IP‐induced cardioprotection, control and FAK KO mice were subjected to the standard infarct protocol of 40 minutes of LCA occlusion followed by 24 hours of reperfusion either in the presence or absence of IP. Control groups consisted of a WT group and a tamoxifen‐treated α‐MHC‐MerCreMer+/−/FAKWT/WT (hereafter referred to as experimental control) group. A total of 123 mice were entered into the infarct study. Forty‐two mice were excluded due to all‐cause mortality during some portion of the experimental protocol. Another 24 mice were excluded due to technical problems: 12 mice were excluded due to inadequate phthalo blue or tetrazolium staining, 6 mice were excluded due to technical failure associated with ventilation, and 6 mice were excluded due to trauma to coronary vessels (total mortality=34% [42 of 143]). Fifty‐seven mice successfully completed the surgical protocol without complications and were included in the final infarct size analysis.
The area‐at‐risk (AAR) was not significantly different between any of the groups (Figure 5A). Figure 5B shows that IP significantly reduced infarct size (expressed as % AAR) in WT mice by 54.7% (43.7% versus 19.8%; control versus IP; n=14 [control], n=8 [IP]; P≤0.001), results similar to other reports of the cardioprotective effect of IP in murine infarct models29 and validating our infarct model system. More importantly, IP also provided significant protection in experimental control animals (tamoxifen‐treated, α‐MHC‐MerCreMer+/−/FAKWT/WT), reducing infarct size by 60.9% (44.7% versus 17.5%; control versus IP; n=9 both groups; P≤0.001), comparable to the level of protection afforded by IP in WT mice. However, in FAK KO mice, IP failed to elicit a significant protective effect (44.3% versus 37.1%; FAK KO control versus FAK KO IP; n=9 [control], n=8 [IP]; P=NS). Moreover, infarct size in preconditioned FAK KO mice was not significantly different than infarct size in nonpreconditioned WT or experimental control mice, indicating that FAK KO abrogated IP‐elicited cardioprotective signaling. Infarct size in nonpreconditioned FAK KO mice was not statistically different from either nonpreconditioned WT or experimental control mice (44.3% versus 43.7% versus 44.7%, KO versus WT versus Exp Ctrl, P=NS). Figure 5C shows representative midventricular cross‐sections of IP and non‐IP hearts from control and FAK KO mice.
Figure 5.
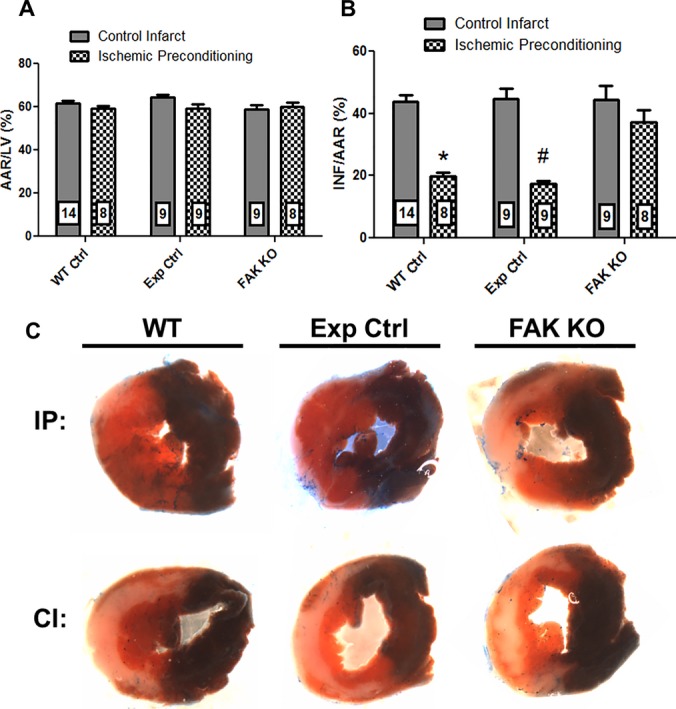
FAK KO abrogates IP‐elicited cardioprotection. A, Bar graph of ratio of myocardial area at risk (AAR) to left ventricular area (LV). B, Bar graph of ratio of infarcted myocardium (INF) to AAR. Numbers within bars indicate sample size of each group. *P≤0.001 vs WT Ctrl IP; #P≤0.001 vs Exp Ctrl IP. C, Representative stained midventricular cross‐sections from hearts of preconditioned (IP) and nonpreconditioned (CI) WT, Exp Ctrl, and FAK KO mice subjected to a 40‐minute episode of ischemia. Nonischemic tissue is stained blue, ischemic but viable tissue is stained red, and infarcted myocardium is white. CI indicates nonpreconditioned control infarct; Exp Ctrl, experimental control (Cre‐positive, floxed FAK‐negative mouse administered tamoxifen); FAK, focal adhesion kinase; IP, ischemic preconditioning; KO, knock out; WT, wild‐type.
FAK KO Prevents IP‐induced PI3K/Akt Signaling
Our previous studies demonstrated that IP and heat stress activate both FAK and Akt7 and that interference with FAK activity prevents heat stress‐elicited Akt activation, indicating that Akt becomes activated downstream of FAK in the hypothesized cytoskeletal‐based survival pathway.11 However, it remains unknown whether the IP‐stimulated activation of Akt is dependent on upstream FAK activity in an in vivo model of IR. If FAK is indeed an integral member of IP‐induced cardioprotective signaling, then reduction of myocyte FAK protein expression (and therefore FAK signaling) should result in a reduction in PI3K/Akt activation and signaling.
To answer this question, FAK KO and WT mice were subjected to either the IP protocol or a sham experimental protocol, and ventricular lysates were harvested and analyzed for expression of tyrosine phosphorylated PI3K p85 as a surrogate for PI3K activation30–31 or phosphorylation of serine residue 473 as a surrogate for Akt activation.32 In WT animals the IP protocol elicited a 67.2% increase in pPI3K p85 expression (P≤0.01; n=5 both groups) and an 88.8% increase in AktpSer473 expression (P≤0.001; n=4 both groups) indicating that IP induced strong PI3K and Akt activation (Figure 6). However, FAK KO mice subjected to IP did not exhibit a significant increase in either pPI3K p85 or AktpSer473 expression, but rather exhibited expression levels comparable to both WT and FAK KO mice subjected to the sham protocol (Figure 6).
Figure 6.
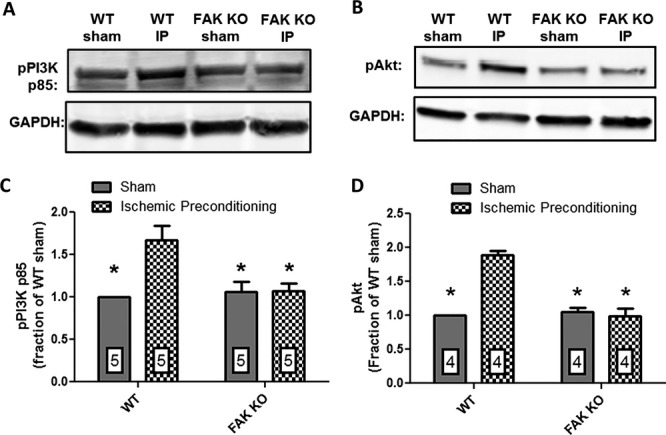
FAK KO abrogates IP‐induced PI3K/Akt signaling. Representative Western blots showing (A) pPI3K p85; (B) pAkt expression in lysates from hearts of WT and FAK KO mice subjected to either the IP protocol or a sham procedure; (C) normalized integrated density data for cardiac pPI3K p85 expression; and (D) pAkt expression. In WT mice IP significantly enhanced pPI3K p85 expression by 67.2% (P≤0.01; n=5 hearts, both groups) and pAkt expression by 88.8% (P≤0.001; n=4 hearts, both groups) versus WT mice subjected to the sham experimental protocol. pPI3K p85 and pAkt expression in FAK KO mice subjected to the IP protocol was not significantly different from their expression in either WT or FAK KO sham hearts. FAK indicates focal adhesion kinase; IP, ischemic preconditioning; KO, knock out; PI3K, phosphatidylinositol‐3‐kinase; WT, wild‐type.
Discussion
Our laboratory's previous findings supported an important role of FAK in stress‐elicited cardioprotective signaling, showing that both IP and heat stress activate FAK7 and interference with FAK activity compromises cardiac myocyte survival in response to simulated IR.4 The current study resulted in 3 important findings that confirm and extend our previous results. First, the results demonstrate for the first time that selective deletion of the essential membrane‐based member of cytoskeletal signaling, FAK, abrogates the protective effect of IP in a well‐established in vivo murine model of myocardial infarction. Second, the results further support a key role for cytoskeletal‐based signaling in mediating a general pathway of IP‐elicited cardioprotection by demonstrating that FAK KO prevents the IP‐induced activation of both PI3K and Akt, well‐established mediators of IP.8,21 And third, the results demonstrate that the α‐MHC‐MerCreMer system may be used to induce the KO of FAK in the adult heart while avoiding the dilated cardiomyopathy previously reported and described in the tamoxifen‐inducible α‐MHC‐MerCreMer system.28
Hypothesized Mechanisms of IP‐induced Cardioprotective Signaling
Most current hypotheses of the mechanism of IP have the protective signaling induced by or through the release of endogenous autocoids which subsequently bind to G‐protein coupled receptors (GPCRs), leading to downstream activation of survival signaling kinases.33 In these schemes, the release of autocoids, such as adenosine and bradykinin, and the additive effects of binding to their respective receptors is thought to serve as the trigger for IP‐elicited protective signaling.33 This concept is supported by studies that have shown that agonists of adenosine, bradykinin, and other GPCRs may mimic the protective effect of IP.34–35 The ability of multiple triggers to mimic the protective effect of IP led to the conclusion that the respective signaling pathways converge downstream at a common point. Furthermore, it has been demonstrated that blockade of a single trigger eliminates the protective signaling elicited by a single cycle of IP, but cannot eliminate the more robust protective signaling stimulated by multiple cycles of IP,35–36 further emphasizing the importance of multiple triggers in the cardioprotective pathway. Protein kinase C (PKC) has been proposed as a point of convergence, as the protective effect of IP and the pharmacological mimicking of IP can be inhibited by PKC inhibitors.33 However, tyrosine kinases have also been hypothesized to play in important role through demonstration that while neither PKC nor tyrosine kinase inhibition alone prevented the protective effect of IP, the inhibition of both together was able to eliminate the protective effect of IP.37 The binding of autocoids to GPCRs is thought to lead to protection from ischemic cell death through the downstream activation of survival kinases, notably ERK1/2, PKC, PI3K, and Akt.8 Akt is thought to help protect against lethal IR injury by delaying/inhibiting mitochondrial permeability pore transition through phosphorylation and inactivation of glycogen synthase kinase‐3β (GSK‐3β) and by reducing apoptosis through inactivation of proapoptotic molecules such as BAD.8,38
Our hypothesis is that IP stimulates a cytoskeletal signaling cascade that begins with activation of the proximal cytoskeletal signaling complex at the sarcolemmal membrane and leads to the downstream activation of survival kinases, most notably Akt. Within this cytoskeletal signaling cascade, FAK is an essential proximal mediator, as numerous studies have shown that interference with FAK activity significantly impairs the cytoskeletal signaling cascade.39–41 Engagement of integrins enhances targeting of FAK into costameres and focal adhesions where FAK becomes auto‐phosphorylated at tyrosine 397 and thereby activated.13 Activated/phosphorylated FAK may subsequently interact with SH2 domain‐containing signaling partners, including PI3K.17,42 PI3K‐FAK interaction may subsequently stimulate the activation of PI3K, ultimately leading to the downstream activation of the survival kinase Akt.
Mechanotransduction, Cytoskeletal Signaling, and Ischemic Preconditioning
The importance of mechanical forces in influencing FAK activity and downstream signaling within the heart is highlighted by a number of studies that have demonstrated the importance of FAK in mediating the hypertrophic response of cardiac myocytes stimulated by mechanical force. Eble et al43 demonstrated that overexpression of the FAK competitive inhibitor FRNK prevented endothelin‐induced myocyte hypertrophy in neonatal rat ventricular myocytes. Chu et al44 showed the importance of FAK serine‐910 (S910) phosphorylation in the hypertrophic response by demonstrating that interference with FAK S910 phosphorylation prevents the normal sarcomere assembly in response to endothelin. Interestingly, Chu et al44 also compared FAK S910 phosphorylation in failing and nonfailing human hearts and found that FAK S910 phosphorylation was substantially reduced in left ventricular tissue from failing hearts. Furthermore, DiMichele et al40 demonstrated that FAK deletion abolished hypertrophic remodeling induced by pressure overload with transverse aortic constriction. Thus, it is evident that while acute activation of FAK and cytoskeletal signaling mediates cell survival, chronic activation of the same pathway plays a prominent role in driving the hypertrophic response of ventricular myocytes. As our current and previous studies have demonstrated that FAK is an important mediator of IP‐elicited cardioprotection, it is possible that mechanical force may serve as an additional trigger for IP‐elicited cardioprotective signaling. Further studies will be needed to better discriminate between these possibilities.
Effect of FAK on Control Infarct Size
Hakim et al45 previously reported that ventricular myocyte‐specific deletion of FAK enhances baseline susceptibility to myocardial infarction. However, our current study did not find enhanced infarct size in non‐preconditioned hearts of FAK KO mice (see Figure 5B), suggesting that FAK signaling is not as important for maintaining cell viability under baseline conditions, but rather is induced in response to stimuli such as cell stress. Reasons for this apparent contradiction may be due to differences in the respective Cre‐lox systems used in each study and the associated timing of Cre‐mediated FAK deletion and/or differences in surgical methodology employed by each study. Whereas the α‐MHC‐MerCreMer system used in the current study allows for the induction of site‐specific recombination in adult mice, the Hakim study utilized a Cre recombinase under the control of the MLC2v gene, which is the earliest expressed ventricular‐restricted marker during mammalian development and, accordingly, Cre enzymes under the control of MLC2v may mediate genetic recombination as early as E8.75.46 Therefore, earlier reduction in FAK protein expression could possibly account for the differences in observations between the 2 studies. Furthermore, while the present study utilized a 40‐minute coronary occlusion as the index period of ischemia, the Hakim study utilized a 30‐minute coronary occlusion. This difference in surgical methodology may also account for the observed differences.
Dilated Cardiomyopathy Associated With α‐MHC‐Cre Expression
It has been previously reported that Cre expression in cardiac myocytes driven by the α‐MHC promoter is associated with the development of a severe dilated cardiomyopathy.47 Koitabashi et al28 also reported a dilated cardiomyopathy in tamoxifen‐inducible α‐MHC‐Cre systems. However, in that study the dilated cardiomyopathy resolved 3 weeks after the final dose of tamoxifen. During our initial assessments of tamoxifen dosing regimens, we did observe occasional acute cardiac failure following administration of high doses of tamoxifen to mice containing the α‐MHC‐MerCreMer allele. However, by modifying the tamoxifen dosing regimen, we were able to achieve significant reductions in FAK expression while circumventing significant effects on baseline cardiac function (see Table and Figure 3).
Summary and Conclusions
The current study describes a tissue‐specific, inducible model of FAK KO in adult mice that results in normal gross and histologic appearance and function at baseline. More importantly, the current study demonstrates for the first time in a well‐characterized in vivo infarction model that loss of FAK both inhibits the IP‐induced stimulation of PI3K/Akt signaling as well as abrogates IP‐elicited cardioprotection. These findings further support our hypothesis that myocardial stress leads to the activation of a cytoskeletal‐based survival signaling pathway that may play a prominent role in protecting the myocardium from ischemia/reperfusion injury.
Sources of Funding
.This work was supported by National Institutes of Health grant RO1 HL‐084405 to Dr Vander Heide.
Disclosures
None.
Acknowledgments
We thank Dr Jessica Bradley, Venkat Subramaniam, and Elizabeth McIlwain for their assistance with echocardiography.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013; 127:e6-e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heron M. Deaths: leading causes for 2008. Natl Vital Stat Rep. 2012; 60:1-94 [PubMed] [Google Scholar]
- 3.Jones RN, Reimer KA, Hill ML, Jennings RB. Effect of hypothermia on changes in high‐energy phosphate production and utilization in total ischemia. J Mol Cell Cardiol. 1982; 14suppl 3:123-130 [DOI] [PubMed] [Google Scholar]
- 4.Wei H, Campbell W, Vander Heide RS. Heat shock‐induced cardioprotection activates cytoskeletal‐based cell survival pathways. Am J Physiol Heart Circ Physiol. 2006; 291:H638-H647 [DOI] [PubMed] [Google Scholar]
- 5.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986; 74:1124-1136 [DOI] [PubMed] [Google Scholar]
- 6.Vander Heide R. Clinically useful cardioprotection: ischemic preconditioning then and now. J Cardiovasc Pharmacol Ther. 2011; 16:251-254 [DOI] [PubMed] [Google Scholar]
- 7.Wei H, Vander Heide RS. Ischemic preconditioning and heat shock activate Akt via a focal adhesion kinase‐mediated pathway in Langendorff‐perfused adult rat hearts. Am J Physiol Heart Circ Physiol. 2010; 298:H152-H157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia‐reperfusion injury. Physiol Rev. 2008; 88:581-609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jennings RB, Sebbag L, Schwartz LM, Crago MS, Reimer KA. Metabolism of preconditioned myocardium: effect of loss and reinstatement of cardioprotection. J Mol Cell Cardiol. 2001; 33:1571-1588 [DOI] [PubMed] [Google Scholar]
- 10.Hausenloy DJ, Yellon DM, Mani‐Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004; 287:H841-H849 [DOI] [PubMed] [Google Scholar]
- 11.Wei H, Vander Heide RS. Heat stress activates Akt via focal adhesion kinase‐mediated pathway in neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008; 295:H561-H568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am J Physiol Heart Circ Physiol. 2005; 289:H2291-H2301 [DOI] [PubMed] [Google Scholar]
- 13.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003; 116:1409-1416 [DOI] [PubMed] [Google Scholar]
- 14.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004; 1692:103-119 [DOI] [PubMed] [Google Scholar]
- 15.Torsoni AS, Marin TM, Velloso LA, Franchini KG. RhoA/ROCK signaling is critical to FAK activation by cyclic stretch in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2005; 289:H1488-H1496 [DOI] [PubMed] [Google Scholar]
- 16.Schaller MD. Paxillin: a focal adhesion‐associated adaptor protein. Oncogene. 2001; 20:6459-6472 [DOI] [PubMed] [Google Scholar]
- 17.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta. 2001; 1540:1-21 [DOI] [PubMed] [Google Scholar]
- 18.van de Water B, Nagelkerke JF, Stevens JL. Dephosphorylation of focal adhesion kinase (FAK) and loss of focal contacts precede caspase‐mediated cleavage of FAK during apoptosis in renal epithelial cells. J Biol Chem. 1999; 274:13328-13337 [DOI] [PubMed] [Google Scholar]
- 19.Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM. GFP‐FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ Res. 2002; 90:1282-1289 [DOI] [PubMed] [Google Scholar]
- 20.Pfister R, Acksteiner C, Baumgarth J, Burst V, Geissler HJ, Margulies KB, Houser S, Bloch W, Flesch M. Loss of beta1D‐integrin function in human ischemic cardiomyopathy. Basic Res Cardiol. 2007; 102:257-264 [DOI] [PubMed] [Google Scholar]
- 21.Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, Malhowski A, Cheng Z, Khan M, McGregor M. Myocardial Akt: the omnipresent nexus. Physiol Rev. 2011; 91:1023-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue‐specific gene manipulations in the adult and embryonic heart using a tamoxifen‐inducible Cre protein. Circ Res. 2001; 89:20-25 [DOI] [PubMed] [Google Scholar]
- 23.Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006; 172:151-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005; 169:941-952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007; 357:271-296 [DOI] [PubMed] [Google Scholar]
- 26.Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S, Izumo S. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol Genomics. 2004; 16:349-360 [DOI] [PubMed] [Google Scholar]
- 27.Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM. Myocardial ischemia and reperfusion: a murine model. Am J Physiol. 1995; 269:H2147-H2154 [DOI] [PubMed] [Google Scholar]
- 28.Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA. Avoidance of transient cardiomyopathy in cardiomyocyte‐targeted tamoxifen‐induced mercremer gene deletion models. Circ Res. 2009; 105:12-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol. 1998; 275:H1375-H1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saward L, Zahradka P. Angiotensin II activates phosphatidylinositol 3‐kinase in vascular smooth muscle cells. Circ Res. 1997; 81:249-257 [DOI] [PubMed] [Google Scholar]
- 31.Cuevas BD, Lu Y, Mao M, Zhang J, LaPushin R, Siminovitch K, Mills GB. Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3‐kinase. J Biol Chem. 2001; 276:27455-27461 [DOI] [PubMed] [Google Scholar]
- 32.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF‐1. EMBO J. 1996; 15:6541-6551 [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010; 24:225-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991; 84:350-356 [DOI] [PubMed] [Google Scholar]
- 35.Goto M, Liu Y, Yang XM, Ardell JL, Cohen MV, Downey JM. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ Res. 1995; 77:611-621 [DOI] [PubMed] [Google Scholar]
- 36.Miki T, Cohen MV, Downey JM. Opioid receptor contributes to ischemic preconditioning through protein kinase C activation in rabbits. Mol Cell Biochem. 1998; 186:3-12 [PubMed] [Google Scholar]
- 37.Vahlhaus C, Schulz R, Post H, Rose J, Heusch G. Prevention of ischemic preconditioning only by combined inhibition of protein kinase C and protein tyrosine kinase in pigs. J Mol Cell Cardiol. 1998; 30:197-209 [DOI] [PubMed] [Google Scholar]
- 38.Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005; 118:5675-5678 [DOI] [PubMed] [Google Scholar]
- 39.Torsoni AS, Constancio SS, Nadruz W, Jr, Hanks SK, Franchini KG. Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circ Res. 2003; 93:140-147 [DOI] [PubMed] [Google Scholar]
- 40.DiMichele LA, Doherty JT, Rojas M, Beggs HE, Reichardt LF, Mack CP, Taylor JM. Myocyte‐restricted focal adhesion kinase deletion attenuates pressure overload‐induced hypertrophy. Circ Res. 2006; 99:636-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young SR, Gerard‐O'Riley R, Kim JB, Pavalko FM. Focal adhesion kinase is important for fluid shear stress‐induced mechanotransduction in osteoblasts. J Bone Miner Res. 2009; 24:411-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen HC, Guan JL. Stimulation of phosphatidylinositol 3′‐kinase association with focal adhesion kinase by platelet‐derived growth factor. J Biol Chem. 1994; 269:31229-31233 [PubMed] [Google Scholar]
- 43.Eble DM, Strait JB, Govindarajan G, Lou J, Byron KL, Samarel AM. Endothelin‐induced cardiac myocyte hypertrophy: role for focal adhesion kinase. Am J Physiol Heart Circ Physiol. 2000; 278:H1695-H1707 [DOI] [PubMed] [Google Scholar]
- 44.Chu M, Iyengar R, Koshman YE, Kim T, Russell B, Martin JL, Heroux AL, Robia SL, Samarel AM. Serine‐910 phosphorylation of focal adhesion kinase is critical for sarcomere reorganization in cardiomyocyte hypertrophy. Cardiovasc Res. 2011; 92:409-419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hakim ZS, DiMichele LA, Rojas M, Meredith D, Mack CP, Taylor JM. FAK regulates cardiomyocyte survival following ischemia/reperfusion. J Mol Cell Cardiol. 2009; 46:241-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Kubalak SW, Chien KR. Ventricular muscle‐restricted targeting of the RXRalpha gene reveals a non‐cell‐autonomous requirement in cardiac chamber morphogenesis. Development. 1998; 125:1943-1949 [DOI] [PubMed] [Google Scholar]
- 47.Buerger A, Rozhitskaya O, Sherwood MC, Dorfman AL, Bisping E, Abel ED, Pu WT, Izumo S, Jay PY. Dilated cardiomyopathy resulting from high‐level myocardial expression of Cre‐recombinase. J Card Fail. 2006; 12:392-398 [DOI] [PubMed] [Google Scholar]