

Abstract
Background
Ischemia and reperfusion (IR) injury remains a major cause of morbidity and mortality and multiple molecular and cellular pathways have been implicated in this injury. We determined whether acute inhibition of excessive mitochondrial fission at the onset of reperfusion improves mitochondrial dysfunction and cardiac contractility postmyocardial infarction in rats.
Methods and Results
We used a selective inhibitor of the fission machinery, P110, which we have recently designed. P110 treatment inhibited the interaction of fission proteins Fis1/Drp1, decreased mitochondrial fission, and improved bioenergetics in three different rat models of IR, including primary cardiomyocytes, ex vivo heart model, and an in vivo myocardial infarction model. Drp1 transiently bound to the mitochondria following IR injury and P110 treatment blocked this Drp1 mitochondrial association. Compared with control treatment, P110 (1 μmol/L) decreased infarct size by 28±2% and increased adenosine triphosphate levels by 70+1% after IR relative to control IR in the ex vivo model. Intraperitoneal injection of P110 (0.5 mg/kg) at the onset of reperfusion in an in vivo model resulted in improved mitochondrial oxygen consumption by 68% when measured 3 weeks after ischemic injury, improved cardiac fractional shortening by 35%, reduced mitochondrial H2O2 uncoupling state by 70%, and improved overall mitochondrial functions.
Conclusions
Together, we show that excessive mitochondrial fission at reperfusion contributes to long‐term cardiac dysfunction in rats and that acute inhibition of excessive mitochondrial fission at the onset of reperfusion is sufficient to result in long‐term benefits as evidenced by inhibiting cardiac dysfunction 3 weeks after acute myocardial infarction.
Keywords: cardiac myocytes, Drp1, heart, mitochondria, protein‐protein interaction inhibitor
Introduction
The first observation that cardiac mitochondria change their size and number through fission and fusion was made more than 4 decades ago,1 and the key role of mitochondrial dysfunction in ischemia and reperfusion (IR) injury and cardiomyopathy has been subsequently recognized.2–4 Mitochondrial fission is required to eliminate damaged or aged mitochondria via autophagy.5 However, excessive mitochondrial fission can contribute to ischemia/reperfusion injury and to heart failure.6–7 Therefore, inhibition of excessive mitochondrial fission might be crucial to maintain proper mitochondrial function and cardiac function.
In yeast, fission is controlled by the translocation of cytosolic Dnm1 (yeast dynamin‐related protein 1; drp1) and its interaction with the outer mitochondrial protein, fission protein 1 (Fis1).8–9 In addition to Fis1, other adaptor proteins, mitochondrial fission factor (Mff)10 and mitochondrial elongation factor 1(MIEF1 [MiD51])11–12 at the surface of mitochondria in mammals, bind dynamin related protein 1 (Drp1) to promote fission or fusion respectively. A study in HL‐1 cells showed that inhibiting mitochondrial fission reduces IR injury. Two hours of ischemia induced mitochondrial fragmentation that persisted during the 5 hours of reperfusion.13 Fragmentation of mitochondria was inhibited by pretreatment with a pharmacological inhibitor of Drp1, mitochondrial division inhibitor (mdivi‐1)14 (given prior to the ischemic event), suggesting that this excessive mitochondrial fission and fragmentation is dependent on Drp1.15 Mitochondrial fragmentation was also observed in mouse hearts after coronary artery occlusion followed by reperfusion. Under these conditions, treatment with mdivi‐1 before the ischemic period reduced infarct size in vivo when measured 2 hours after myocardial infarction (MI).15 These reports provide evidence that inhibiting the mitochondrial fission machinery prior to ischemic insult protects the heart against ischemia and reperfusion injury.
Although myocardial ischemia per se causes tissue damage due to increased acidity, sodium, and calcium overload, and to adenosine triphosphate (ATP) depletion, the return of blood flow, termed reperfusion, aggravates these injurious effects due to increased reactive oxygen species (ROS) levels and uncoupling of mitochondrial oxidative phosphorylation.16–17 Here, we set out to determine whether inhibition of mitochondrial fragmentation can produce benefit when given after MI and the consequence of acute inhibition of mitochondrial fragmentation on long‐term cardiac functions.
We made use of Drp1 inhibitor, P110, a peptide that we recently rationally designed to selectively inhibit Fis1/Drp1 interaction.18 This 7‐amino‐acid peptide represents a homologous sequence between Fis1 and Drp1. We demonstrated that P110 does not affect Drp1 interaction with the two other mitochondrial adaptors of Drp1, Mff, and MIEF1(MiD51), or with the mitochondrial fusion proteins, Mfn1 or Mfn2.18 In a model of Parkinson's disease in culture, we also showed that inhibiting Drp1/Fis1 interaction reduced excessive mitochondrial fragmentation and ROS production as well as improved mitochondrial membrane potential and mitochondrial integrity.18 In the current study, we used P110 peptide to determine the role of Drp1/Fis1 interaction in reperfusion‐induced injury using primary rat cultured cardiac myocytes, an ex vivo rat heart model, and an in vivo rat model of myocardial infarction. We determined also the long‐term consequences of acute inhibition of Drp1 after the ischemic event on development of post‐MI cardiac dysfunction.
Methods
Peptide Synthesis
Peptides were synthesized using microwave chemistry on a Liberty Microwave Peptide Synthesizer (CEM Corporation) as previously described.18 Briefly, peptides were synthesized as one polypeptide with TAT47‐57 carrier in the following order: N‐terminus–TAT–spacer (Gly‐Gly)–cargo–C‐terminus.
Cell Culture
Primary cultures of cardiac myocytes were prepared from the heart of 1‐day‐old rats by gentle digestion at 37°C using a cell isolation kit protocol (Cellutron). Cells were cultured in the presence of 0.1 mmol/L bromodeoxyuridine on primary tissue culture dishes (BD Falcon) or on laminin‐coated slides in Dulbeco's modified Eagle's medium with 10% fetal bovine serum for 4 days. The cells were washed with PBS and incubated at 37°C for 2 hours in ischemia buffer (NaCl 125 mmol/L, KCl 8 mmol/L, KH2PO4 1.2 mmol/L, MgSO4 1.25 mmol/L, CaCl2 1.2 mmol/L, NaHCO3 6.25 mmol/L, sodium lactate 5 mmol/L, Hepes 20 mmol/L, pH 6.6) in a GasPak pouch (Becton Dickinson). Then the cells were reexposed to oxygen and the buffer was replaced by reoxygenation Krebs‐Henseleit buffer (NaCl 110 mmol/L, KCl 4.7 mmol/L, KH2PO4 1.2 mmol/L, MgSO4 1.25 mmol/L, CaCl2 1.2 mmol/L, NaHCO3 25 mmol/L, glucose 15 mmol/L, Hepes 20 mmol/L, pH 7.4) for the indicated time. Treatments with control peptide (TAT47‐57) or P110 commenced 30 minutes before ischemia and continued during ischemia and reoxygenation (1 μmol/L).
Isolation of Mitochondrial‐enriched Fraction and Lysate Preparation
Cardiac myocyte cells were washed with cold PBS and incubated on ice in lysis buffer (300 mmol/L sucrose, 10 mmol/L Hepes, 2 mmol/L EDTA, pH 7.2) for 30 minutes in the presence of protease inhibitor cocktail and phosphatase inhibitor cocktail. Cells were scraped and then disrupted 10 times by repeated aspiration through a 27‐gauge needle. The homogenates were spun at 800g for 5 minutes at 4°C and the resulting supernatants were spun at 10 000g for 20 minutes at 4°C. The pellets were washed with lysis buffer and spun at 10 000g again for 20 minutes at 4°C. The final pellets were suspended in lysis buffer containing 1% Triton X‐100 and were noted as mitochondrial‐rich lysate fractions. The mitochondrial membrane protein voltage‐dependent anion channel (VDAC) was used as marker and loading control for mitochondrial fractions and enolase was used as a marker and loading control for total fractions.
Co‐immunoprecipitation
Coimmunoprecipitation experiments were performed as already described.18–19 Briefly, proteins from cardiac myocytes were cross‐linked in 1% paraformaldehyde followed by washing in PBS containing 100 mmol/L glycine. The cells were then lysed by sonication in PBS with 1% Triton X‐100 and incubated with the respective antibodies and protein A/G agarose. The immunoprecipitates were loaded on a SDS‐PAGE and probed with Drp1 antibody, as described above.
Immunocytochemistry
Cells cultured on chamber slides were treated with respective peptides as above and subjected to ischemia and reoxygenation (IRO). At the end of the incubation the cells were washed with cold PBS, fixed in 4% formaldehyde then blocked for 2 hours with 2% normal goat serum in PBS containing 0.1% Triton‐X (blocking buffer). The cells were then incubated overnight at 4°C with antibodies against Tom‐20 (Santa Cruz Biotechnology, 1:500). Cells were washed with blocking buffer and incubated for 2 hours with Alexa546‐labeled goat antirabbit antibody (1:500, Invitrogen). Slides were mounted and imaged by Leica SP5 multiphoton/confocal Laser Scanning Microscope using a ×63 oil immersion objective. To determine superoxide production in cultures, cells plated on laminin‐coated 96 well plates were incubated after 2 hours of ischemia and 3 hours of reoxygenation with 5 μmol/L CellRox for 45 minutes or MitoSOX red mitochondrial superoxide indicator (Invitrogen) for 10 minutes at 37°C. Staining was carried out in presence of DAPI to enable counting cell numbers. The level of fluorescence staining was analyzed using a fluorescent plate reader (High‐Throughput Bioscience Center, Stanford) at 640 nm excitation and 665 nm emission or 510 nm excitation and 590 nm emission, for CellRox and MitoSOX, respectively and normalized to DAPI staining level.
Ex vivo Model of Cardiac Ischemia‐reperfusion
All protocols were approved by the Stanford University Institutional Animal Care and Use Committee. Male Wistar rats (270–300 g) were heparinized (1000 U/kg IP) and then anesthetized with Beuthanasia‐D, (pentobarbital sodium 6240 mg/kg and phenytoin sodium 800 mg/kg IP) (Schering‐Plough Animal Health Crop). Then, the hearts were perfused via the aorta at 10 mL/min with oxygenated Krebs–Henseleit buffer containing NaCl (120 mmol/L), KCl (5.8 mmol/L), NaHCO3 (25 mmol/L), NaH2PO4 (1.2 mmol/L), MgSO4 (1.2 mmol/L), CaCl2 (1 mmol/L), and dextrose (10 mmol/L) at pH 7.4 in a Langendorff coronary perfusion system at 37°C. The hearts were subjected to 30 minutes global, no‐flow ischemia followed by 90 minutes of reperfusion. TAT47‐57 (control peptide) and P110 peptide (1 μmol/L) were perfused during the entire equilibration and reperfusion period (Figure 3A). Normoxic control hearts were subjected to 120 minutes perfusion in the absence of ischemia. At the end of the reperfusion period, hearts were sliced into 1‐mm‐thick transverse sections and incubated in triphenyltetrazolium chloride solution (TTC, 1% in phosphate buffer, pH 7.4) at 37 °C for 15 minutes then fixed in 10% formalin. Infarct size was expressed as a percentage of the risk zone (equivalent to total LV muscle mass) as we previously described.20 We used 6 animals per group (control and treated‐group).
Figure 3.

Localization of Drp1 at the mitochondria and mitochondrial function on IR injury in an ex vivo Langendorff model. A, Isolated heart protocol of IR. B, Hearts subject to IR injury were treated with TAT or P110 at 1 μmol/L during equilibration period and for 20 minutes at reperfusion. P110 reduces mitochondrial fragmentation after IR injury. Mitochondrial morphology was analyzed by electron microscopy of the indicated groups. Mitochondrial size and arrangement is shown at ×4000 magnification of respective heart sections (bar=0.5 μm). C, Lower magnification (×800) of EM sections showed mitochondria arranged along the myofibrils after P110 treatment (bar=2 μm). D, Flow cytometry analysis (FACS) of isolated cardiac mitochondria size (forward scatter; FCS) in respective groups after IR. The mean of each group is shown in histogram, on the right (*P<0.05 vs normoxia, **P<0.05 vs IR). E, Translocation of Drp1 to the mitochondria upon IR is blocked by P110 peptide compared to normoxia and control (n=6). Quantitative data of the Western blot demonstrating Drp1 translocation is provided in histogram, on the right (*P<0.05 vs normoxia, **P<0.05 vs IR). Drp1 indicates dynamin related protein 1; EM, electron microscopy; IR, ischemia and reperfusion.
Measurement of Cardiac ATP Levels
ATP levels were measured in total extract samples from heart after ex vivo IR injury using ATP determination kit protocol (Invitrogen). Briefly, 100 to 150 mg cross sections of heart tissue were weighed then lysed in 1% TCA after the weight of each section was determined for further normalization. The tissue debris was spun down and the supernatants were brought to pH 7. Ten μL of each lysate was used in the assay in a total volume of 200 μL reaction buffer. The level of ATP was measured using a luminometer plate reader and the amount was normalized to the wet tissue weight (nmol/mg).
Electron Microscopy of Heart Ex vivo
Thin sections of respective heart tissue after IR were fixed in 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer, pH=7.4. The fixed material was sectioned at the Stanford Electron Microscopy Facility. Sections were taken between 75 and 80 nm, picked up on formvar/Carbon coated 75 mesh Ni grids and stained for 20 seconds in 1:1 saturated uracetate (≈7.7%) in acetone followed by staining in 0.2% lead citrate for 3 to 4 minutes for contrast. Mitochondrial samples were observed in a JEOL 1230 transmission electron microscope at 80 kV and photos were taken using a Gatan Multiscan 791 digital camera.
In vivo Rat Acute Myocardial Infarction Model
Acute myocardial infarction (AMI) was induced by ligation of the left anterior descending (LAD) coronary artery for 30 minutes, as previously described.20–22 Male Wistar rats were anesthetized with ketamine (50 mg/kg IP) and xylazine (10 mg/kg IP), endotracheally intubated, and mechanically ventilated with room air (respiratory rate of 80 breaths/min and tidal volume of 2.5 mL). Body temperature was maintained at 37°C using a rectal probe linked to a thermocoupled thermometer and an appropriate heating blanket. The heart was exposed by a left thoracotomy between the fourth and fifth ribs. After a 10‐minute period of stabilization, the LAD coronary artery was ligated close to its origin from the aortic root. The normoxia control animals (sham) were exposed to the same procedure with no ligation. The free ends of the ligature were used to form a noose around a syringe plunger which was placed flat on the myocardium. Coronary occlusion was achieved by tightening the noose around the plunger for 30 minutes. Occlusion was determined by observation of immediate pallor of the LV free wall and reflow was achieved by release of the ligature just after injecting an intraperitoneal injection (IP) 0.5 mg/kg of the respective peptides. A 4‐0 vicryl absorbance suture was used to close the chest and a nylon suture to stitch the skin. Buprenorphine (0.05 mg/kg) was given subcutaneously every 8 hours for 2 days postoperatively. Fractional shortening was determined in 6 to 7 rats per group at 3 days and 3 weeks after MI by M‐mode echocardiography.
Echocardiographic Measurements
Noninvasive cardiac function was assessed by two‐dimensional (2D)–guided M‐mode echocardiography in 1% vaporized isoflurane‐anesthetized rats after the experimental period as described previously.22 Transthoracic echocardiography was performed using an Acuson Sequoia model 512 echocardiographer equipped with a 14‐MHz linear transducer. LV systolic function was estimated by fractional shortening (FS) as follows: FS (%)=[(LVEDD–LVESD)/LVEDD]×100, where LVEDD is the LV end‐diastolic diameter, and LVESD is the LV end‐systolic diameter. For the data shown in Table, 2D parasternal short‐axis views of the LV were obtained at the level of the papillary muscles. M‐mode tracings were obtained from the short‐axis views of the LV at or just below the tip of the mitral‐valve leaflets and at the level of the aortic valve and left atrium. All LV structures were manually measured by the same blinded observer, using the leading‐edge method of the American Society of Echocardiography that has been validated for the infarcted rat model. The measurements obtained were the mean of at least 5 cardiac cycles on the M‐mode tracings. All echocardiographic evaluations were performed by the same examiner who was blinded to the experimental conditions. The number of animals used for the analyses is mentioned at the top of each respective group in the table.
Table 1.
Analysis of In vivo Cardiac Function by Echocardiography
| Parameter | 3 days | 21 days | |||
|---|---|---|---|---|---|
| Norcon | IRcon | IRP110 | IRcon | IRP110 | |
| n=6 | n=7 | n=6 | n=6 | n=6 | |
| EF, % | 86±4 | 63±3* | 73±3*,* | 58±3* | 71±4*,* |
| LVEdD, mm | 6.6±0.2 | 6.8±0.3 | 6.6±0.4 | 7.4±0.3 | 6.7±0.5 |
| LVEsD, mm | 3.3±0.1 | 4.8±0.2* | 4.2±0.3* | 5.4±0.3* | 4.3±0.3*,* |
| dSEPth, mm | 0.9±0.1 | 0.9±0.1 | 0.9±0.1 | 1.0±0.1 | 1.0±0.1 |
| sSEPth, mm | 1.6±0.2 | 1.3±0.2 | 1.5±0.2 | 1.6±0.1 | 1.6±0.1 |
| dPWth, mm | 1.0±0.1 | 1.1±0.1 | 1.1±0.1 | 1.2±0.1* | 1.2±0.1 |
| sPWth, mm | 2.0±0.1 | 1.9±0.2 | 1.7±0.2 | 1.8±0.1 | 1.8±0.1 |
| HR, bpm | 327±26 | 278.1±16 | 296±15 | 278±10 | 290±12 |
dPWth, diastolic posterior wall thickness; dSEPth, diastolic septum thickness; EF, ejection fraction; HR, heart rate; IRcon, ischemia/reperfusion control; IRP110, ischemia/reperfusion treated with P110 peptide; LVEdD, left ventricle end diastolic dimension; LVEsD, left ventricle end systolic dimension; n, at top of table, indicates the number of animals used for the echocardiography study in the respective group; Norcon, normoxia (control); NorP110, Normoxia (treated with P110); sPWth, systolic posterior wall thickness; sSEPth, systolic septum thickness.
P<0.05 vs Norcon.
P<0.05 vs IRcon.
Mitochondrial Isolation
Heart mitochondria were isolated as described elsewhere23 from 6 rats per group. Briefly, cardiac samples from a remote area were minced and homogenized in isolation buffer (300 mmol/L sucrose, 10 mmol/L Hepes, 2 mmol/L EGTA, pH 7.2, 4°C) containing 0.1 mg/mL of type I protease (bovine pancreas) to release mitochondria from within muscle fibers and later washed in the same buffer in the presence of 1 mg/mL bovine serum albumin. The suspension was homogenized in a 40‐mL tissue grinder and centrifuged at 950g for 5 minutes. The resulting supernatant was centrifuged at 9500g for 10 minutes. The mitochondrial pellet was washed, resuspended in isolation buffer and centrifuged again (9500g for 10 minutes). The mitochondrial pellet was washed and the final pellet was resuspended in a minimal volume of isolation buffer. The respective fractions were analyzed by Western blot using antibodies against Drp1 (BD), microtubule‐associated protein 1 light chain 3 (LC3‐II) (Cell Signaling), Jun kinase (JNK), and p‐JNK (Cell Signaling). The distinct cellular fractions were identified by their respective loading control; VDAC (MitoSciences), was used to identify mitochondria and enolase (Santa Cruz Biotechnology) for total fraction.
Analysis of Mitochondrial Size
Flow cytometric analysis of mitochondrial size was determined in isolated cardiac mitochondria.24 The mitochondria were stained with 400 nmol/L MitoTraker Red CMXRos (Invitrogen) for 15 minutes at 37°C then fixed in 4% formaldehyde for 15 minutes in warmed 5% sucrose in PBS to avoid fission25 before analyzing mitochondrial size. Mitochondria size was measured by recording forward scatter measurement using R‐Phycoerythrin (PE) channel on LSRII BD flow cytometer.
Mitochondrial O2 Consumption
Mitochondrial O2 consumption was monitored in a 0.125 mg mitochondrial protein/mL suspension in a buffer containing 125 mmol/L sucrose, 65 mmol/L KCl, 10 mol/L Hepes, 2 mmol/L inorganic phosphate, 2 mmol/L MgCl2, 100 μmol/L EGTA and 0.01% bovine serum albumin, pH 7.2 using a computer‐interfaced Clark‐type electrode (OROBOROS Oxygraph‐2k) operating with continuous stirring at 37°C.23 Succinate, malate and glutamate (2 mmol/L of each) were used as substrates and adenosine diphosphate (ADP) (1 mmol/L) was added to induce state‐3 respiratory rate. A subsequent addition of oligomycin (1 μg/mL) was used to determine state‐4 rate. RCR by dividing state‐3 by state‐4 oxygen consumption rates, which demonstrates the tightness of the coupling between mitochondrial respiration and phosphorylation was calculated.
Mitochondrial H2O2 Release
Mitochondrial H2O2 release was measured, as described elsewhere,23 in 0.125 mg protein/mL mitochondrial suspensions in buffer under the same conditions as oxygen consumption measurements. Amplex Red (25 μmol/L) oxidation was followed in the presence of 0.5 U/mL horseradish peroxidase and using succinate, malate, and glutamate (2 mmol/L of each) as substrates. Amplex Red is oxidized in the presence of extramitochondrial horseradish peroxidase bound to H2O2, generating resorufin, which can be detected using a fluorescence spectrophotometer. Excitation/emission wavelengths were 563/587 nm. Calibration was conducted by adding known amounts of H2O2 (A240=43.6 (mol L−1)−1 cm−1) to the experimental buffer.
Statistical Analysis
Data are presented as means±standard error of the mean (SEM). One‐way analysis of variance (ANOVA) was used to analyze the data shown in Figures 2A, 2B, 3D, 3E, 4B through 4F, and 5. Two‐way ANOVA was used to analyze data depicted in Table and Figures 2C and 6. Whenever significant F‐values were obtained, Tukey adjustment was used for multiple comparison purposes. Kruskal Wallis and Dunn's test were used to analyze data presented in Figures 1, 2D and 2E. Statistical significance was considered achieved when the value of P <0.05. For the in vitro studies, we ran 3 independent experiments performed in duplicate for each point.
Figure 1.

Mitochondrial Drp1 translocation in cardiac myocytes under ischemia‐reoxygenation injury is blocked by P110. A, Primary culture cardiomyocytes were subjected to 2 hour of ischemia followed by 2 to 60 minutes of reoxygenation. Western blot analysis of Drp1 in total and mitochondrial fractions was determined by anti‐Drp1 antibodies. VDAC was used as a loading control. Quantitation of the levels of Drp1 is provided in a histogram. B, Cells were treated with peptide TAT47‐57 and P110 (1 μmol/L) for 30 minutes prior 2 hours ischemia and during 30 minutes reoxygenation before analyzing Drp1 levels as above. P110 decreased Drp1 levels at the mitochondria. Enolase and VDAC were used as a loading controls and subcellular compartment controls. C, Cross‐linked proteins from total lysate cardiomyocytes after IRO treated with control or P110 peptide were immunoprecipitated using anti‐Fis1, anti‐Mff and anti‐MIEF1, respectively. Drp1 co‐immunoprecipitated was analyzed by Western blot analysis. Antibody control and 5% input is shown for respective proteins. The amounts of pulled‐down Drp1 are shown under the respective blots, normalized to the respective protein. Data are expressed as mean±SE of three independent experiments performed in duplicate for each group. (*P<0.05 vs normoxia, **P<0.05 vs IRO). Drp1 indicates dynamin related protein 1; Fis1, fission protein 1; IRO, ischemia and reoxygenation; VDAC, voltage‐dependent anion channel.
Figure 2.

Mitochondrial fragmentation and function in cardiac myocytes under IRO. A, Mitochondria of cardiac myocytes after 2 hours of ischemia followed by 2 hours of reoxygenation in the presence or absence of P110 are labeled with anti‐Tom20 antibody to determine the number of cells with mitochondria fragmentation. Bar scale=20 μm. Boxed area under each micrograph is enlarged to determine mitochondria fragmentation. B, Cytochrome c release was determined in the cytosolic fraction after IRO injury in the presence and absence of the peptide. Enolase and VDAC were used as cytosolic and mitochondrial loading control, respectively. C, P110 treatment decreased the number of TUNEL‐positive cells after IR. D and E, Cellular reactive oxygen species and mitochondria reactive oxygen species are measured in intact cells after IRO using a fluorescent plate reader. Data are expressed as mean±SE of three independent experiments performed in duplicate for each group. (*P<0.05 vs normoxia, **P<0.05 vs IRO). IR indicates ischemia and reperfusion; IRO, ischemia and reoxygenation; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Results
Translocation of Drp1 and Binding to Fis1 in Cardiac Myocytes is Inhibited by P110 Peptide
Cytosolic Drp1 associates with the mitochondrial adaptor protein, Fis1, to trigger Drp1 activation and further mitochondrial fission.26–28 We first determined whether simulated ischemia results in mitochondrial translocation and activation of Drp1. We used the previously described protocol for heart mitochondrial isolation21,29 and observed that 2 hours of ischemia followed by IRO for 2 to 60 minutes resulted in Drp1 translocation to the mitochondria of rat neonatal cardiomyocytes in culture. There was a 200±10% increase in the amount of mitochondrial Drp1 after 10 minutes of reoxygenation as compared with cells left in normoxic conditions (Figure 1A, middle and right panels). After 60 minutes of reperfusion, mitochondrial levels of Drp1 returned to basal levels, while the total cellular levels of Drp1 remained unchanged throughout the treatment (Figure 1A, left). Treatment with the Drp1/Fis1 protein/protein interaction inhibitor, P110 (1 μmol/L), 30 minutes before ischemia and during ischemia and reperfusion reduced IRO‐induced increase of mitochondrial Drp1 by 50±3% (Figure 1B). P110 does not change mitochondrial Drp1 levels under normoxic condition relative to untreated cells (0.23±0.02 versus 0.2±0.03, respectively).
A number of adaptor proteins bind mitochondrial Drp1.10–12 However, IRO increases Drp1 association only with Fis1 and P110 selectively inhibited only this interaction, not affecting the interaction of Drp1 with its other adaptor proteins, Mff or MIEF1, in cardiomyocytes (Figure 1C). Therefore, P110 is a selective inhibitor of Drp1 binding to Fis1.
IRO‐induced Mitochondrial Fragmentation, Cytochrome c Release and ROS Elevation are Inhibited by P110 Treatment
We next determined mitochondrial fragmentation following IRO in cultured rat cardiac myocytes. Less than 10% of the cells have fragmented mitochondria under normoxic conditions. Following IRO, 30±5% of the cells exhibited excessive mitochondrial fragmentation (dot‐like mitochondria), an effect that was inhibited by 58±4% in the presence of P110 (Figure 2A). IRO‐induced fission was associated with a 2‐fold increase in the release of mitochondrial cytochrome c to the cytosolic fraction, indicating a loss of mitochondrial integrity; this increase was blocked by treatment with P110 (Figure 2B). We determined apoptosis in cells subjected IRO by measuring the number of Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)‐positive cells. IRO caused an increase of 67±3% of TUNEL‐positive cells and P110 treatment decreased TUNEL‐positive cells by 38±2% (Figure 2C). Note that P110 treatment did not affect the number of TUNEL‐positive cells under normoxic condition. IRO also increased cellular and mitochondrial ROS production (measured by CellRox and Mitosox, respectively) 70±8% and 41±7%, and these were reduced to basal levels following P110 treatment (Figure 2D and 2E). These data demonstrated that inhibition of Drp1 association with the mitochondria by P110 greatly inhibited mitochondrial fragmentation and ROS production and maintained mitochondrial integrity.
P110 Treatment Reduced Infarct Size and Restored Mitochondrial Morphology and Function in an Ex vivo Model of MI
We next examined mitochondrial fragmentation in rat heart subjected to IR injury, using the Langendorff preparation30 (Figure 3A). Electron micrographs show well‐organized mitochondria along the sarcomeres (Figure 3B, left panel). After IR, the intersarcomeric mitochondria were smaller, and treatment with P110 (1 μmol/L), as described in Figure 3A, greatly restored mitochondrial morphology and organization along the contractile elements in these hearts (Figure 3B right versus middle panels); Figure 3C provides lower magnification images, demonstrating the structural benefits of P110 treatment at reperfusion.
To provide a better quantitation of mitochondrial fragmentation by IR and the effect of the treatment with the Drp1 inhibitor P110, we determined mitochondrial size using fluorescence‐activated cell sorting (FACS) analysis.24 After IR, there was a 38±3% reduction in mitochondrial size relative to mitochondria isolated from normoxic hearts, shown by the reduced forward scatter of the mitochondria (Figure 3D). Treatment with P110 at reperfusion resulted in normalization of mitochondrial size; the distribution of size of mitochondria isolated from normoxic heart and from hearts subjected to ischemia that were treated with P110 at reperfusion was almost indistinguishable (Figure 3D).
Using isolated mitochondrial fractions prepared from the ex‐vivo‐model hearts, we next confirmed that Drp1 association with mitochondria increased following IR in this model. There was a 2.2±0.1 fold increase in mitochondrial Drp1 after 90 minutes of reperfusion and P110 treatment blocked this IR‐induced increase in Drp1 association with the mitochondria to 0.88±0.05‐fold of basal (Figure 3E).
Two hours after IR, infarct size measured by triphenyl tetrazolium chloride (TTC) staining was 60±5% of the tissue and P110 treatment reduced the damage by about 30% (to 43±3% of the heart; Figure 4A). We reasoned that the cardioprotective effect of this inhibitor may reflect its action on a pathway related to improved mitochondrial function. Indeed, treatment before and after reperfusion also reduced IR‐induced increases in H2O2 release from the mitochondria (from 160±5% in IR hearts treated with control peptide to 125±4% after treatment with P110; Figure 4B). ATP levels in total heart tissue declined from 24.4±2.0 to 4.0±0.3 nmol/mg tissue by IR. The Drp1 inhibitor P110, increased ATP levels as compared to IR (from 4.0±0.3 to 6.8±0.3 nmol/mg tissue, respectively; Figure 4C).
Figure 4.

Cardiac damage and mitochondrial functions in heart subjected to IR ex vivo. A, Infarct size was determined by TTC staining (insert). B, Measurement of mitochondrial H2O2 release was determined in mitochondrial fraction of hearts after IR injury (100 μg each) using Amplex Red oxidation as a fluorescent marker. C, Level of ATP was measured in total heart extract after IR injury. Cleaved caspase 3 was determined as a marker of apoptosis (D). Autophagy (E) and JNK phosphorylation (F) as markers of cell stress are apparent in ex vivo heart after IR injury, as measured by increase in LC3‐II (E) and p‐JNK (F) in total lysates of heart subjected to IR in an ex vivo model. The effect of treatment with P110 (1 μmol/L) before and after reperfusion decreased autophagy and JNK phosphorylation as compared with normoxia and IR control hearts. (*P<0.05 vs normoxia, **P<0.05 vs IR; n=6/group). IR indicates ischemia and reperfusion; JNK, Jun kinase; LC3‐II, microtubule‐associated protein 1 light chain 3; TTC, triphenyltetrazolium chloride.
Autophagy and Apoptosis/cell Stress are Greatly Reduced by the Drp1 Inhibitor, P110
In Figure 4D, we determined the level of apoptosis in hearts subjected to IR by measuring the levels of caspase 3 activation by proteolysis (measuring caspase 3 fragments 15 and 19 kDa). IR increased the levels of cleaved caspase 3 by 28±2% as compared with the levels in normoxic hearts, indicating increased apoptosis under these experimental conditions. P110 decreased the levels of cleaved caspase 3 close to basal (normoxic) levels. Autophagy is activated after excessive mitochondrial fission and loss of mitochondrial membrane potential to remove damaged mitochondria.31–33 A marker of autophagy is the generation of a lipidated LC3‐II, a microtubule‐associated protein 1 light chain 3, also known as ATG8.34 The levels in the hearts of the autophagy marker, LC3‐II increased 5‐fold (from 0.08±0.01 to 0.40±0.02) after IR in this ex vivo model. Direct inhibition of either Drp1 at the onset of reperfusion decreased this increase by ≈50% (to 0.25±0.02; Figure 4E). Similarly, the levels of the stress marker, phosphorylated JNK35–36 decreased by 55±2% after P110 treatment (Figure 4F).
Acute and Chronic Effect of P110 Peptide Using an In vivo MI Rat Heart Model
In the final study, we measured the long‐term benefits of acute inhibition of mitochondrial fragmentation. As depicted in Figure 5A, after transient (30 minutes) LAD occlusion, the indicated peptides were injected intraperitoneally and cardiac functions were measured by echocardiogram. The ejection fraction of control rats was 86±4% and MI reduced it to 63±3% and 58±3% 3 days and 3 weeks after MI, respectively (Table). MI rats also displayed reduced fractional shortening (Figure 5B) and LV end‐systolic diameter compared to controls. P110 treatment did not affect these values under control conditions. Rats treated with P110 peptide (a single intraperitoneal injection; 0.5 mg/kg) at the onset of reperfusion showed improved cardiac function as measured by ejection fraction and fractional shortening 3 days after the onset of reperfusion (to 37±2%) and this was sustained when measured 3 weeks later (to ≈35±3%). In addition to providing FS as a systolic index, Table summarizes additional echocardiographic measurements, including ejection fraction, to support our conclusion that acute P110 treatment improved cardiac function. No changes in cardiac structure were seen in MI rats (Table).
Figure 5.
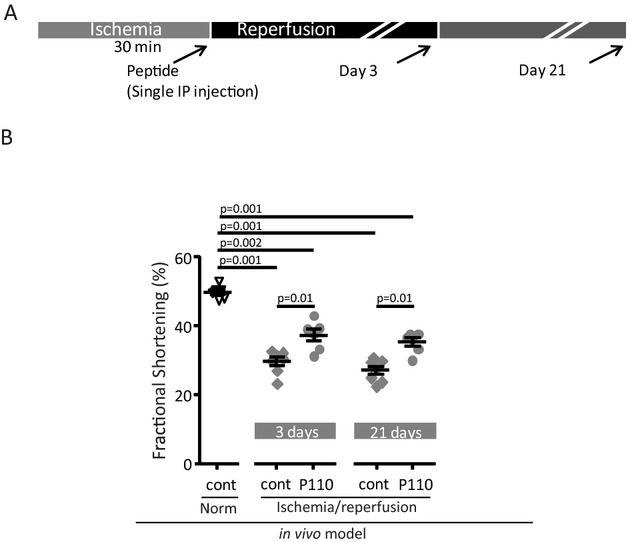
Cardiac function in myocardial infarction‐induced heart failure. A, Protocol of the treatment. B, Fractional shortening was measured in heart after LAD occlusion for 30 minutes followed by 3 days and 3 weeks of reperfusion in the presence of the respective peptides (0.5 mg/kg IP). Echocardiogram results are shown as individual rat heart results with respective P values between groups of control sham operated rats, and rats subjected to 30 minutes LAD occlusion and treated at the onset of reperfusion with cont (control peptide) or P110 using IP injection of 0.5 mg/kg. (n=7/control MI group, n=6/Normoxia and MI treated group). LAD indicates left anterior descending; MI, myocardial infarction.
We next determined whether the acute treatment with P110 after reperfusion resulted in long‐term benefit of mitochondrial functions. Oxygen consumption was measured in isolated mitochondria from hearts 3 weeks after myocardial infarction. We found a clear disruption of mitochondrial bioenergetics 3 weeks after MI, as depicted by reduced oxygen consumption at state‐3 (ADP‐dependent state) and upon carbonyl cyanide m‐chlorophenyl hydrazone (CCCP)‐induced mitochondrial uncoupling (Figure 6A). There was also a significant decrease in the efficiency of mitochondrial oxidative phosphorylation, as measured by RCR (Figure 6B). Acute treatment with P110 peptide only during the first few minutes of reperfusion resulted in preserved mitochondrial function when measured 3 weeks after IR injury.
Figure 6.

Mitochondrial bioenergetics in cardiac isolated mitochondria from post‐myocardial infarction dysfunction animals. Mitochondrial respiratory rates upon addition of ADP (state 3), oligomycin (state 4) and CCCP (uncoupling state) (A) and respiratory control rate (state 3/state 4) (B). Measurements were performed in cardiac isolated mitochondria after 3 weeks of in vivo rat acute myocardial infarction model. P110 treatment at the onset of reperfusion improved mitochondrial functions (*P<0.05 vs normoxia; **P<0.05 vs MI control peptide; n=6/normoxia group, n=7/MI control and treated group). CCCP indicates carbonyl cyanide m‐chlorophenyl hydrazone; MI; myocardial infarction.
Reduction of oxygen consumption has been associated with excessive mitochondrial ROS release in both acute and chronic cardiac diseases.37–38 We therefore determined the effect of acute P110 treatment on mitochondria ROS release as measured 3 weeks later. Three weeks after MI, hearts displayed increased mitochondria H2O2 release at state‐3, state‐4, and in the uncoupled state as compared to control rats subjected to sham surgery 3 weeks earlier (Figure 6C). Importantly, the presence of the Drp1 inhibitory peptide, P110, during the first few minutes of reperfusion decreased H2O2 release from the mitochondria upon the addition of ADP (state‐3), oligomycin (state‐4) and CCCP (uncoupled state), demonstrating an improvement in mitochondria bioenergetics (Figure 6C). P110 treatment did not affect either mitochondrial respiration or H2O2 release in control rats (Figure 6).
Taken together, our results show that 3 weeks after IR, cardiac mitochondria have a prominent uncoupling between the mitochondrial electron transport chain and oxidative phosphorylation. Obviously, the half‐life of the peptide is short; however, such a P110 treatment was sufficient to reduce pathological cardiac remodeling and mitochondrial dysfunction as a result of a smaller initial injury to the myocardium that was subjected to IR.
Discussion
There is much interest in the role of mitochondrial dynamics and functions in cardiovascular diseases.7,24 The majority of the mitochondria in heart tissue are tightly packed between the cardiac myofibers, such that the fusion and fission process may be restricted. Some reports show that decreased mitochondrial fission prior to the ischemic event is cardioprotective4,15 and that excessive fission and fragmentation contribute to cell death in other cell types.31,39 Yet, fission, especially under stress, is also thought to be required to enable the elimination of dysfunctional mitochondria, thus preserving the healthy population of mitochondria in a variety of cell types and in the myocardium during heart failure, for example.40
Here, we describe the beneficial effects of an inhibitor of excessive mitochondria fission and fragmentation, P110, in cultured rat cardiomyocytes, an ex vivo model of cardiac ischemia and an in vivo rat model of myocardial infarction. P110 inhibitory peptide is composed of a 7‐amino‐acid peptide (DLLPRGT; Drp149‐55), representing a homology sequence between Drp1 and Fis1 (ELLPKGS; Fis160‐66; the three homologous differences relative to Drp1 are provided in italics).18 The Drp1‐derived peptide was bound to the cell permeable peptide, TAT47‐57.41 We previously showed that P110 inhibits excessive activation of Drp1 by inhibiting its GTPase activity and the interaction between Drp1 and Fis1 on the mitochondria under stress conditions in neuronal cells and in a Parkinson's disease model.18 We also showed that P110 has no effect on any other mitochondrial fission or fusion proteins or on other members of the dynamin family (see also Figure 1) and that it does not cause any effects in nonstress conditions.18 Therefore, P110 selectively inhibits pathological (excessive) mitochondrial fission and fragmentation.
Mitochondrial fragmentation results in loss of ATP synthesis and increased ROS production that lead to tissue damage. Using an ex vivo Langendorff model and in vivo LAD occlusion model, we found that treating with P110 both before and after reperfusion inhibited reperfusion‐induced excessive fission, reduced H2O2 release, and increased ATP production and O2 consumption in heart after IR. P110 decreased infarct size and improved cardiac function in an in vivo MI model.
Previous work has shown that metabolically impaired mitochondria are preferably targeted by autophagy and that inhibition of fission reduces autophagy in a hepatocyte model of mitochondrial stress and in yeast.42–43 Similarly, we found that P110 treatment of the heart before and after reperfusion inhibits the increase in the autophagy marker, LC3‐II, after IR as well as the increase of cleaved caspase 3 and JNK phosphorylation, markers of apoptosis and cell death, respectively.35–36 Finally, we demonstrated that the effect of P110 peptide, administered only at the onset of reperfusion, inhibits the excessive pathological fragmentation driven by Drp1 binding to Fis1 that contributes further to ROS production and cell death, thus improving cardiac and mitochondrial functions even when measured 3 weeks after MI. Since the half‐life of these peptides is likely less than one hour, these data demonstrate that acute excessive mitochondrial fission results in long‐term impairment of the myocardium and that inhibiting this acute impairment may reduce the subsequent deterioration of cardiac function after myocardial infarction.
The effect of the pharmacological inhibitor of Drp1, mdivi‐114 was previously determined in an in vivo MI model in mice15 and in rats.44 Mdivi‐114 has an IC50 of ≈10 μmol/L14 and in the in vivo studies that showed a reduction in infarct size after MI, mdivi‐1 was used at a dose that was equivalent to 50 μmol/L administered before MI.15 (10 μmol/L mdivi‐1 was ineffective in reducing cardiac injury in the ex vivo and in vivo models15). A recent report demonstrates that mdivi‐1 also decreases the activity of delayed‐rectifier K+ current in murine cardiac HL‐1 cells, with the same IC50 value (≈12 μmol/L).45 Further, in that study, mdivi‐1 was shown to prolong the duration of action potentials and increased spontaneous action potentials in HL‐1 cells. The observed effects of mdivi‐1 on K+ channels were direct (as evidenced, for example, by patch‐clamp experiments where the drug was given through the pipette) and were not associated with mdivi‐1 inhibition of mitochondrial fission. It remains to be determined whether the benefit of mdivi‐1 reported represents, in part, the suppressed K+ channels during MI, in vivo.
Mdivi‐1 benefits were determined only after 2 hours of reperfusion15 and the long‐term benefits on cardiac mitochondrial functions and on cardiac functions were not determined. Further, the drug was administered prior to ischemia. Here, we showed that acute inhibition of fission at the onset of reperfusion is sufficient to provide benefits to the myocardium as measured both within an hour of treatment, but also after 3 weeks. Therefore, our study shows that mitochondrial fragmentation is part of the reperfusion injury.
Our study shows two previously unreported benefits of inhibiting mitochondrial fragmentation. First, we demonstrated here that P110 treatment was sufficient to restore mitochondrial functions in three different heart models of IR injury. Second, we showed that the mitochondrial fragmentation is triggered by reperfusion, as an inhibitor of fission given at reperfusion was sufficient to restore cardiac tissue integrity and cardiac functions. We conclude that controlling Drp1‐mediated excessive mitochondrial fission after MI, at the onset of reperfusion, contributes to increased cardiac protection and prevention of dysfunction.
Sources of Funding
This work was supported by NIH grant HL52141 to Dr Mochly‐Rosen, by Sao Paulo Research Foundation (FAPESP 2012/05765‐2) and The National Council for Scientific and Technological Development (CNPq 470880/2012‐0) grants to Dr Ferreira.
Disclosures
None.
Acknowledgments
We thank Dr. Nir Qvit for peptide synthesis, Dr. Adrienne Gordon for helpful discussions and Marcele Coelho for technical assistance (in vivo MI model). We thank the Stanford Shared FACS Facility specially Dr. David R Parks for helpful support and discussion and John J. Perrino at Stanford electron microscopy facility as well as Kitty Lee at Stanford cell sciences imaging facility. We thank the department of Anatomy, Institute of Biomedical Sciences, University of São Paulo, for the travel grant to M.‐H.D.
References
- 1.Tandler B, Hoppel CL. Possible division of cardiac mitochondria. Anat Rec. 1972; 173:309-323 [DOI] [PubMed] [Google Scholar]
- 2.Stanley WC, Hoppel CL. Mitochondrial dysfunction in heart failure: potential for therapeutic interventions? Cardiovasc Res. 2000; 45:805-806 [DOI] [PubMed] [Google Scholar]
- 3.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001; 33:1065-1089 [DOI] [PubMed] [Google Scholar]
- 4.Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal. 2013; 19:400-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim I, Rodriguez‐Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007; 462:245-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hom J, Sheu SS. Morphological dynamics of mitochondria–a special emphasis on cardiac muscle cells. J Mol Cell Cardiol. 2009; 46:811-820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010; 88:16-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fannjiang Y, Cheng WC, Lee SJ, Qi B, Pevsner J, McCaffery JM, Hill RB, Basanez G, Hardwick JM. Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 2004; 18:2785-2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki M, Neutzner A, Tjandra N, Youle RJ. Novel structure of the N terminus in yeast Fis1 correlates with a specialized function in mitochondrial fission. J Biol Chem. 2005; 280:21444-21452 [DOI] [PubMed] [Google Scholar]
- 10.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010; 191:1141-1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011; 12:565-573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011; 30:2762-2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brady NR, Hamacher‐Brady A, Gottlieb RA. Proapoptotic BCL‐2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL‐1 cells and GFP biosensors. Biochim Biophys Acta. 2006; 1757:667-678 [DOI] [PubMed] [Google Scholar]
- 14.Cassidy‐Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak‐dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008; 14:193-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010; 121:2012-2022 [DOI] [PubMed] [Google Scholar]
- 16.Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003; 35:339-341 [DOI] [PubMed] [Google Scholar]
- 17.Halestrap AP. Mitochondria and reperfusion injury of the heart–a holey death but not beyond salvation. J Bioenerg Biomembr. 2009; 41:113-121 [DOI] [PubMed] [Google Scholar]
- 18.Qi X, Qvit N, Su YC, Mochly‐Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013; 126:789-802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajek P, Chomyn A, Attardi G. Identification of a novel mitochondrial complex containing mitofusin 2 and stomatin‐like protein 2. J Biol Chem. 2007; 282:5670-5681 [DOI] [PubMed] [Google Scholar]
- 20.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly‐Rosen D. Inhibition of delta‐protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003; 108:2304-2307 [DOI] [PubMed] [Google Scholar]
- 21.Churchill EN, Disatnik MH, Mochly‐Rosen D. Time‐dependent and ethanol‐induced cardiac protection from ischemia mediated by mitochondrial translocation of varepsilonPKC and activation of aldehyde dehydrogenase 2. J Mol Cell Cardiol. 2009; 46:278-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campos JC, Queliconi BB, Dourado PM, Cunha TF, Zambelli VO, Bechara LR, Kowaltowski AJ, Brum PC, Mochly‐Rosen D, Ferreira JC. Exercise training restores cardiac protein quality control in heart failure. PLoS ONE. 2012; 7:e52764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue‐, substrate‐, and site‐specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009; 46:1283-1297 [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Liu Y, Dorn GW., II Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011; 109:1327-1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song W, Bossy B, Martin OJ, Hicks A, Lubitz S, Knott AB, Bossy‐Wetzel E. Assessing mitochondrial morphology and dynamics using fluorescence wide‐field microscopy and 3D image processing. Methods. 2008; 46:295-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H, Chan DC. Mitochondrial dynamics in mammals. Curr Top Dev Biol. 2004; 59:119-144 [DOI] [PubMed] [Google Scholar]
- 27.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007; 8:870-879 [DOI] [PubMed] [Google Scholar]
- 28.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin‐like protein DLP1. Mol Cell Biol. 2003; 23:5409-5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Budas GR, Churchill EN, Disatnik MH, Sun L, Mochly‐Rosen D. Mitochondrial import of PKCepsilon is mediated by HSP90: a role in cardioprotection from ischaemia and reperfusion injury. Cardiovasc Res. 2010; 88:83-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hondeghem LM, Cotner CL. Reproducible and uniform cardiac ischemia: effects of antiarrhythmic drugs. Am J Physiol. 1978; 235:H574-H580 [DOI] [PubMed] [Google Scholar]
- 31.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008; 1777:1092-1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wikstrom JD, Twig G, Shirihai OS. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol. 2009; 41:1914-1927 [DOI] [PubMed] [Google Scholar]
- 33.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008; 27:433-446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000; 19:5720-5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H(2)O(2) regulates cardiac myocyte phenotype via concentration‐dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003; 35:615-621 [DOI] [PubMed] [Google Scholar]
- 36.El‐Mowafy AM, White RE. Resveratrol inhibits MAPK activity and nuclear translocation in coronary artery smooth muscle: reversal of endothelin‐1 stimulatory effects. FEBS Lett. 1999; 451:63-67 [DOI] [PubMed] [Google Scholar]
- 37.Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, Sluse FE, Souza‐Pinto NC, Vercesi AE. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013; 18:2029-2074 [DOI] [PubMed] [Google Scholar]
- 38.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010; 88:40-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank S, Gaume B, Bergmann‐Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin‐related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001; 1:515-525 [DOI] [PubMed] [Google Scholar]
- 40.Dorn GW., II Mitochondrial dynamism and cardiac fate. Circ J. 2013; 77:1370-1379 [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly‐Rosen D. Molecular transporters for peptides: delivery of a cardioprotective epsilonPKC agonist peptide into cells and intact ischemic heart using a transport system, R(7). Chem Biol. 2001; 8:1123-1129 [DOI] [PubMed] [Google Scholar]
- 42.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001; 15:2286-2287 [DOI] [PubMed] [Google Scholar]
- 43.Priault M, Salin B, Schaeffer J, Vallette FM, di Rago JP, Martinou JC. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005; 12:1613-1621 [DOI] [PubMed] [Google Scholar]
- 44.Zhang N, Wang S, Li Y, Che L, Zhao Q. A selective inhibitor of Drp1, mdivi‐1, acts against cerebral ischemia/reperfusion injury via an anti‐apoptotic pathway in rats. Neurosci Lett. 2013; 535:104-109 [DOI] [PubMed] [Google Scholar]
- 45.So EC, Hsing CH, Liang CH, Wu SN. The actions of mdivi‐1, an inhibitor of mitochondrial fission, on rapidly activating delayed‐rectifier K(+) current and membrane potential in HL‐1 murine atrial cardiomyocytes. Eur J Pharmacol. 2012; 683:1-9 [DOI] [PubMed] [Google Scholar]