

Introduction
Cardiovascular calcification is a prominent feature of chronic inflammatory disorders that associate with significant morbidity and mortality. Vascular calcification is highly prevalent in patients with chronic kidney disease (CKD), a multifactorial disorder. CKD often results from hypertension and diabetes, and patients with CKD are among the highest‐risk groups for cardiovascular events. Notably, CKD accelerates the development of atherosclerosis. Our group and others have demonstrated that CKD causes excessive vascular inflammation and calcification.1 CKD is characterized by increased serum phosphate levels, which is, in turn, associated with the progression of calcification. But phosphate binder therapy reportedly has no effect on vascular calcification in CKD patients,2 suggesting that targeting phosphate alone cannot improve vessel stiffness. A variety of other therapies, which have produced mixed results at best, have been tested to prevent or hinder the progression of vascular calcification in CKD.3 A greater understanding of the mineralization process itself may therefore lead to new and effective therapeutics for vascular calcification in CKD.
In vitro and in vivo models of cardiovascular calcification have helped generate a greater mechanistic understanding of the vascular calcification process. A common model of CKD is apolipoprotein E‐deficient (ApoE−/−) or low‐density lipoprotein receptor‐deficient (ldlr−/−) mice subjected to 5/6 nephrectomy and fed a high‐fat, high‐cholesterol diet.4 Similar to dysmetabolic CKD patients, both arterial intimal and medial calcification occurs in these animals. The use of 5/6 nephrectomy in mice with varying diets allows for the investigation of medial calcification, which represents patients with renal failure undergoing hemodialysis. Each model recapitulates the formation of calcification in distinct vascular layers, which may lead to different clinical outcomes (Figure).
Figure 1.
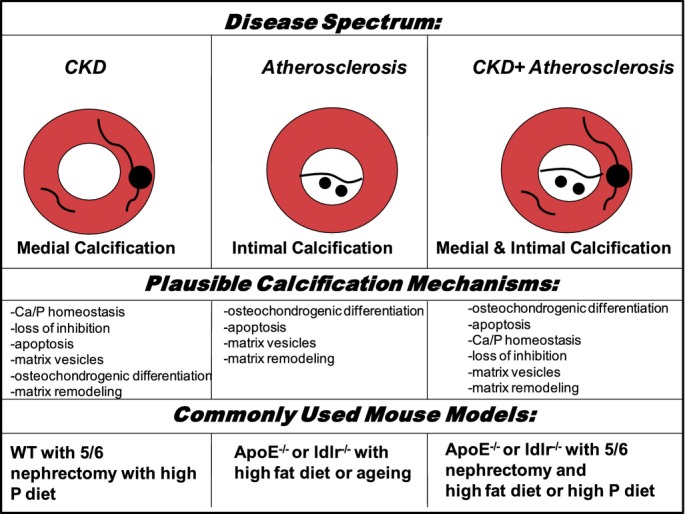
Modeling human vascular calcification in mice. Human disease associated intimal and medial vascular calcification diagramed with plausible mechanisms and commonly used mouse models. ApoE−/− indicates apolipoprotein E‐deficient; Ca, calcium; CKD, chronic kidney disease; ldlr−/−, low‐density lipoprotein receptor‐deficient; P, phosphate; WT, wild type.
Clinically, vascular calcification occurs in the vessel intima or media and often overlaps, particularly in a growing population of patients with CKD‐related atherosclerosis. The process of intimal calcification associates with atherosclerotic vascular disease, observed as spotty calcifications of the atherosclerotic plaques, where small hydroxyapatite mineral clefs (microcalcifications) seem to associate with cholesterol crystals observed in early lesions.5 These microcalcifications in the plaque could provoke a rupture of vulnerable plaque.6 Medial calcification occurs primarily in association with CKD and diabetes, and is independent of hypercholesterolemia. The sheet‐like calcifications of the tunica media can lead to increased vascular stiffness and reduced compliance of vessels. The differences between medial and intimal calcification stress the importance of choosing appropriate disease models for mechanistic and therapeutic studies.
Pathogenesis of vascular calcification is complex—beyond just a simple precipitation of calcium and phosphate, it is instead an active, cell‐regulated process. Several cell types likely play a role in the vascular calcification process, including smooth muscle cells (SMCs),7 circulating bone marrow‐derived cells,8 and macrophages.5 Vascular calcification mechanistic studies have focused on cellular differentiation, calcium and phosphate homeostasis, loss of inhibition, apoptosis, the release of calcifying vesicles, and changes in the extracellular matrix (ECM) among several other cellular processes3 (Figure). Osteogenic transition of vascular SMCs or stem cells is induced by bone morphogenetic proteins,9 high phosphate levels,10 inflammation,11–12 and oxidative stress,13 and leads to a unique molecular pattern marked by osteogenic transcription factors. Emerging evidence also suggests that alternative mechanisms independent of osteogenic differentiation, including the release of matrix vesicles by SMCs or macrophages, may contribute to vascular calcification in CKD.5,14 Understanding the connections between these mechanisms and signaling pathways could provide novel mechanistic insight into the calcification process, and potentially help lead to cardiovascular disease therapeutics.
In this issue of JAHA, Masuda et al15 build on previous studies linking tumor necrosis factor α (TNFα) to vascular calcification, while providing a novel link between inflammation, endoplasmic reticulum stress, and calcification. Their study found that administering TNFα to mouse vascular SMCs dose‐dependently induced the protein kinase RNA‐like endoplasmic reticulum kinase (PERK)—eukaryotic initiation factor 2α (eIF2α)‐activating transcription factor 4 (ATF4)—C/EBP homologous protein (CHOP) axis of the endoplasmic reticulum stress response.
Suggesting that inflammation may regulate vascular calcification through an endoplasmic reticulum stress pathway, the authors also showed short hairpin RNA‐mediated knockdowns of PERK, ATF4, and CHOP led to significant reductions in both TNFα‐induced calcium deposition and alkaline phosphatase activity. Supporting their in vitro findings, both phospho‐ATF4 and total ATF4, along with CHOP expression levels, increased significantly in the aortas of 5/6 nephrectomy ApoE−/− mice on a high‐fat, high‐cholesterol diet. When treated with infliximab to block TNFα, or with 2 different chemical chaperones to reduce endoplasmic reticulum stress, calcified lesions decreased significantly in these CKD mice. The infliximab results provide further support for targeting inflammation as a means of blocking vascular calcification, which had been previously reported by another group using ldlr−/− diabetic mice.16
On a mechanistic level, treatment of these CKD mice with the chemical chaperones also led to a significant reduction in the expression of a type III sodium‐dependent phosphate transporter, PiT‐1, possibly mediated through a reduction of CHOP expression. This study also demonstrated that controlling the expression of PiT‐1 through CHOP overexpression correlated with the amount of inorganic phosphate uptake in SMCs. Of importance relating to this result, PiT‐1 was shown previously to participate in the vascular calcification process.17
The findings of Masuda et al suggest that inflammation at least partly regulates vascular calcification through activating an endoplasmic reticulum stress pathway, which in turn may increase inorganic phosphate uptake, leading to increased mineralized calcium deposition and osteogenic differentiation. The growing connection of endoplasmic reticulum stress to vascular calcification is intriguing, considering that this stress response process has also been connected to bone formation.18
Besides inflammation, many additional mechanistic factors have been proposed and likely play a role in vascular calcification in combination with or independently of the inflammation driven process. For example, previous studies19 have demonstrated that some components of the ECM (elastin, collagen) could be important factors in the regulation of calcification progression.1 Exactly how the ECM components and the factors that regulate ECM remodeling, such as matrix metalloproteases and cathepsins, could lead to medial calcification is not yet fully understood. Furthermore, additional unreported factors may also be involved in this mechanistic pathway.
A report by Purnomo et al20 in this issue of JAHA examined the role of ECM in the development of vascular calcification, by highlighting the role of glycosaminoglycans (GAG). The study found that exostosin‐like glycosyltransferase 2 (EXTL2) deficiency caused increased heparin sulfate and chondroitinsulfate in the aortas of EXTL2‐deficienct mice. When CKD was induced in mice by 5/6 nephrectomy along with a high‐phosphate diet—a different CKD model than that used by Masuda et al—elevated calcification was observed in the media layer of the aorta, which is characterized by increased GAG levels. The use of 5/6 nephrectomy along with a high‐phosphate diet allowed other aspects of CKD, in addition to vascular calcification, to be modeled. EXTL2‐deficient CKD mice had also higher systolic blood pressure compared to wild‐type mice, which might be due to increased vascular calcification accompanied by increased stiffness and decreased compliance of the arteries.
Importantly, CKD itself increased heparin sulfate and chondroitinsulfate in wild‐type mice, which might accelerate the progression of vascular calcification. In line with this evidence, treatment with heparitinase and chondroitinase prevented calcification in aorta explants. As such, changes in the sulfation pattern of GAG could conceivably contribute to the vascular calcification process. Further study is required, however, to dissect the full influence of specific GAG sulfation patterns in triggering pathways involved in the development of vascular calcification.
In the EXTL2‐deficient mice, Runx2 and collagen 1A1 were increased, whereas SMC markers were decreased. Purnomo et al suggested that under high‐phosphate conditions heparin sulfate and chondroitinsulfate trigger a signaling pathway that further induces SMC osteogenic transition. Whether similar results occur in EXTL2‐deficient mice under other disease‐relevant conditions remains to be determined. For example, testing this pathway in dyslipidemic conditions, such as those used by Masuda et al, may help reveal additional information about the disease process (eg, whether ECM‐based calcification effects intimal calcification).
Taken together, the results of this study provide novel insights into the role of GAG in calcification, and further suggest that modulation of the ECM might be a therapeutic target in the prevention of vascular calcification in CKD.
While these two studies depicting the role of two potentially independent mechanisms of calcification in CKD help to connect previous findings from other groups, several questions remain unanswered—notably, how inflammatory cytokines like TNFα activate endoplasmic reticulum stress, and whether GAG are increased in the aortas of CKD patients. Just how these proposed mechanisms will ultimately translate to the human disease condition remains to be seen. While mouse models are useful, an important next step to furthering these results will be to test these working hypotheses in human cells and tissues.
In summary, the work of Masuda et al and Purnomo et al adds to several existing working hypotheses on how vascular calcification occurs, particularly in CKD. Of paramount importance in interpreting the results of these and other studies involves understanding the model used and the type of calcification it reflects. Differences between intimal and medial calcification may also be reflected in the mechanisms behind their development. Each animal and cell culture model has limits; no one model fully encapsulates the human disease process. Each new study, however, incrementally adds toward our growing understanding of the process of vascular calcification.
Disclosure
None.
Sources of Funding
Dr Aikawa is supported by grants from the National Institutes of Health (R01HL114805, R01HL109506).
Acknowledgments
The authors thank Sara Karwacki for her excellent editorial assistance.
References
- 1.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009; 119:1785-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seifert ME, de Las Fuentes L, Rothstein M, Dietzen DJ, Bierhals AJ, Cheng SC, Ross W, Windus D, Dávila‐Román VG, Hruska KA. Effects of phosphate binder therapy on vascular stiffness in early‐stage chronic kidney disease. Am J Nephrol. 2013; 38:158-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu M, Rementer C, Giachelli CM. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int. 2009. 10.1007/s00223‐013‐9712‐z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shobeiri N, Adams MA, Holden RM. Vascular calcification in animal models of CKD: a review. Am J Nephrol. 2010; 31:471-481 [DOI] [PubMed] [Google Scholar]
- 5.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage‐derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Cir Res. 2013; 113:72-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly‐Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L, Weinbaum S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci USA. 2013; 110:10741-10746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wada T, McKee MD, Steitz S, Giachelli CM. Calcification of vascular smooth muscle cell cultures: inhibition by osteopontin. Cir Res. 1999; 84:166-178 [DOI] [PubMed] [Google Scholar]
- 8.Khosla S, Eghbali‐Fatourechi GZ. Circulating cells with osteogenic potential. Ann N Y Acad Sci. 2006; 1068:489-497 [DOI] [PubMed] [Google Scholar]
- 9.Boström K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993; 91:1800-1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuichi J, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morri H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Cir Res. 2000; 87:e10-e17 [DOI] [PubMed] [Google Scholar]
- 11.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor‐alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000; 102:2636-2642 [DOI] [PubMed] [Google Scholar]
- 12.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jafferer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early‐stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007; 116:2841-2850 [DOI] [PubMed] [Google Scholar]
- 13.Goettsch C, Rauner M, Hamann C, Sinningen K, Hempel U, Bornstein SR, Hofbauer LC. Nuclear factor of activated T cells mediates oxidised LDL‐induced calcification of vascular smooth muscle cells. Diabetologia. 2011; 54:2690-2701 [DOI] [PubMed] [Google Scholar]
- 14.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell‐derived matrix vesicles to enhance mineralization. Cir Res. 2011; 109:e1-e12 [DOI] [PubMed] [Google Scholar]
- 15.Masuda M, Miyazaki‐Anzai S, Levi M, Ting T, Miyazaki M. PERK‐eIF2α‐ATF4‐CHOP signaling contributes to TNFα‐induced vascular calcification. J Am Heart Assoc. 2013; 2:e00023810.1161/JAHA.113.000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al‐Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic Msx2‐Wnt calcification cascade is regulated by TNF‐alpha‐dependent signals in diabetic Ldlr−/− mice. Aterioscler Thromb Vasc Biol. 2007; 27:2589-2596 [DOI] [PubMed] [Google Scholar]
- 17.Li X, Yang HY, Giachelli CM. Role of the sodium‐dependent phosphate cotransporter, Pit‐1, in vascular smooth muscle cell calcification. Circ Res. 2006; 98:905-912 [DOI] [PubMed] [Google Scholar]
- 18.Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, Saitoh M, Nishimura R, Yoneda T, Kou I, Furuichi T, Ikegawa S, Ikawa M, Okabe M, Wanaka A, Imaizumi K. Signaling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009; 11:1205-1211 [DOI] [PubMed] [Google Scholar]
- 19.Pai AS, Giachelli CM. Matrix remodeling in vascular calcification associated with chronic kidney disease. J Am Soc Nephrol. 2010; 21:1637-1640 [DOI] [PubMed] [Google Scholar]
- 20.Purnomo E, Emoto N, Nugrahaningsih DA, Nakayama K, Yagi K, Heiden S, Nadanaka S, Kitagawa H, Hirata K. Glycosaminoglycan overproduction in the aorta increases aortic calcification in murine chronic kidney disease. J Am Heart Assoc. 2013; 2:e00040510.1161/JAHA.113.000405 [DOI] [PMC free article] [PubMed] [Google Scholar]