

Abstract
Background
Failure of colony PCRs in green microalga Chlorella vulgaris is typically attributed to the difficulty in disrupting its notoriously rigid cell walls for releasing the genetic materials and therefore the development of an effective colony PCR procedure in C. vulgaris presents a challenge.
Results
Here we identified that colony PCR results were significantly affected by the accumulated lipids rather than the rigid cell walls of C. vulgaris. The higher lipids accumulated in C. vulgaris negatively affects the effective amplification by DNA polymerase. Based on these findings, we established a simple and extremely effective colony PCR procedure in C. vulgaris. By simply pipetting/votexing the pellets of C. vulgaris in 10 ul of either TE (10 mM Tris/1 mM EDTA) or 0.2% SDS buffer at room temperature, followed by the addition of 10 ul of either hexane or Phenol:Chloroform:Isoamyl Alcohol in the same PCR tube for extraction. The resulting aqueous phase was readily PCR-amplified as genomic DNA templates as demonstrated by successful amplification of the nuclear 18S rRNA and the chloroplast rbcL gene. This colony PCR protocol is effective and robust in C. vulgaris and also demonstrates its effectiveness in other Chlorella species.
Conclusions
The accumulated lipids rather than the rigid cell walls of C. vulgaris significantly impede the extraction of genetic materials and subsequently the effective colony PCRs. The finding has the potential to aid the isolation of high-quality total RNAs and mRNAs for transcriptomic studies in addition to the genomic DNA isolation in Chlorella.
Keywords: Colony PCR protocol, Microalgae, Chlorella vulgaris
Background
Due to their diversity, robustness of growing under brackish water, easiness of cultivation, minimum nutrition requirement and high algal oil content, microalgae are heralded as the 3rd generation feedstock for biofuels production [1-18]. Among those eukaryotic microalgae, the green microalga Chlorella vulgaris is the most promising species for industrial applications. In comparison to other microalgae, C. vulgaris has relatively high growth rates, photosynthetic efficiencies and capable of accumulating over 50% lipids (dry cell weight) under nitrogen limited conditions [4,5].
Those advantages have led to the great interest in this species for algal oil production and significant efforts have been exercised towards the molecular level understanding and characterization of the lipids pathways and genetic manipulation of Chlorella genes for maximizing its oil contents and optimizing the lipid profiles [4,6]. With these goals in consideration, it is essential to develop transformation tools which require a simple procedure for the rapid identification of microbial transformants by simple PCR analysis in routine studies [7]. Due to its rapidness, simplicity and minimal amounts of cells required, colony PCR is an essential and also expeditious technique for such a purpose [5,8]. Besides the identification of microbial transformants, colony PCRs are frequently employed in gene amplification, identification of recombinant integrants and plasmid constructs [9,10], taxonomy screening [11] and diagnostics [12]. In addition, diversity studies of microalgae demands a rapid colony PCR protocol for identification of a collection of algal strains by the 18S rRNA analysis [13].
Few colony PCR procedures have been specifically developed for some microalgae. These methods involve the resuspending of cells using either chelating buffers, 10 nM Ethylenediaminetetraacetic acid (EDTA) [5,14], 5-6% Chelex-100 [5,14] and Tris/EDTA (TE) [5] or in surfactants 0.2% Triton X-100 [14], followed by an essential boiling step to facilitate cell lysis. However, no existing protocols have been successfully applied to C. vulgaris. Although Yeast Protein Extraction buffer (Y-PER) was recently employed for colony PCR in C. vulgaris[8], it is however more effective in colony PCR of other microalgal strains than C. vulgaris and typically requires an additional boiling step for C. vulgaris. Furthermore, this additional boiling step leads to the hydrolysis of the cell walls, which results over-release of polysaccharides and subsequently inhibit PCR amplification in C. vulgaris. Therefore subsequent dilution of the resulting extract is necessary for effective PCR amplification [8]. Overall, the use of Y-PER in colony PCR of C. vulgaris managed to work but required a few additional steps and the colony PCR results are not consistently reproducible.
The rigidity of C. vulgaris cell walls is well documented [19,20] and characterized as robust and resilient [21]. In addition, it has been described as a “recalcitrant” and “cumbersome barrier” against oil extraction [22], genetic manipulation plus cell fusion [19,23] and DNA extraction [13], thus requiring the utilization of mechanical homogenization, organic solvents, enzymatic digestion or a combination of these methods [19,20]. Together with the accumulation of lipids in aging C. vulagris cultures, its cell walls have been reported to be more resistant to detergents as they age [24]. Therefore, the failure of colony PCR in C. vulgaris has been typically attributed to its notoriously rigid cell walls which are considered as the main barrier for effectively releasing of genomic DNA [5,8]. In contrast, here we identified that the accumulated lipids rather than the rigid cell walls affect the effective PCR amplification. Based on it, we established a simple and extremely effective colony PCR procedure for C. vulgaris and also demonstrated its effectiveness for colony PCR in other species in the Chlorella genus.
Materials and methods
Microalgal strains
Algae culture and the medium used are identical to those ascribed in the UTEX Culture Collection of Algae (http://web.biosci.utexas.edu/utex/) and are as follows. Chlorella protothecoides UTEX 256, Chlorella minutissima UTEX 2341, Chlorella kessleri UTEX 262, and Chlorella luteoviridis UTEX 258 were grown in proteose medium, Chlorella sorokiniana UTEX 1666 in volvox-dextrose medium, Chlorella anitrata UTEX 1798 in modified Bold 3N medium, Chlorella vulgaris UTEX 30 in algae culture broth Fluka 17124, Chlorella desiccata UTEX 2526 in artificial seawater medium, Chlorella stigmatophora UTEX LB993 in Erdschreiber’s medium, Chlorella saccharophila UTEX 2911 in Trebouxia Medium, and Chlorella sphaerica UTEX LB2485 and Chlorella spp. in Bold 3N medium. The detailed compositions of these culture media were described in the UTEX culture collection of Algae (http://web.biosci.utexas.edu/utex/).
Lysis buffers for cell disruption
Three different lysis buffers, 10 mM Tris (T4661 Sigma Trizma® base) /1 mM Ethylenediaminetetraacetic acid (EDTA) E6758 Sigma (TE buffer), 0.2% Sodium dodecyl sulfate (SDS) (Cat# 102918, MP Biomedicals) and Yeast Protein Extraction buffer (Y-PER) (Product# 78990, Thermo Scientific) are individually examined for their genomic DNA extraction efficiency for colony PCRs. 95% n-hexane (Cat# 650552, Sigma) and Phenol:Chloroform:Isoamyl Alcohol (P:C:I 25:24:1) (P2069, Sigma) are used for lipid extraction.
Colony PCR procedures
Algal cells collected from a colony or liquid culture were resuspened into 10 μl of the above mentioned buffers in a PCR tube. To facilitate cell disruption, the mixture was pipetted/vortexed for a few seconds, followed by the addition of 10 μl of hexane in the initial testing and later replaced by Phenol:Chloroform:Isoamyl Alcohol (PCI) in the same tube. The lysate was micro-centrifuged at room temperature 25°C and 13.4 × 1000 rpm using Eppendorf Minispin® (Cat# 5452 000.018) and 1-2 μl of the resulting aqueous layer was directly subjected to PCR analysis.
The extraction efficiency of genomic DNA by various buffers was analyzed by PCR amplification of the nuclear 18S rRNA and the chloroplast rbcL gene. The universal oligos for amplification of the 18S rRNA include the forward oligo (5′-CCT GGT TGA TCC TGC CAG-3′) and the reverse oligo (5′-A/TTG ATC CTT CT/CG CAG GTT CA-3′) [5]. Amplification of the chloroplast rbcL gene was carried out using the oligo pair, the forward primer (5′-GCG GGT GTT AAA GAC TAC CG-3′) and the reverse primer (5′-CCT AAA GTA CCA CCG CCA AA-3′). PCRs were carried out in 20 μl of 1× high fidelity buffer B (New England Biolabs, Ipswich, MA), 2 pmol of each primer, 4 μmol of each dNTP, 1-2 μl of diluted genomic DNA templates extracted previously, and 1 units of Phusion DNA polymerase with high fidelity for 30 cycles using i-Cycler from BioRad (Hercules, CA, USA). Each cycle consisted of 10 s at 98°C, 30 s at 56°C, and 45 s at 72°C, with a final extension step of 5 mins at 72°C. The PCR products were subjected to 0.8% agarose gel electrophoresis for analysis.
Algal cell culture and total lipid analysis
C. vulgaris cells from a colony were picked and inoculated in 100 ml freshwater algae media (Fluka 17124) under sterile conditions and grown in 100 lumens fluorescence lighting using a ratio of 16:8 light to dark cycle and agitation of 100 rpm at 25°C. After 14-days growth, this initial seed culture was re-inoculated into four 200 ml cultures in 1 litre shake flasks and grown in the same conditions above.
Cell density was measured using the difference in absorbance at 735 nm and 680 nm. Cell lysis and triglyceride extraction of C. vulgaris were carried out by centrifuging 5 ml of algae culture which was followed by re-suspension of the algae cell pellet in 20 ul of 30% (v/v) Triton-× 100 in ethanol. Post incubation at 100°C for 5 minute, the mixture was microcentrifuged at 13,400 rpm for 2 minutes and the resulting supernatant used for cellular lipid analysis with a commercial serum triglyceride kit (Sigma TR0100), according to the manufacturer’s instructions. Cellular triglyceride was quantified using a glyceryl trioleate (T7140 Sigma) standard curve obtained using the above kit and shown here as relative triglyceride per cell concentration using the above OD values.
Results
Our recent efforts on the development of transformation protocols in C. vulgaris require a procedure for rapid identification of transformants by PCR screening. We previously demonstrated that using of Y-PER with additional boiling step (98°C for 10 mins) led to the effective release of genetic materials from C. vulgaris[8] regardless of growth stage. However, we observed that the PCRs frequently failed with the released genomic DNA, especially from the aged cells. Microscopic examination (Figure 1) of the Y-PER treated algal cells harvested from different growth phases further illustrated no morphological differences of the un-deformed rigid cell walls following the elution of DNA. This suggests other factors rather than the rigid cell walls impeding genetic material extraction. Based on those observations, we postulated that factors other than the rigid cell walls impeding genetic material extraction and subsequent PCR amplification. Since C. vulgaris is known to accumulate significant lipids in the stationary growth phase, this prompts us to examine if these accumulated lipids impede effective PCR amplification.
Figure 1.
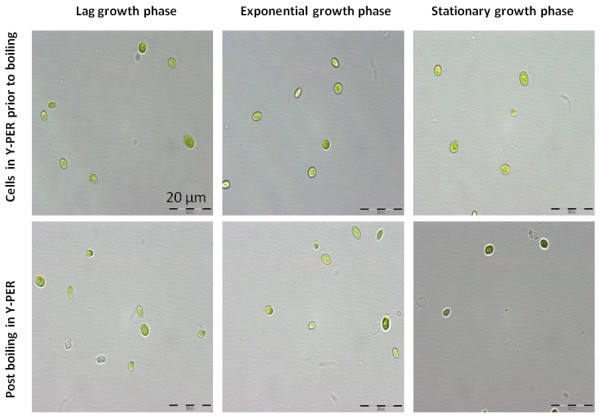
Microsopoic examination of C. vulgaris cells treated with Y-PER reagent.C. vulgaris harvested at the different growth stages (upper panel) and post the additional boiling step at 98°C for 5 min (lower panel).
Using hexane, a solvent known specifically for lipids extraction in microalgae, crude cell lysates of the aged C. vulgaris colonies from an agar culture (past 14 days) are disrupted by pipette mixing in TE, 0.2% SDS and Y-PER buffers prior to lipid extraction. Post micro-centrifugation, the resulting aqueous phases were analyzed by PCR amplification. Sequences encoding the 18S rRNA was successfully amplified from the aqueous layer in the respective buffers as indicated in Figure 2, while no PCR products were detected from the crude cell lysates using the various buffers without the additional hexane extraction step. This distinctly demonstrated that the further extraction of the crude cell lysate with hexane was essential. It also suggests that lipid removal by hexane extraction is a necessary step for successful PCRs.
Figure 2.
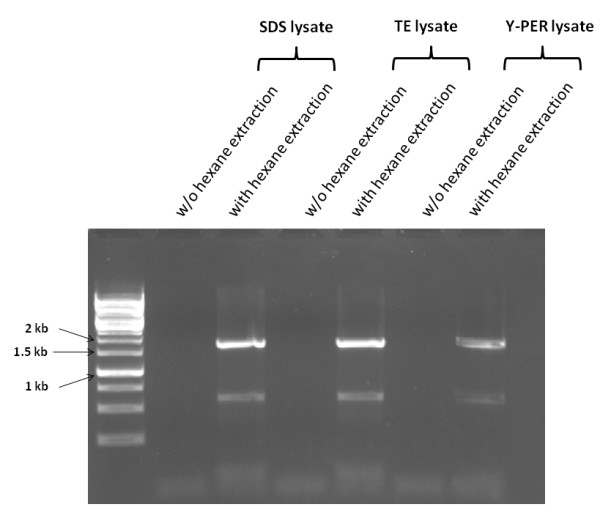
Colony PCR amplification of the 18S rRNA from the aqueous layer of hexane extracted cell lysate of C. vulgaris .
In addition, we observed that amongst those aqueous buffers for initial cell disruption by pipetting, the aqueous phases from the initial 10 mM TE and 0.2% SDS treatment produced better results than Y-PER when used as DNA templates for PCRs. Furthermore, in consideration of the unknown compositions of Y-PER reagent, we employed TE and 0.2% SDS for the cell disruption in all the subsequent experiments. Secondly, due to its high flammability hexane was replaced with PCI for lipid removal. Additionally in contrast to hexane, PCI produces an immiscible layer denser than water, thus allowing simpler and more precise sampling from the aqueous layer. Further removal of residual phenol from the aqueous layer is not required for successful PCR.
To further evaluate the impact of lipid accumulation in C. vulgaris on the efficacy of PCR amplification, we performed PCRs on algal cells extracted under various growth stages. Consistent with the above study, the additional step with PCI extraction led to successful PCR amplifications of the 18S rRNA sequences regardless of the growth stages (Figure 3). We also noticed that further PCI extraction is not required for successful PCR amplification of the 18S rRNA sequences from crude cell lysate disrupted in TE harvested in both the lag phase and the exponential growth phase, which corresponded to the trace lipid accumulation as indicated in Figure 3. However, the removal of total lipids by the additional PCI extraction was essential for the cell lysates from the stationary growth phase which accumulated relatively higher lipids (Figure 3). These results collectively demonstrate that the removal of accumulated lipids is essential for successful colony PCRs.
Figure 3.
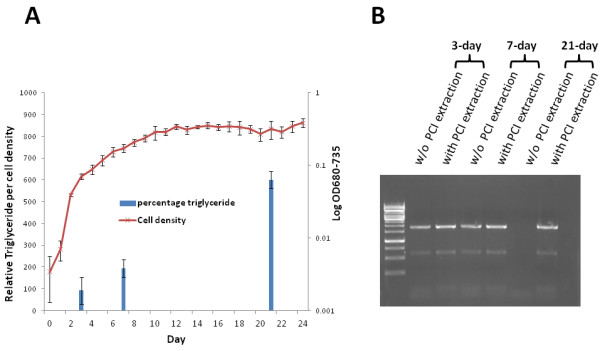
Total lipids accumulated in various growth stages of C. vulgaris (A) and their impact on the effective colony PCR amplifications (B). PCI denotes Phenol:Chloroform:Isoamyl Alcohol (Cat# P2069, Sigma).
In addition to the nuclear 18S rRNA analysis, we next examined the effectiveness of PCR amplifying the chloroplast rbcL gene in C. vulgaris from the aqueous layer of the extracts. As indicated in Figure 4, rbcL gene was successfully amplified from the aqueous layer of the TE crude lysate. This suggests the overall effectiveness of the initial cell disruption by agitation in TE buffer and the subsequent lipid removal by PCI extraction on releasing both the nuclear and chloroplast DNAs.
Figure 4.
PCR amplification of the chloroplast rbcL gene from the aqueous layer of phenol-chloroform extracted cell lysate of C. vulgaris. PCI denotes Phenol:Chloroform:Isoamyl Alcohol (Cat# P2069, Sigma).
We next asked if this procedure is effective in other species of Chlorella. By following the above protocol, strains from various Chlorella species were agitated in either TE or 0.2% SDS and followed by lipids removal using PCI extraction, the extracted genomic DNAs in the aqueous layers were subjected to the 18S rRNA PCR amplification. In contrast with those crude cell lysates without further PCI extraction, the 18S rRNAs sequences were successfully amplified from all the test strains as depicted in Figure 5. This result further demonstrated the effectiveness of the additional PCI treatment of the crude cell lysates on the isolation of PCR-quality genomic DNA from a wide range of Chlorella species. Based on Figure 5, we also observed that the initial step of disruption of algal cells in either TE or 0.2% SDS were comparably inadequate for the releasing genomic DNAs by agitation and this further suggests that the subsequent phenol-chloroform extraction results the isolation of PCR-quality genomic DNAs.
Figure 5.
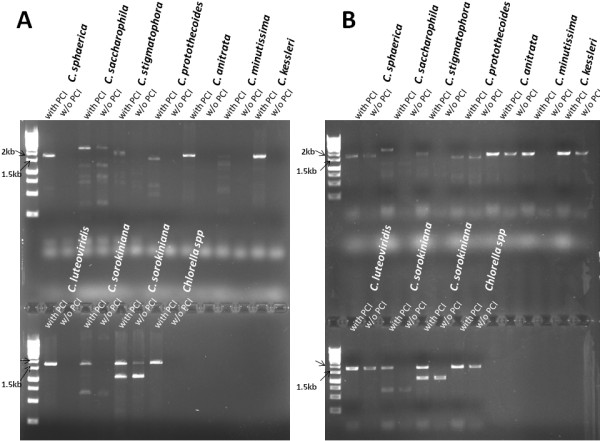
Colony PCR analysis of the 18S rRNA from other species of Chlorella. Cells were disrupted in either 10 mM TE (A) or 0.2% SDS (B) and followed by PCI extraction for lipid removal. PCI denotes Phenol:Chloroform:Isoamyl Alcohol (Cat# P2069, Sigma).
Discussion
As the most promising eukaryotic green microalga with the potential for industrial application for algal oil production, intensive research efforts have been attempted on C. vulgaris amongst other oleaginous algal species and therefore it is essential to develop a simple PCR protocol for rapid identification of recombinants with desired phenotypes. In this work, we identified the accumulated lipids (removed via solvent extraction) rather than the rigid cell walls which are often considered in previous researches [13,19,23] as the major barriers impeding DNA extraction. Based on this finding, we established a robust protocol for simple, effective and robust colony PCR in C. vulgaris.
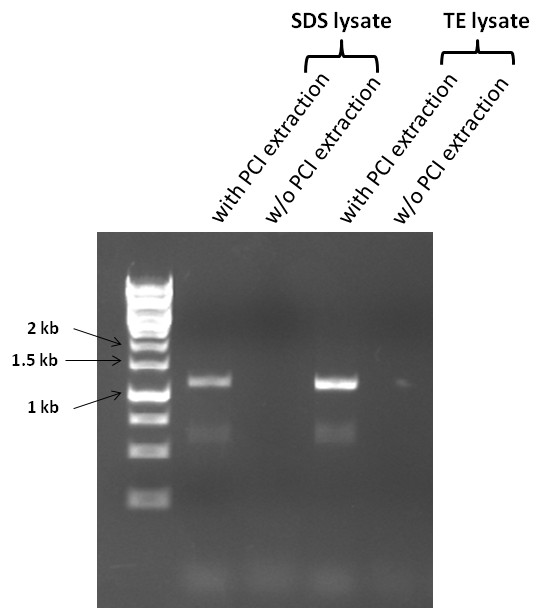
Previous protocols [5,14] reported the use of the chelating buffers, 10 mM EDTA, 5-6% Chelex-100 and TE buffer, together with the use of the surfactants, 0.2% Triton X-100. The ionic detergent SDS and Triton X-100 are used to cause disruption of the cell membrane, resulting in the lysis and release of cellular components [25,26], while the use of chelating agents prevent cellular nuclease degradation of nucleic acids during cell disruption [25,27]. These extraction buffers do not have the properties for oil removal. The organic solvent Phenol:Chloroform [27] were used mainly to separate cellular and nucleoproteins from DNA. By contrast, hexane [28] and chloroform–methanol [22] have been previously used for lipid extraction from microalgae. In the manuscript presented here, hexane in conjunction with TE buffer was initially used for lipid extraction/separation from C. vulgaris without prior mechanical disruption and DNA was obtained from the lower aqueous TE buffer phase of the immiscible mixture post centrifugation. This was later substituted with Phenol:Chloroform:Isoamyl Alcohol due to it being denser than the aqueous extraction buffers, allowing a cleaner and more rapid removal of the aqueous phrase containing DNA. An additional comparison of rapid DNA extraction without boiling comparing TE buffer, 0.2% SDS, Y-PER [8] and PCI was carried out on aged C. vulgaris cultures. Of these, PCR amplification was successful only with the use of PCI extraction as described in this work (Additional file 1: Figure S1).
In addition, Modifications of the DNA extraction using Chelex-100 [14] involve additional mechanical homogenization for higher plants [29] or high temperature incubation in ethanol following homogenization for Chlamydomonas reinhardtii and Arabidopsis thaliana[30]. The protocol presented here dispenses with these procedures, involves fewer steps and equipment. Compared with the use of Chelex-100, a chelating resin in combination with incubation in ethanol, the method here clearly segregates the genetic material from cellular lipids and non polar cell components. Additionally, incubation at high temperatures (100°C) for at least 10 minutes was necessary for the release of chloroplastic genetic material for the above method [5]. In the protocol presented here, DNA from chloroplast is obtained in a single step and without the high temperature or boiling step. DNA degradation can thus be reduced [31] and the procedure may be readily adaptable for the extraction of other cellular components. The study [5] described the use of Chelex-100 for extracting PCR-quality genomic DNAs from Chlorella species. Its effectiveness was demonstrated in the Chlorella species, C. sorokiniana, C. protothecoides, C. zofingiensis and C. kessleri. However, DNA extraction of C. vulgaris was not carried out nor described in the references [5,14].
To the best of our knowledge the only example of rapid DNA extraction in C. vulgaris[8] requires boiling incubation, subsequent centrifugation of the cell pellet in the surfactant, Y-PER and dilution of the supernatant in water. No other procedure involving the use of Chelex-100 on DNA extraction of C. vulgaris has been reported [14]. Fundamental differences exist in the cell wall composition [19,20], rigidity and cellular components of Chlamydomonas reinhardtii, a model organism for microalgae study and C. vulgaris with its lipid accumulating potential, thus requiring the use of different regime for DNA extraction.
In summary, we have established here, a simple and extremely efficient method for routine colony PCR analysis in C. vulgaris that can be extended to other species in Chlorella genus. Additionally, the finding that the accumulated lipids rather than the rigid cell walls negatively affecting effective DNA manipulation has the potential to aid the isolation of high quality and large quantity of total RNAs and mRNAs for transcriptomic studies in addition to the genomic DNA isolation in Chlorella.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CJYT and CL performed laboratory work. JW analyzed the data. HZ conceived the experiment and analyzed the data. CL and HZ drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
A comparison of rapid DNA extraction from C. vulgaris harvested in stationary growth phase without boiling using individual buffers for PCR amplification. The buffers, 10 mM Tris/1 mM EDTA (TE), 0.2% Sodium dodecyl sulfate (SDS), Yeast Protein Extraction buffer (Y-PER) (Cat# 78990, Thermo Scientific), Chelex-100, and TE treated followed by PCI extraction were examined for their genomic DNA extraction efficiency for colony PCRs.
Contributor Information
Crystal Jing Ying Tear, Email: crystal_tear@ices.a-star.edu.sg.
Chanyuen Lim, Email: lim_chan_yuen@ices.a-star.edu.sg.
Jinchuan Wu, Email: wu_jinchuan@ices.a-star.edu.sg.
Hua Zhao, Email: zhao_hua@ices.a-star.edu.sg.
Acknowledgements
We thank the IB group in ICES for their insights and discussion. This work was funded by A*STAR in Singapore ICES/12-574A01.
References
- Mata TM, Martins AA, Caetano NS. Microalgae for biodiesel production and other applications: a review. Renew Sustain Energy Rev. 2010;14:217–232. doi: 10.1016/j.rser.2009.07.020. [DOI] [Google Scholar]
- Chisti Y. Biodiesel from microalgae. Biotechnol Adv. 2007;25:294–306. doi: 10.1016/j.biotechadv.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Yu WL, Ansari W, Schoepp NG, Hannon MJ, Mayfield SP, Burkart MD. Modifications of the metabolic pathways of lipid and triacylglycerol production in microalgae. Microb Cell Fact. 2011;10:91. doi: 10.1186/1475-2859-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Sarkany N, Cui Y. Biomass and lipid productivities of Chlorella vulgaris under autotrophic, heterotrophic and mixotrophic growth conditions. Biotechnol Lett. 2009;31:1043–1049. doi: 10.1007/s10529-009-9975-7. [DOI] [PubMed] [Google Scholar]
- Wan M, Rosenberg JN, Faruq J, Betenbaugh MJ, Xia J. An improved colony PCR procedure for genetic screening of Chlorella and related microalgae. Biotechnol Lett. 2011;33:1615–1619. doi: 10.1007/s10529-011-0596-6. [DOI] [PubMed] [Google Scholar]
- Stephenson AL, Dennis JS, Howe CJ, Scott SA, Smith AG. Influence of nitrogen-limitation regime on the production by Chlorella vulgaris of lipids for biodiesel feedstocks. Biogeosciences. 2010;1:47–58. [Google Scholar]
- Radakovits R, Jinkerson RE, Darzins A, Posewitz MC. Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell. 2010;9:486–501. doi: 10.1128/EC.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packeiser H, Lim CY, Balagurunathan B, Wu JC, Zhao H. An extremely simple and effective colony PCR procedure for bacteria, yeasts, and microalgae. Appl Biochem Biotechnol. 2013;169:695–700. doi: 10.1007/s12010-012-0043-8. [DOI] [PubMed] [Google Scholar]
- Luhe AL, Gerken H, Tan L, Wu JC, Zhao H. Alcohol tolerance of Escherichia coli acrR and marR regulatory mutants. J Mol Cat B: Enzymatic. 2012;76:89–93. [Google Scholar]
- Cairney J, Xu N, Pullman GS, Ciavatta VT, Johns B. Natural and somatic embryo development in loblolly pine. Appl Biochem Biotechnol. 1999;77:5–17. doi: 10.1385/ABAB:77:1-3:5. [DOI] [Google Scholar]
- Niemi RM, Heiskanen I, Wallenius K, Lindstrom K. Extraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia. J Microbiol Methods. 2001;45:155–165. doi: 10.1016/S0167-7012(01)00253-6. [DOI] [PubMed] [Google Scholar]
- Müller FM, Werner KE, Kasai M, Francesconi A, Chanock SJ, Walsh TJ. Rapid extraction of genomic DNA from medically important yeasts and filamentous fungi by high speed cell disruption. J Clin Microbiol. 1998;36:1625–1629. doi: 10.1128/jcm.36.6.1625-1629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawley KP, Fawley MW. A simple and rapid technique for the isolation of DNA from microalgae. J Phycol. 2004;40:223–225. doi: 10.1111/j.0022-3646.2004.03-081.x. [DOI] [Google Scholar]
- Cao M, Fu Y, Guo Y, Pan JM. Chlamydomonas (Chlorophyceae) colony PCR. Protoplasma. 2009;235:107–110. doi: 10.1007/s00709-009-0036-9. [DOI] [PubMed] [Google Scholar]
- Schultz DJ, Craig R, Cox-Foster DL, Mumma RO, Medford JI. RNA isolation from recalcitrant plant tissue. Plant Mol Bio Rep. 1994;12:310–316. doi: 10.1007/BF02669273. [DOI] [Google Scholar]
- Snirc A, Silberfeld T, Bonnet J, Tillier A, Tuffet S, Sun JS. Optimization of DNA extraction from brown algae (Phaeophyceae) based on a commercial kit. J Phycol. 2010;46:616–621. doi: 10.1111/j.1529-8817.2010.00817.x. [DOI] [Google Scholar]
- Falcao VDR, Tonon AP, Oliveira MC, Colepicolo P. RNA Isolation method for polysaccharide rich algae: agar producing Gracilaria tenuistipitata(Rhodophyta) J Appl Phycol. 2008;20:9–12. doi: 10.1007/s10811-007-9174-7. [DOI] [Google Scholar]
- Lim DKY, Garg S, Timmins M, Zhang ESB, Thomas-Hall SR, Schuhmann H, Li Y, Schenk PM. Isolation and evaluation of oil-producing microalgae from subtropical coastal and brackish waters. PLoS ONE. 2012;7:e40751. doi: 10.1371/journal.pone.0040751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll JM. Review-methodolgies for transferring DNA into eukaryotic microalgae. Spanish J Agr Res. 2006;4:316–330. [Google Scholar]
- Gerken HG, Donohoe B, Knoshaug EP. Enzymatic cell wall degradation of Chlorella vulgaris and other microalgae for biofuels production. Planta. 2013;237:239–53. doi: 10.1007/s00425-012-1765-0. [DOI] [PubMed] [Google Scholar]
- Atkinson JAW, Gunning BES, John PCL. Sporopollenin in the cell wall of Chlorella and other algae: ultrastructure, chemistry, and incorporation of 14C-acetate, studied in synchronous cultures. Planta. 1972;107:1–32. doi: 10.1007/BF00398011. [DOI] [PubMed] [Google Scholar]
- Zheng H, Yin J, Gao Z, Huang H, Ji X, Dou C. Disruption of Chlorella vulgaris Cells for the release of biodiesel-producing lipids: a Comparison of grinding, ultrasonication, bead milling, enzymatic lysis, and microwaves. Appl Biochem Biotechnol. 2011;164:1215–1224. doi: 10.1007/s12010-011-9207-1. [DOI] [PubMed] [Google Scholar]
- Honjoh KI, Suga K, Shinohara F, Maruyama I, Miyamoto T, Hatano S, Iio M. Preparation of protoplasts from Chlorella vulgaris K-73122 and cell wall regeneration of protoplasts from C. vulgaris K-73122 and C-27. J Fac Agric Kyushu Univ. 2003;47:257–266. [Google Scholar]
- Corre G, Templier J, Largeau C, Rousseau B, Berkaloff C. Influence of cell wall composition on the resistance of two Chlorella species (chlorophyta) to detergents. J Phyco. 1996;32:584–590. doi: 10.1111/j.0022-3646.1996.00584.x. [DOI] [Google Scholar]
- Marmur J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol. 1961;3:208–218. doi: 10.1016/S0022-2836(61)80047-8. [DOI] [Google Scholar]
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:6. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale JW, Greenaway PJ. Preparation of chromosomal DNA from E. coli. Methods Mol Biol. 1985;2:197–200. doi: 10.1385/0-89603-064-4:197. [DOI] [PubMed] [Google Scholar]
- Halim R, Gladman B, Danquah MK, Webley PA. Oil extraction from microalgae for biodiesel production. Bioresour Technol. 2011;102:178–85. doi: 10.1016/j.biortech.2010.06.136. [DOI] [PubMed] [Google Scholar]
- HwangBo K, Son SH, Lee JS, Min SR, Ko SM, Liu JR, Choi DS, Jeong WJ. Rapid and simple method for DNA extraction from plant and algal species suitable for PCR amplification using a chelating resin Chelex 100. Plant Biotechnol Rep. 2010;4:49–52. doi: 10.1007/s11816-009-0117-4. [DOI] [Google Scholar]
- Berthold DA, Best BA, Malkin R. A rapid DNA preparation for PCR from Chlamydomonas reinhardtii and Arabidopsis thaliana. Plant Mol Biol Report. 1993;11:338–344. doi: 10.1007/BF02905336. [DOI] [Google Scholar]
- Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10:506–513. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A comparison of rapid DNA extraction from C. vulgaris harvested in stationary growth phase without boiling using individual buffers for PCR amplification. The buffers, 10 mM Tris/1 mM EDTA (TE), 0.2% Sodium dodecyl sulfate (SDS), Yeast Protein Extraction buffer (Y-PER) (Cat# 78990, Thermo Scientific), Chelex-100, and TE treated followed by PCI extraction were examined for their genomic DNA extraction efficiency for colony PCRs.