

Abstract
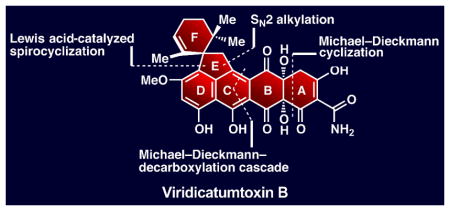
Will the real viridicatumtoxin B please stand up
Total synthesis of viridicatumtoxin B resulted in its structural revision and opens the way for analogue construction and biological evaluation of this complex tetracycline antibiotic. The highly convergent strategy employed allows for swift construction of the entire carbocyclic framework of the molecule.
Keywords: antibiotics, natural products, structural revision, tetracyclines, total synthesis
The discovery and development of antibiotics in the first half of the 20th century revolutionized medicine and led to a dramatic enhancement in life expectancy.[1] However, the emergence of drug-resistant bacterial strains caused by the extensive use of antibiotics in both humans and livestock risks rendering the world’s arsenal of antibiotics ineffective.[2] The development of novel antibacterial agents is therefore of high priority.
The nefarious molecular architectures of tetracyclines have challenged synthetic chemists for over six decades,[3] with impressive results being reported, most recently from the Myers group.[4] Structurally related to the tetracycline antibiotics are the viridicatumtoxins (1 – 3, Figure 1), which constitute an intriguing subclass of naturally occurring potent antibacterial agents. Originally isolated in 1973 from a Penicillium species,[5] viridicatumtoxin A (1) was structurally elucidated through the aid of an X-ray crystallographic analysis in 1976.[6] In 2008, Kim and coworkers reported the re-isolation of viridicatumtoxin A (1) as well as the isolation of viridicatumtoxin B, whose structure was assigned as 2 (Figure 1).[7] Finally, spirohexaline (3) was reported in early 2013.[8] Viridicatumtoxins A (1) and B (2) were shown to exhibit antibacterial activity against a variety of Gram-positive bacterial strains, including methicillin-resistant Staphylococcus aureus (MRSA) [MIC = 0.25 and 0.5 μg/mL, respectively; compare tetracycline (4, Figure 1) and vancomycin MIC = 8 and 0.25–1 μg/mL, respectively].[7] Their mode of action was implied to involve disruption of bacterial peptidoglycan biosynthesis through inhibition of UPP synthase.[9] Despite their impressive antibacterial activities, no synthetic approaches toward the viridicatumtoxins have been reported to date.
Figure 1.

Molecular structures of viridicatumtoxin A (1), B (2), spirohexaline (3), and tetracycline (4).
Inspired by the structural complexity, incomplete and curious structural assignment containing an epoxy hemiacetal structural motif, and potent antibiotic properties of viridicatumtoxin B (2), we embarked on its total synthesis. Our objectives were to fully elucidate the structure of viridicatumtoxin B (2) and establish the foundation for the synthesis and biological evaluation of designed analogues within this family of antibiotics. A number of distinct structural features add significantly to the challenge of synthesizing viridicatumtoxin B (2) as compared to the tetracyclines (e.g. 4, Figure 1). These features include viridicatumtoxin B’s EF spirosystem, its hindered quaternary carbon center at C15, its highly oxidized AB ring system, and the extensive substitution situated on the ABCD tetracyclic core of the molecule. Based on biosynthetic considerations[10] and the known structure of viridicatumtoxin A (1),[6] we tentatively assigned the relative configuration of viridicatumtoxin B (2) as shown in Figure 2. Our highly convergent strategy toward 2 was derived from the retrosynthetic analysis shown in Figure 2. Thus, disconnection of the indicated six carbon–carbon bonds through the reactions shown revealed four key building blocks: allylic bromide 5, cyclic anhydride 6, quinone monoketal 7 (known),[11] and isoxazole phenyl ester 8. Their construction and assimilation into the targeted molecule would involve three cyclizations (as indicated in Figure 2), the extrusion of carbon dioxide (from 6), and the rupture of the isoxazole ring (8).
Figure 2.

Retrosynthetic analysis of viridicatumtoxin B (2).
The constructions of building blocks 5, 6, and 8 are summarized in Scheme 1. The synthetic route to allylic bromide 5, the precursor for the EF spirocyclic ring system, commenced with a modification of a known, four-step sequence to convert geranic acid (9) to methyl ester allylic alcohol 10 (Scheme 1a).[12] Silylation (TBSCl, imid.) of 10 followed by ester reduction (DIBAL-H) produced allylic alcohol 11 in 91% overall yield. Low-temperature mesylation of the latter (MsCl, Et3N) followed by displacement of the resulting mesylate moiety with LiBr produced the desired fragment 5 in quantitative yield (Scheme 1a). Synthesis of the cyclic anhydride 6 commenced from intermediate 13 [available in two steps from 4-chlororesorcinol (12) by a literature procedure, Scheme 1b].[13] Selective mono-demethylation of 13 (directed by the nearby ester moiety on the aromatic ring) was achieved through the use of BBr3. Reprotection of the liberated phenolic group to give intermediate 14 was accomplished using Ag2O/BnBr (66% yield for 2 steps). bis-Saponification (aq. NaOH) and anhydride formation (Ac2O) produced the desired cyclic anhydride 6 (90% yield for 2 steps). The synthesis of isoxazole fragment 8 commenced from known Stork–Hagedorn isoxazole 16[3i,14] (Scheme 1c), which was prepared following a modified literature process involving sequential acylation of dimethylmalonate (AcCl, Et3N, MgCl2, 96% yield),[15] methylation of the resulting enol (Me2SO4, K2CO3, 54% yield), cyclization (H2NOH•HCl, NaOMe, 48% yield), and subsequent benzylation of the free hydroxyl group (BnBr, Ag2O, 67% yield). Saponification of the so-produced methyl ester then produced carboxylic acid 17 (aq. NaOH, 99% yield). Conventional esterification methods to prepare the phenyl ester 18 such as through the intermediacy of the corresponding chloride or the mixed anhydride failed, as did the use of peptide coupling reagents. The desired phenyl ester formation, however, could be achieved under Mitsunobu conditions (PPh3, PhOH, DIAD, 78% yield).[16] The need to install an electron-withdrawing—and ideally easily removable—group at the isoxazole methyl group was predicated on model systems of the AB-ring system; such a group was found to be required to facilitate a conjugate addition later. To this end, deprotonation of the methyl group of phenyl ester 18 (LiHMDS) and quenching of the resulting anionic species with TeocCl produced the desired fragment 8 (86% yield). Quinone monoketal 7 was readily available in one step following a literature procedure.[11]
Scheme 1.

a) Synthesis of allylic bromide 5; b) synthesis of cyclic anhydride 6; and c) synthesis of isoxazole 8. Reagents and conditions: a) a) H3PO4 (0.2 equiv), toluene, reflux, 90 min; b) MeI (3.9 equiv), K2CO3 (2.0 equiv), acetone, 25 °C, 15 h; c) mCPBA (1.2 equiv), CH2Cl2, 0→25 °C, 3 h; d) NaOMe (1.5 equiv), MeOH, reflux, 17 h, 70% yield for 4 steps; e) TBSCl (1.6 equiv), imidazole (2.0 equiv), CH2Cl2, 25 °C, 12 h; f) DIBAL-H (2.7 equiv), CH2Cl2, −78→0 °C, 70 min, 91% for 2 steps; g) Et3N (2.0 equiv), MsCl (1.7 equiv), CH2Cl2, −50 °C, 1 h; then LiBr (3.5 equiv), THF, −50→−20 °C, 1 h, quant.; b) a) MeI (4.0 equiv), K2CO3 (8.0 equiv), acetone, reflux, 15 h, 91%; b) NaH (6.0 equiv), DEM (4.0 equiv), THF, 0 °C, 2.5 h; then LDA (1.0 equiv), THF, 0 °C, 3.5 h, 65%; c) BBr3 (1.35 equiv), CH2Cl2, −78→25 °C, 30 min; d) BnBr (1.1 equiv), Ag2O (1.9 equiv), DMF, 25 °C, 15 h, 66% for 2 steps; e) NaOH (27 equiv), H2O:EtOH 5:7, reflux, 15 h; f) Ac2O (1.1 equiv), toluene, reflux, 1 h, 90% for 2 steps; c) a) MgCl2 (1.0 equiv), Et3N (2.0 equiv), AcCl (1.0 equiv), MeCN, 0→25 °C, 23 h, 96%; b) Me2SO4 (1.3 equiv), K2CO3 (1.3 equiv), DMF, 0→25 °C, 17 h, 54%; c) H2NOH•HCl (1.4 equiv), NaOMe (3.1 equiv), MeOH, 0→25 °C, 24 h, 48%; d) BnBr (1.2 equiv), Ag2O (1.5 equiv), DMF, 25 °C, 18 h, 67%; e) NaOH (1.9 equiv), H2O:EtOH 3:10, 25 °C, 3 h, 99%; f) PPh3 (1.05 equiv), PhOH (1.05 equiv), DIAD (1.05 equiv), THF, reflux, 3 h, 78%; g) LiHMDS (2.2 equiv), THF, −78 °C, 30 min; then TeocCl (2.2 equiv), −78 °C, 2 h, 86%. TBS = tert-butyldimethylsilyl, DIBAL-H = diisobutylaluminum hydride, Ms = methanesulfonyl, DEM = diethylmalonate, LDA = lithium diisopropylamide, DMF = dimethylformamide, DIAD = diisopropylazodicarboxylate, PhOH = phenol, THF = tetrahydrofuran, LiHMDS = lithium hexamethyldisilazide, Teoc = 2-(trimethylsilyl)ethoxycarbonyl.
With all four building blocks (5–8) in hand, the stage was now set for their union as shown in Scheme 2a. Annulation of cyclic anhydride 6 with quinone monoketal 7 to afford tricyclic compound 19 was achieved using a one-step Michael–Dieckmann/decarboxylation protocol (DBU, 65 °C, 41% yield). However, a two-step protocol involving exposure of 6 and 7 to NaH to induce anion generation and cycloaddition, followed by work-up and subsequent treatment with DBU to promote decarboxylation was preferred, as it proved more efficient in producing 19 (54% yield overall).[17] Reaction of ketal 19 with catalytic amounts of CSA caused extrusion of methanol, furnishing the corresponding anthrone (99% yield), whose alkylation with allylic bromide 5 proceeded under mild basic conditions (i.e. Na2CO3) to give intermediate 20 (77% yield, d.r. ca. 1:1). Much to our delight, ionization of the allylic TBS ether was readily achieved with catalytic quantities of BF3•OEt2, presumably forming highly stabilized but fleeting allylic cation 21, whose intramolecular trapping by the nearby electron-rich arene led to spirocycle 22 as a single diastereomer (73% yield). The connectivity and relative stereochemical relationships within pentacycle 22 were assigned by comparing its NMR spectroscopic data with those of the related compound 23 (10-methoxy as opposed to 10-benzyloxy, Scheme 2b), whose structure was secured through X-ray crystallographic analysis [m.p. = 114–116 °C (CHCl3/EtOAc), see ORTEP representation, Scheme 2b].[18]
Scheme 2.

a) Synthesis of heptacycle 27 and b) ORTEP representation of spirocycle 23 (thermal ellipsoids at 30% probability; grey = C, red = O, green = H). Reagents and conditions: a) 7 (3.0 equiv), NaH (3.0 equiv), THF, 0 °C, 45 min; then 25 °C, 1 h; b) DBU (5.0 equiv), toluene, 65 °C, 4.5 h, 54% for 2 steps; c) CSA (0.02 equiv), CH2Cl2, 25 °C, 30 min, 99%; d) 5 (1.1 equiv), Na2CO3 (10 equiv), DMF, 25 °C, 1 h, 77%, d.r. ca. 1:1; e) BF3•OEt2 (0.15 equiv), CH2Cl2, 0 °C, 20 min, 73%; f) PIDA (1.2 equiv), MeOH:CH2Cl2 1:1, 0→25 °C, 1 h; g) CSA (0.07 equiv), CH2Cl2, 0 °C, 5 min, 85% for 2 steps; h) PIDA (1.2 equiv), MeOH:CH2Cl2 10:1, 25 °C, 1.5 h, 90%; i) 8 (1.1 equiv), tBuOK (1.2 equiv), toluene, 25 °C, 15 min, 91%, d.r. ca. 2:1; j) TBAF (10 equiv), NH4F (20 equiv), degassed THF, 25 °C, 5 min, 86%. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, CSA = (±)-camphor-10-sulfonic acid, PIDA = iodobenzene diacetate; TBAF = tetra-n-butylammonium fluoride.
In order to ready the newly acquired pentacycle 22 for cyclization with isoxazole 8, and thus forge ring A of the growing molecule, the former was treated with PhI(OAc)2 (PIDA, phenolic oxidation) to produce B-ring enone 24, which, however, did not undergo the desired Michael–Dieckmann reaction with isoxazole 8. We hypothesized that this inertness was due to the adjacent electron-rich naphthalene ring, which decreases the electrophilicity of the enone moiety. Treatment of 24 with catalytic amounts of CSA (elimination of methanol, 85% yield for 2 steps) and subsequent exposure of the resulting C-ring quinomethide/B-ring p-methoxyphenol to PhI(OAc)2 (PIDA) gave quinomethide 25 (90% yield), whose superior electrophilicity now allowed its facile fusion with its intended nucleophilic partner. Thus, the anticipated Michael–Dieckmann reaction between highly reactive enone 25 and isoxazole 8 was achieved using a slight excess of potassium tert-butoxide in toluene affording heptacycle 26 and its isomer 15-epi-26 [91% yield, d.r. ca. 2:1 favoring the natural C15 epimer (26), vide infra]. The relative stereochemical assignment of C4 and C4a in 26 was based on the observed 3J4,4a = 10.0 Hz coupling constant, indicative of an H4,H4a anti-relationship. From this point on, the two C15 diastereomers were carried forward as a mixture through several additional steps until separation became convenient. The use of a phenyl ester for the Michael–Dieckmann reaction was critical for success in the union of enone 25 and isoxazole 8, in agreement with previous observations by White et al.[19] and Myers et al.[4d] Indeed, the methyl ester counterpart of isoxazole fragment 8 failed to enter this cyclization, leading instead to the initially formed conjugate addition adduct whose subsequently-attempted ring closure under a variety of basic conditions failed.
The next task, that of removing the Teoc group with concomitant decarboxylation, proved problematic, initially leading to decomposition[20a,b] or low yield[20c] of the desired product (i.e. 27) under various conditions. After considerable experimentation it was found that using a freshly prepared solution of TBAF buffered with NH4F[21] in degassed THF achieved the desired goal of removing the Teoc group in 86% yield. We hypothesized that these buffered conditions served to keep the C1,C12-β-diketone moiety of 26 in its fully protonated state, thereby allowing for smooth anion formation during the decarboxylation step. This transformation marked the successful completion of the entire carbon framework of viridicatumtoxin B (2).
Soon after arriving at compound 27, we realized that its conversion to the more advanced intermediates of obligatory higher oxygenation (at C4a and C12a) would present challenges. Among the most serious issues were the multitude of functional groups present in the substrate, a problem exacerbated by the scarcity of protons around the AB ring system which made spectroscopic analysis rather tedious, and the practical problems arising from the insoluble nature of the intermediates. Upon extensive experimentation, and as shown in Scheme 3a, hydroxylation of compound 27 at C12a was finally achieved with dimethyldioxirane (DMDO) in the presence of catalytic amounts of Ni(acac)2[22] at −78 °C in methylene chloride,[23] leading to the desired hydroxylated product in 50% yield after one recycle of the 40% recovered starting material. Reduction of the quinomethide moiety of the latter with NaCNBH3, as a soft hydride source, was then carried out to afford naphthalene derivative 28 (+ 15-epi-28) in 58% combined yield for the two diastereomers (28:15-epi-28 ca. 2:1). At this stage, the two isomers were separated chromatographically, with the natural diastereomer taken through the remaining steps. The assignment of the structural configurations of 28 and 15-epi-28 were based on comparisons of the NMR spectroscopic data of the two isomers with those of the 10-methoxy counterpart 29 (Scheme 3b), whose structure was unambiguously proven by X-ray crystallographic analysis [m.p. = 213–215 °C dec. (EtOAc), see ORTEP representation, Scheme 3b].[18]
Scheme 3.

a) Unsuccessful attempts to install the C4a hydroxyl group and synthesis of C1 hydroxy compound 33 and b) ORTEP representation of heptacycle 29 (thermal ellipsoids at 30% probability; grey = C, red = O, blue = N, green = H). Reagents and conditions: a) Ni(acac)2 (0.2 equiv), DMDO (5.1 equiv), CH2Cl2, −78→−60 °C, 6.5 h, 36%, 60% brsm, 50% after one recycle; b) NaCNBH3 (10 equiv), THF, −78→−60 °C, 90 min, 39% for 28, 19% for 15-epi-28, chromatographically separated; c) 2 N aq. HCl:THF 1:10, 25 °C, 5 h, quant.; d) NaBH(OAc)3 (1.2 equiv), EtOAc:acetone 1:1, 40 °C, 105 min, 47%; acac = acetylacetonate, DMDO = dimethyldioxirane.
From intermediate 28, a number of strategies and tactics were envisioned for the installment of the C4a hydroxyl group (see Scheme 3a). Our initial efforts were directed at the generation of enol ether 30 and its hydroxyl-directed α-epoxidation, followed by hydrolysis of the resulting epoxide to obtain compound 31. However, all attempts to remove a molecule of methanol from ketal 28 under a variety of both Lewis and Brønsted acidic conditions did not lead to any detectable enol ether product (30).[24] Faced with this predicament, we proceeded to hydrolyze the dimethyl ketal moiety of 28 under acidic conditions (aq. HCl, quant.) with the intention of converting the resulting triketone (32) to the desired product (31) through oxygenation of its enolate (at C4a). All attempts, however, to achieve this conversion were met with failure (see 32 to 31, Scheme 3a). The reluctance of these substrates to react as projected may be attributed to unwanted rearrangement pathways, including β-elimination/aromatization or ring-expansion/lactone-formation involving the C12a hydroxyl group.[4a,25]
Speculating that the C4a,C5 diol could be introduced through dihydroxylation, we next attempted to reduce the C5-ketone within 32 (see Scheme 3a) to its corresponding alcohol by employing 1,3-directed reduction reagents[26] such as NaBH(OAc)3, expecting that we may eventually be able to dehydrate the latter to the desired C4a,C5 olefinic bond within ring B. Surprisingly, however, treatment of triketone 32 with a slight excess of NaBH(OAc)3 in EtOAc:acetone (1:1) at 40 °C produced the 1,2-directed reduction product 33 (47% yield) instead, as evident from diagnostic HMBC correlations. This result seemingly stands in contrast to the recently reported 1,3-reduction of a similar system by Tatsuta et al.[27], but is most likely a result of the electron-withdrawing effect exerted by the adjacent isoxazole system which renders the C1 ketone highly electrophilic. Interestingly, the use of solvents other than EtOAc:acetone (1:1) resulted in intractable mixtures of reduction products. Recognizing the potential implications of this unexpected reduction, and building on the intelligence we had accumulated thus far on the reactivity of our system, we reasoned that a substrate such as 33 could be less prone to the suspected undesired reactions such as β-elimination followed by a thermodynamically downhill aromatization of ring A.
Following this reasoning, and in order to exploit the fortuitous formation of 33, the C1 hydroxyl group of the latter was silylated, affording TBS ether 34 (TBSOTf, 2,6-lutidine, 61% yield, Scheme 4). Much to our delight, the desired C4a hydroxylation was successfully performed by generating the trianion (35) of 34 with excess KHMDS at −78 °C and quenching it with the Davis oxaziridine reagent,[28] leading to C4a hydroxylated compound 36 (20% yield + 45% recovered starting material, 36% brsm). Key to the success of this reaction were the free phenol at C11, unprotected alcohol at C12a (which allows alkoxide formation, thereby preventing β-elimination), and protection of the secondary alcohol at C1. Structural deviations from this substrate resulted in no conversion, rearrangement pathways, or β-elimination of the C12a oxygen atom. The stereochemical outcome of this hydroxylation was supported by the revealing NOESY correlations of product 36 (see Supporting Information), and was in agreement with our expectations based on steric considerations (see structure 35, Scheme 4). Removal of the TBS group from compound 36 with HF•py then led to the expected hydroxy compound 37 (61% yield). From NMR studies, we surmised that compound 37 exists in equilibrium with its cyclic hemiacetal isomer 37′, with the two isomers interconverting slowly as evidenced from their unusually broad 1H NMR signals at ambient temperature. In support of this notion was the fact that the 1H NMR spectrum of this mixture acquired at −40 °C displayed two sets of signals. Finally, oxidation of 37/37′ with DMP[29] furnished the corresponding C1 ketone (66% yield), which was subjected to hydrogenolysis[4d] (two benzyl ethers and N–O bond; H2, Pd black, 98% yield) to afford compound (±)-38 as shown in Scheme 4. The physical properties of (±)-38 matched those reported for viridicatumtoxin B[7] except for the presence of the C5 13C NMR signal at 194.1 ppm for (±)-38 and the absence of the reported[7] C5 13C NMR signal at 116.4 ppm.[30] No spectroscopic evidence for the existence of an epoxy hemiacetal structural motif was observed in the 13C NMR spectrum of (±)-38, compelling us to revise the structure of natural viridicatumtoxin B to that of 38. Pleasantly, synthetic viridicatumtoxin B (38) crystallized in crystals suitable for X-ray analysis from CH2Cl2/EtOH (m.p. = 245–247 °C dec., see ORTEP representation, Figure 3), which provided unambiguous proof of its structure.[18]
Scheme 4.

C4a oxidation and completion of the synthesis. Reagents and conditions: a) TBSOTf (40 equiv), 2,6-lutidine (60 equiv), CH2Cl2, 0→25 °C, 1 h, 61%; b) KHMDS (3.4 equiv), THF, −78 °C, 1 h; then Davis ox. (3.9 equiv), −78 °C, 1.7 h, 20% + 45% recovered 34; c) HF•py (excess), MeCN, 0→50 °C, 25 h, 61%; d) DMP (3.0 equiv), DCE, 0→50 °C, 7.5 h, 66%; e) H2, Pd black (4.9 equiv), 1,4-dioxane:MeOH 1:1, 25 °C, 8 min, 98%; TBSOTf = tert-butyldimethylsilyl trifluoromethanesulfonate; KHMDS = potassium hexamethyldisilazide, Davis ox. = (±)-trans-2-(phenylsulfonyl)-3-phenyloxaziridine, py = pyridine, DMP = Dess–Martin periodinane, DCE = 1,2-dichloroethane.
Figure 3.

ORTEP representation of synthetic viridicatumtoxin B (38) (thermal ellipsoids at 30% probability; grey = C, red = O, blue = N, green = H).
In conclusion, the total synthesis of racemic viridicatumtoxin B has been achieved through a highly convergent synthetic strategy from four easily accessible building blocks. This accomplishment led to the revision of the originally assigned epoxy hemiacetal structure of viridicatumtoxin B (2, Figure 1) to its hydroxy ketone form (38, Scheme 4). Efforts currently in progress aim to achieve an enantioselective total synthesis of viridicatumtoxin B (38) and confirm its absolute configuration. The developed chemistry sets the stage for further advances to occur in the field, including the design, synthesis, and biological evaluation of analogues of the viridicatumtoxins as potential leads for drug discovery to combat bacterial infections.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health, USA (grant AI055475-11) and the Skaggs Institute of Research. Fellowships to C. N. (Feodor Lynen Research Fellowship, Alexander von Humboldt Foundation), C. R. H. H. (NSF Graduate Research Fellowship), and A. E. (Fundación Alfonso Martín Escudero) are gratefully appreciated. We thank Dr. R. K. Chadha for X-ray crystallographic assistance and Drs. D. H. Huang and L. Pasternack for NMR spectroscopic assistance. We thank Prof. W.-G. Kim for graciously providing scanned NMR spectra of natural viridicatumtoxin B.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D. Angew Chem. 2006;118:5194–5254. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:5072–5129. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Chen JS, Edmonds DJ, Estrada AA. Angew Chem. 2009;121:670–732. doi: 10.1002/anie.200801695. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:660–719. doi: 10.1002/anie.200801695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Walsh C. Antibiotics, Actions, Origins, Resistance. 1. ASM; Washington, D. C: 2003. [Google Scholar]; b) Rachakonda S, Cartee L. Curr Med Chem. 2004;11:775–793. doi: 10.2174/0929867043455774. [DOI] [PubMed] [Google Scholar]; c) Levy SB, Marshall B. Nature Medicine. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]; d) Nathan C. Nature. 2004;431:899–902. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- 3.a) Conover LH, Butler K, Johnston JD, Korst JJ, Woodward RB. J Am Chem Soc. 1962;84:3222–3224. [Google Scholar]; b) Muxfeldt H, Rogalski W. J Am Chem Soc. 1965;87:933–934. doi: 10.1021/ja01082a056. [DOI] [PubMed] [Google Scholar]; c) Gurevich AI, Karapetyan MG, Kolosov MN, Korobko VG, Onoprienko VV, Popravko SA, Shemyakin MM. Tetrahedron Lett. 1967;8:131–134. doi: 10.1016/s0040-4039(00)90501-x. [DOI] [PubMed] [Google Scholar]; d) Korst JJ, Johnston JD, Butler K, Bianco EJ, Conover LH, Woodward RB. J Am Chem Soc. 1968;90:439–457. [Google Scholar]; e) Muxfeldt H, Hardtmann G, Kathawala F, Vedejs E, Mooberry JB. J Am Chem Soc. 1968;90:6534–6536. doi: 10.1021/ja01025a063. [DOI] [PubMed] [Google Scholar]; f) Barton DHR, Magnus PD, Hase T. J Chem Soc (C) 1971:2215–2225. doi: 10.1039/j39710002215. [DOI] [PubMed] [Google Scholar]; g) Muxfeldt H, Haas G, Hardtmann G, Kathawala F, Mooberry JB, Vedejs E. J Am Chem Soc. 1979;101:689–701. doi: 10.1021/ja01025a063. [DOI] [PubMed] [Google Scholar]; h) Wasserman HH, Lu TJ, Scott AI. J Am Chem Soc. 1986;108:4237–4238. [Google Scholar]; i) Stork G, La Clair JJ, Spargo P, Nargund RP, Totah N. J Am Chem Soc. 1996;118:5304–5305. [Google Scholar]; j) Tatsuta K, Yoshimoto T, Gunji H, Okado Y, Takahashi M. Chem Lett. 2000:646–647. [Google Scholar]; k) Wzorek JS, Knöpfel TF, Sapountzis I, Evans DA. Org Lett. 2012;14:5840–5843. doi: 10.1021/ol302691j. [DOI] [PubMed] [Google Scholar]
- 4.a) Charest MG, Siegel DR, Myers AG. J Am Chem Soc. 2005;127:8292–8293. doi: 10.1021/ja052151d. [DOI] [PubMed] [Google Scholar]; b) Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. Science. 2005;308:395–398. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]; c) Brubaker JD, Myers AG. Org Lett. 2007;9:3523–3525. doi: 10.1021/ol071377d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sun C, Wang Q, Brubaker JD, Wright PM, Lerner CD, Noson K, Charest M, Siegel DR, Wang YM, Myers AG. J Am Chem Soc. 2008;130:17913–17927. doi: 10.1021/ja806629e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wright PM, Myers AG. Tetrahedron. 2011;67:9853–9869. doi: 10.1016/j.tet.2011.09.143. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kummer DA, Li D, Dion A, Myers AG. Chem Sci. 2011;2:1710–1718. doi: 10.1039/C1SC00303H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutchison RD, Steyn PS, Van Rensburg SJ. Toxicol Appl Pharmacol. 1973;24:507–509. doi: 10.1016/0041-008x(73)90057-4.We propose that viridicatumtoxin be renamed “viridicatumtoxin A.” It has been suggested that viridicatumtoxin A was isolated from Penicillium aethiopicum and not the originally reported Penicillium viridicatum; see: Frisvad JC, Filtenborg O. Mycologia. 1989;81:837–861.
- 6.a) Kabuto C, Silverton JV, Akiyama T, Sankawa U, Hutchison RD, Steyn PS, Vleggaar R. J Chem Soc, Chem Commun. 1976:728–729. [Google Scholar]; b) Silverton JV, Kabuto C, Akiyama T. Acta Cryst B. 1982;38:3032–3037. [Google Scholar]
- 7.Zheng CJ, Yu HE, Kim E-H, Kim WG. J Antibiot. 2008;61:633–637. doi: 10.1038/ja.2008.84. [DOI] [PubMed] [Google Scholar]
- 8.Inokoshi J, Nakamura Y, Hongbin Z, Uchida R, Nonaka K, Masuma R, Tomoda H. J Antibiot. 2013;66:37–41. doi: 10.1038/ja.2012.83. [DOI] [PubMed] [Google Scholar]
- 9.Koyama N, Inokoshi J, Tomoda H. Molecules. 2013;18:204–224. doi: 10.3390/molecules18010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) de Jesus AE, Hull WE, Steyn PS, van Heerden FR, Vleggaar R. J Chem Soc, Chem Commun. 1982:902–904. [Google Scholar]; b) Horak RM, Maharaj VJ, Marais SF, van Heerden FR, Vleggaar R. J Chem Soc, Chem Commun. 1988:1562–1564. [Google Scholar]; c) Zhou H, Li Y, Tang Y. Nat Prod Rep. 2010;27:839–868. doi: 10.1039/b911518h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chooi YH, Cacho R, Tang Y. Chem Biol. 2010;17:483–494. doi: 10.1016/j.chembiol.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Li Y, Chooi YH, Sheng Y, Valentine JS, Tang Y. J Am Chem Soc. 2011;133:15773–15785. doi: 10.1021/ja206906d. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chooi YH, Wang P, Fang J, Li Y, Wu K, Wang P, Tang Y. J Am Chem Soc. 2012;134:9428–9437. doi: 10.1021/ja3028636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prepared in one step from 4-methoxyphenol: Pelter A, Elgendy S. Tetrahedron Lett. 1988;29:677–680.
- 12.Kadás I, Árvai G, Horváth G. Org Prep Proced Int. 1998;30:79–85. [Google Scholar]
- 13.Bauta WE, Lovett DP, Cantrell WR, Jr, Burke BD. J Org Chem. 2003;68:5967–5973. doi: 10.1021/jo034165c. [DOI] [PubMed] [Google Scholar]
- 14.Stork G, Hagedorn AA., III J Am Chem Soc. 1987;100:3609–3611. [Google Scholar]
- 15.Rathke MW, Cowan PJ. J Org Chem. 1985;50:2622–2624. [Google Scholar]
- 16.Fitzjarrald VP, Pongdee R. Tetrahedron Lett. 2007;48:3553–3557. [Google Scholar]
- 17.Mechanistic studies of the one-pot procedure using DBU on related systems suggested a stepwise pathway. The use of a strong base (i.e. NaH or LDA) with homophthalic anhydrides and quinones (Tamura Diels–Alder reaction) is typically depicted as a concerted [4+2] reaction, but the synchronicity of the reaction has been questioned: Cox CD, Siu T, Danishefsky SJ. Angew Chem. 2003;115:5783–5787.Angew Chem Int Ed. 2003;42:5625–5629. doi: 10.1002/anie.200352591.González-López M, Shaw JT. Chem Rev. 2009;109:164–189. doi: 10.1021/cr8002714.
- 18.CCDC 941202, 941203, and 945867 contain the supplementary crystallographic data for compounds 23, 29, and 38, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Compounds 23 and 29 were prepared by similar routes to those described herein and will be reported in the full account of this work.
- 19.a) White JD, Nolen EG, Jr, Miller CH. J Org Chem. 1986;51:1150–1152. [Google Scholar]; b) White JD, Demnitz FWJ, Xu Q, Martin WHC. Org Lett. 2008;10:2833–2836. doi: 10.1021/ol8009732. [DOI] [PubMed] [Google Scholar]
- 20.a) Gioeli C, Balgobin N, Josephson S, Chattopadhyaya JB. Tetrahedron Lett. 1981;22:969–972. [Google Scholar]; b) Nicolaou KC, Reddy KR, Skokotas G, Sato F, Xiao XY, Hwang CK. J Am Chem Soc. 1993;115:3558–3575. [Google Scholar]; c) Denmark SE, Kobayashi T, Regens CS. Tetrahedron. 2010;66:4745–4759. doi: 10.1016/j.tet.2010.03.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fürstner A, Weintritt H. J Am Chem Soc. 1998;120:2817–2825. [Google Scholar]
- 22.Adam W, Smerz AK. Tetrahedron. 1996;52:5799–5804. [Google Scholar]
- 23.Gibert M, Ferrer M, Sánchez-Baeza F, Messeguer A. Tetrahedron. 1997;53:8643–8650. [Google Scholar]
- 24.Electron-rich α-methoxy styrenes are known to be very labile, even at near-neutral pH; see, for example: Richard JP, Williams KB. J Am Chem Soc. 2007;129:6952–6961. doi: 10.1021/ja071007k.
- 25.Scott AI, Yamaguchi E, Chung SK. Tetrahedron Lett. 1975;16:1369–1372. [Google Scholar]
- 26.Evans DA, Chapman KT, Carreira EM. J Am Chem Soc. 1988;110:3560–3578. [Google Scholar]
- 27.Tatsuta K, Fukuda T, Ishimori T, Yachi R, Yoshida S, Hashimoto H, Hosokawa S. Tetrahedron Lett. 2012;53:422–425. [Google Scholar]
- 28.Davis FA, Stringer OD. J Org Chem. 1982;47:1774–1775. [Google Scholar]
- 29.Dess DB, Martin JC. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 30.We thank Professor W.-G. Kim for kindly providing us with the 1H and 13C NMR spectra of natural viridicatumtoxin B obtained at 300 MHz and 226 MHz, respectively, from a small sample of natural material available. These spectroscopic data were in accord with those obtained by us at 600 MHz and 151 MHz for synthetic material, confirming the identity of the two samples (except for the racemic nature of ours). Furthermore, we noted certain discrepancies in the reported chemical shift values in ref [7] as compared to those actually found in the provided spectra of the natural product. In particular, the originally unreported carbonyl resonance near 194 ppm which we assigned as C5 was indeed present in the scanned authentic spectrum of the natural product. For the spectra of the natural and synthetic materials and further details, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.