

Abstract
Fludarabine is a nucleoside analog routinely used in conditioning regimens of pediatric allogeneic stem cell transplantation to promote stem cell engraftment. In children, it remains a challenge to accurately and precisely quantify the active intracellular triphosphate species of fludarabine in vivo, primarily due to limitations on blood volume and inadequate assay sensitivity. Here we report a liquid chromatography tandem mass spectrometry (LC-MS/MS) method for determination of fludarabine triphosphate in human peripheral blood mononuclear cells (PBMC). PBMC (~5 million cells) were collected and lysed in 1 mL 70% methanol containing 1.2 mM tris buffer (pH7.4). The lysate (80 uL) was mixed with internal standard (2-chloro-adenosine triphosphate, 150ng/mL, 20 µL) and injected onto an API5000 LC-MS/MS system. Separation was achieved on a hypercarb column (100 × 2.1 mm, 3 µm) eluted with 100mM ammonium acetate (pH9.8) and acetonitrile in a gradient mode at a flow rate of 0.4 mL/min. Multiple reactions monitoring (MRM) and electrospray ionization in negative mode (ESI−) were used for detection. The ion pairs 524.0/158.6 for the drug and 540.0/158.8 for the IS were selected for quantification and 524.0/425.7 used for confirmation. Retention time was 3.0 and 3.4 min for fludarabine triphosphate and the IS, respectively. The concentration range for the calibration curve was 1.52 to 76 nM. Our method is simple, fast, and has been successfully applied in a clinical dose-concentration study in children to quantify intracellular fludarabine in low volume clinical samples. The median concentration was 1.03 and 3.19 pmole/million PBMC at trough and peak time points, respectively. Fludarabine triphosphate is degraded in water within hours but relatively stable in 70% methanol-tris (1.2mM, pH7.4). One limitation is that the hypercarb column takes a longer time to equilibrate than conventional reverse phase columns, and peaks become broad and distorted if the column is not washed and stored properly.
Keywords: LC-MS/MS, fludarabine triphosphate, PBMC, hypercarb column
1. Introduction
Allogeneic hematopoietic cell transplantation (alloHCT) has become the standard-of-care treatment for a variety of pediatric diseases including leukemias, immunodeficiencies and hemoglobinopathies [1–10]. Although major advancements have been made in pediatric alloHCT, high rates of treatment-related death and engraftment failure remain prominent clinical problems. Fludarabine (9-β-D-arabinofuranosyl-2-fluoroadenine monophosphate), a nucleoside analog with potent antitumor and immunosuppressive properties, is routinely used in conditioning regimens of pediatric alloHCT to promote stem cell engraftment [11,12]. To date, no pharmacokinetic (PK) data are available for fludarabine in the setting of pediatric alloHCT: data critical to guide dosing and inform optimal treatment. The value of understanding therapeutic differences in drug response is dependent on the ability to define a dose-concentration relationship [13]. Unfortunately, barriers unique to the pediatric population can make performing PK studies difficult, particularly in infants and young children. Often drug-concentration trials are inhibited by limitations on blood volumes and inadequate assay sensitivity [14].
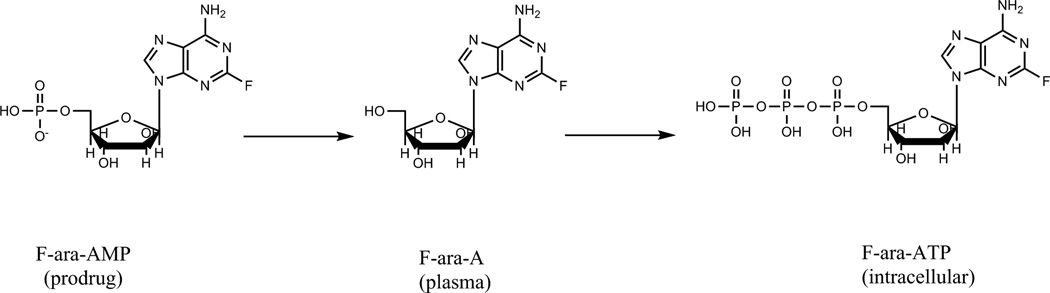
Administered as a prodrug, fludarabine phosphate undergoes rapid dephosphorylation to the systemically circulating compound, F-ara-A (Figure1). F-ara-A is then phosphorylated intracellularly by several kinases to active 9-beta-D-arabinofuranosyl-2-fluoroadenine triphosphate (F-ara-ATP), which is ultimately responsible for inhibition of DNA synthesis and RNA production and resulting in apoptosis [11,12,15]. Bioanalytical assay constraints and restrictions for blood volume, especially in pediatric patients, have limited the evaluation of intracellular drug concentrations of fludarabine in target tissues (e.g. peripheral blood mononuclear cells). In adults, methods to quantify the intracellular species have historically required blood volumes ranging from 10 to 60 ml of blood per sample and therefore are not useful for studies in children [16–21]. Additionally, two major challenges specific to the quantification of intracellular nucleoside triphosphate drugs are the separation of the active triphosphate anabolite and the sensitivity of the analytical method. Since these compounds contain phosphate groups, triphosphate anabolites exhibit a very low retention factor (k) on the widely used reverse phase analytical columns. Previously, ion pair reagents were used to improve separation but sensitivity was compromised [22–26]. Indirect methods (dephosphorylation of the separated triphosphate compound followed by quantification of the parent drug) have also been reported and previously used by our group, but these methods are labor intensive [27–30].
Figure 1.
Chemical structures of fludarabine and fludarabine triphosphate
Understanding relationships between dose and exposure of the active intracellular species is critical to optimizing fludarabine therapy in pediatric alloHCT recipients. Therefore, the aim of this study was to develop and validate a highly sensitivity method for the direct determination of intracellular F-ara-ATP in low volume clinical samples using liquid chromatography-tandem mass spectrometry (LC-MS/MS).
2. Experimental
2.1 Chemicals and reagents
Fludarabine triphosphate tetra-ammonium salt (purity by HPLC-UV peak height, 90.3%) was purchased from Moravek Biochemicals Inc. (Brea, CA, USA). The internal standard (IS) 2-Chloro-adenosine triphosphate tetrasodium hydrate (Cl-ara-ATP) was obtained from Sigma-Aldrich (Saint Louis, MO, USA).
Tris(hydroxymethyl)aminomethane (Tris), ammonium acetate (NH4AC) and ammonium hydroxide (all certified ACS reagents), acetonitrile (MeCN), methanol (MeOH), and other common solvents (HPLC grade) were purchased from Fisher Scientific Inc (Pittsburgh, PA, USA). Blank PBMC were obtained from the laboratory of Dr. Jay Levy of University of California at San Francisco and lysed in 70% MeOH-tris solution (1.2mM, pH7.4) at a concentration of 5×106 cells/mL. The blank PBMC extract was used for preparation of quality control (QC) samples.
Tris buffer (0.8M, pH7.4) was prepared by dissolving 0.968g tris(hydroxymethyl)aminomethane in 10 mL water and adjusting pH to 7.4 with concentrated HCl (12M); 70% MeOH-tris (1.2mM, pH7.4) solution was prepared by adding 35 mL MeOH and 75 µL 0.8 M tris buffer at pH7.4 to 15 mL water (HPLC grade). Mobile phase A (100 mM NH4AC, pH9.8) was prepared by adding 7.71g NH4AC in 985 mL water and adjusting pH to 9.8 with ammonium hydroxide (~15M, 15–17mL).
2.2 LC-MS/MS conditions
The AB Sciex API5000 was coupled with Shimadzu Prominence 20ADXR UFLC pumps and SIL-20ACXR autosampler and managed with the software Analyst® 1.5.1. The gases for the MS system were supplied by an LC-MS gas generator (Source 5000™, Parker Balston Inc., Haverhill, MA, USA). LC conditions were tested with UPLC BEH C18 (2.1×50 mm, 1.7µm, Waters Inc., Milford, MA, USA), Zorbax Extend C18 (2.1×150 mm, 5µm, Agilent Tech. Inc., Santa Clara, CA, USA), Sb-Aq (2.1×150 mm, 3.5µm, Agilent Tech. Inc., Santa Clara, CA, USA), and Synergi polar RP column (2.0×50 mm, 4µm, Phemonenex Inc. Torrance, CA, USA), and Hypercarb column (100 × 2.1 mm, 3 µm, Thermo Scientific Inc. Madison, WI, USA). The final optimized chromatographic separation was achieved on a Hypercarb column (100 × 2.1 mm, 3 µm) equipped with a guard column (10× 2.1 mm, 3 µm) from the same source (Thermo Scientific Inc. Madison, WI, USA). The LC setting was as follows: Solvent A was 100 mM ammonium acetate at pH9.8; solvent B was MeCN. The F-ara-ATP was eluted at a flow rate of 0.4 mL/min in a gradient program consisting of 15% solvent B (0 –1min), from 15 to 50% B (1– 4 min), from 50 to 80% B (4– 5 min), 80% B (5–7 min), 80%–15% B (7.0–7.1 min), and 15% B (7.1–9.5min). Retention times for F-ara-ATP and the IS were 3.0 min and 3.4 min, respectively. The divert valve was set to direct LC eluent to MS source at 2 min and to waste line at 5.9min. The MS conditions for F-ara-ATP and the IS were optimized by separate infusion of 400ng/mL F-ara-ATP or IS into the MS at a flow rate of 10 µL/min constantly while adjusting MS parameters to achieve maximal signal (Table 1). Data was processed with Analyst 1.5.1. (AB Sciex, Foster City, CA, USA).
Table 1.
Inter-day average back-calculated standard concentrations (n = 3)
| Nominal, ng/mL | 0.50 | 1.00 | 2.5 | 5.0 | 10.0 | 25 | 50 | Slope | Intercept | R |
|---|---|---|---|---|---|---|---|---|---|---|
| Mean, ng/mL | 0.540 | 1.01 | 2.34 | 4.84 | 10.1 | 21.9 | 53.3 | 0.0397 | −0.0007 | 0.996 |
| SD | 0.027 | 0.01 | 0.04 | 0.25 | 0.4 | 0.5 | 0.1 | 0.0025 | 0.0028 | 0.0008 |
| Precision (RSD,%) | 5.02 | 1.09 | 1.54 | 5.25 | 3.96 | 2.37 | 0.19 | 0.08 | ||
| Accuracy (% dev) | 7.9 | 0.93 | −6.40 | −3.13 | 0.63 | −12.40 | 6.60 |
2.3. Preparation of F-ara-ATP standard and quality control samples
Two sets of F-ara-ATP stock solutions at 1 mg/mL were prepared in water with separately weighed F-ara-ATP (in salt form). One solution was spiked in 70% MeOH-tris (1.2mM, pH7.4) solution to prepare standard samples and the other spiked in blank PMBC extract to prepare quality control (QC) samples. Calibration standard samples consisted of 0.5, 1, 2.5, 5, 10, 25, and 50 ng/mL, and QC samples consisted of 1.5, 15, and 40 ng/mL. Final concentrations were adjusted by purity (90.3%) and molecular weight (MW 593.2) and converted to molar concentration by the following formula: [nM]=[ng/mL]×0.903/0.593. The IS solution was prepared by dissolving Cl-ara-ATP in 70% MeOH-tris (1.2mM, pH7.4) to produce a final concentration of 150 ng/mL. The stock solutions, standards, QC samples, and the IS solution were aliquoted and stored at – 70°C freezer between uses.
2.4. Method validation
The assay was validated in terms of precision, accuracy, and stability. The method was considered acceptable if precision, expressed as relative standard deviation (RSD), was less than 15% for intra and inter-day variation. RSD for lower limit of quantitation (LLOQ) was set at <20%. RSD was calculated as follows: 100×SD/mean. The accuracy compared to the nominal value (% deviation) was required to be within 15% for intra- and inter-day comparison (% deviation for LLOQ was set at <20%). The LLOQ was determined with signal intensity being ≥ 5-fold blank response or having a RSD<20% if there is no blank signal. The calibration curve was required to have a correlation coefficient (R) of 0.995 or better. The back-calculated values for standards were required to be within 15% deviation of target with the exception of the LLOQ where 20% deviation was permitted.
Calibration curves were obtained by linear regression of the peak area ratio of F-ara-ATP to the IS (Y-axis) versus the nominal F-ara-ATP concentrations (X-axis) with a weighting factor of 1/x. Intra-day precision and accuracy were determined by analysis of at least 5 replicates of each QC sample at low (1.5 ng/mL), medium (15 ng/mL), and high (40 ng/mL) concentration levels along with a set of standards in one batch. The same procedure was repeated on 2 different days with new samples to determine inter-day precision and accuracy (total: n = 18 per concentration level). Precision was reported as RSD and accuracy as percent deviation from the nominal concentration (% deviation). Due to the limitation of the blank PBMC source during validation, the standards and QC samples were made in 70% MeOH-tris (1.2mM, pH7.4) for inter- and intra-day precision and accuracy experiments. QC samples were also prepared in blank PBMC extract (70% MeOH-tris solution containing 5×106 PBMC/mL) to determine LLOQ and evaluate matrix effect and stability. Matrix effect was evaluated by comparing peak area and peak area ratio of samples in PBMC extract with those of samples in 70% MeOH-tris (1.2mM, pH7.4). Carry-over was tested by injecting solvent or double blank PBMC extract after the upper limit of quantification (ULOQ).
The stability of F-ara-ATP was evaluated in PBMC extract at −70 °C for 6 months and at room temperature (22 °C) for overnight (12hr) by comparing to freshly spiked and processed QC samples. The processed samples in the autosampler vials (10 °C) were also tested for 24hr stability by comparing to the values determined immediately after processing. Each condition was tested with QC samples at low (1.5ng/mL) and high (40ng/mL) concentration levels in triplicate. The stability of stock solution was tested for 6 months in −70 °C freezer in water and over a day in room temperature (22 °C) in water and 70% MeOH-tris by comparing with the peak area from freshly prepared stock solution. The stability of IS was tested at room temperature (22 °C) over a day in water and 70%MeOH-tris.
2.5. Clinical sample collection and processing
Blood samples for intracellular quantification at each PK time point were collected in one (1) 4mL cell separation tube with sodium citrate (BD Vacutainer ® CPT Tube) and processed for recovery of PBMCs. CPT tubes were gently inverted within one minute of blood collection to thoroughly mix the anti-coagulant and keep upright at room temperature until centrifugation. All samples were processed within 45 minutes of collection time.
Using a centrifuge equipped with a horizontal rotor, CPT tubes were spun for 20 minutes at 1500×g. After centrifugation, the CPT tube was gently inverted to thoroughly disperse the PMBCs in the plasma layer without disruption of the gel and underlying red blood cells. From the cell suspension 20uL was removed and used for cell counting and viability determination. The remaining suspension was removed, placed into one 15mL conical plastic tube and centrifuged at 750×g for 10 minutes for pellet formation. Next the cells were washed with phosphate buffer saline (without calcium and magnesium) and centrifuge at 500×g for 15 minutes. The process of washing was repeated for a total of three times. After the final wash, the supernatant was discarded, the cells lysed with 1mL of solution containing 70% MeOH and 1.2mM tris buffer (pH7.4), vortexed and immediately store at −70°C until analysis.
2.6 Sample preparation
On the day of analysis, the lysed PBMC samples were thawed, vortexed and centrifuged along with QC samples and a set of calibrators. An 80 µL aliquot of the supernatant was taken and mixed with 20 µL IS (150 ng/mL Cl-ara-ATP) on a vortex mixer for 5 sec. An aliquot (50 µL) of the mixed solution was injected onto LC-MS/MS system. All samples needed to be processed immediately once they were thawed at room temperature.
2.7. Data analysis
The final concentration was reported as pmole/million cells calculated using the following formula:
[pmole/million cells]=[ng/mL]×(0.903/593)×1000×1mL/number of cells in millions. Statistical analysis was performed with Stata 12.1.
3. Results
3.1. LC-MS/MS optimization
Due to its high polarity, F-ara-ATP is easily dissolved in water, with minimal retention on a reverse phase column even when using a highly aqueous mobile phase. Among the columns tested, UPLC BEH C18 (2.1×50 mm, 1.7µm), Zorbax Extend C18 (2.1×150 mm, 5µm), Sb-Aq (2.1×150 mm, 3.5µm), and Synergi polar RP column (2.0×50 mm, 4µm) all yielded very short retention times (k<1). The ZIC®-pHILIC column (2.1×100 mm, 5µm) had a better retention factor but gave a broad peak and high baseline. The hypercarb column (2.1×100 mm, 3µm) had a capacity factor of ~4. Therefore, we chose the hypercarb column for further analytical development. With the commonly used mobile phase A including 0.1% formic acid, 1–10mM ammonium hydroxide, and 5–10 mM NH4AC, broad peaks were observed, and the chromatograms are not reproducible. Because a higher NH4AC concentration sharpened the peak and reduced the equilibration time, 100 mM NH4AC at pH 9.8 was used as solvent A. Solvent B was pure acetonitrile. Under these conditions, the retention time for F-ara-ATP was (3.0 ± 0.2) min. The dead volume (V0) for the hypercarb column (100 × 2.1 mm) was 0.22 mL (0.5 × 0.212 × 10 = 0.22) and the dead time was 0.55 min (0.22/0.4). Thus the k value was estimated to be 4.
Both electrospray ionization (ESI) modes (positive and negative) were tested. ESI− was slightly more sensitive than ESI+ and the background signal was very low in ESI− mode. Multiple reaction monitoring (MRM) mode with ion pair 524/159 for F-ara-ATP and 540/159 for the IS Cl-ara-ATP was selected for quantification. Ion pair 524/426 for F-ara-ATP was used for confirmation. The optimized compound-dependent MS parameters were as follows: for ion pair 524/159, DP, CE and CXP were −95v, −50v, − 15v, respectively; for 524/426, DP, CE and CXP were −95v, −32v, −23v, respectively; for the IS ion pair 540/159, DP, CE and CXP were −95v, −44v, −17v, respectively; EP was 10v for all ion pairs; the dwell times were 200ms for the quantification ion pair and IS and 80ms for the confirmation ion pair. The optimized instrument-dependent parameters were as follows: Turbo (Heater) set at 600 °C; Curtain gas (CUR), 30 psi; Nebulizer Gas (Gas 1), 60 psi; Auxiliary (turbo) Gas (Gas2), 50 psi; Collision-Activated Dissociation(CAD)Gas: 12; IonSprayVoltage, −4500 v. Representative product ion mass spectra of F-ara-ATP and the IS were shown in Figure 2.
Figure 2.

Representative product ion spectra of fludarabine triphosphate (upper panel) and the IS (lower panel).
3.2. Method validation
In 70% MeOH-tris (1.2mM, pH7.4), no baseline signal was observed with the API5000 system. The calibration curve was linear over the concentration range of 0.5 – 50 ng/mL with a mean correlation coefficient r of 0.9960 ± 0.0008 (Table 1). No carry over was observed. In PBMC extract, there was weak baseline signal at the retention time of F-ara-ATP. Based on the criteria that S/N ratio should be ≥5 and accuracy and precision should be ≤20%, The LLOQ in PBMC extract was determined to be 1ng/mL, corresponding to 1.52 nM after purity (90.3%) adjustment and conversion to mole concentration. Representative MRM ion chromatograms of blank PBMC extract and PBMC extract spiked with I.S. and F-ara-ATP at LLOQ were shown in Figure 3.
Figure 3.

Representative chromatograms of F-ara-ATP (upper panel) and Cl-ara- ATP (IS) (lower panel): grey line, blank PBMC extract; black line, QCsamples at LLOQ (1ng/mL) level.
The intra-day precisions (n = 6) over 3 days ranged from 1.4 to 4.7 at the three concentration levels (1.5, 15, and 40 ng/mL), and inter-day precisions were ranged from 6.8 to 8.3, all of them within 15%. The intra- and inter-day accuracy ranged from −11.1 to 9.2 and −1.7 to 1.9, respectively. (Table 2).
Table 2.
Intra- and inter-day precision and accuracy
| Intra-day | Inter-day | |||||
|---|---|---|---|---|---|---|
| Nominal, ng/mL | 1.5 | 15 | 40 | 1.5 | 15 | 40 |
| Mean, ng/mL | 1.33–1.61 | 13.6–15.7 | 36.7–43.7 | 1.48 | 14.8 | 40.8 |
| SD | 0.03–0.06 | 0.3 – 0.6 | 0.5 –1.6 | 0.12 | 1.0 | 3.2 |
| RSD, % | 2.3 – 3.6 | 3.3 – 4.7 | 1.4 – 3.5 | 8.3 | 6.8 | 7.9 |
| % dev. | −11.1- 7.1 | −9.3 - 4.3 | −8.4 – 9.2 | −1.7 | −1.4 | 1.9 |
| No. of samples | 6 | 6 | 6 | 18 | 18 | 18 |
Matrix effect was determined by comparing the peak area of F-ara-ATP in PBMC extract with that in 70%MeOH-tris solvent (Table 3). F-ara-ATP and IS were spiked into a blank PBMC extact in triplicate. At 0.5ng/mL matrix effect was over 120%. Although the IS corrected the matrix effect partially, the LLOQ was finally set at 1 ng/mL.
Table 3.
Matrix effect in PBMC extract (n=3).
| Conc (ng/mL) | 70% MeOH-tris (mean±SD) |
PBMC extract (mean±SD) |
ME,% | |
|---|---|---|---|---|
| 0.5 | Peak area | 0.403±0.006 | 0.498±0.011 | 123 |
| Ratio | 0.0220±0.0005 | 0.0255±0.0006 | 116 | |
| 1.5 | Peak area | 1.08±0.04 | 1.24±0.05 | 115 |
| ratio | 0.0589±0.0005 | 0.0642±0.0016 | 109 | |
| 40 | peak area | 28.7±1.5 | 28.1±0.7 | 98 |
| ratio | 1.62±0.05 | 1.39±0.05 | 86 |
Note: peak area is expressed as ×104 cps. Ratio is calculated with peak area of drug devided by peak area of IS.
The stability of F-ara-ATP in the tested conditions was showed in Table 4. Our results demonstrated that F-ara-ATP was stable in PBMC extract at −70 °C for at least 6 months, in autosampler at 10 °C for 24 hr, and in PBMC extract at room temperature (22 °C) for overnight (12hr). The stock solution was also stable at −70 °C for at least 6 months. Stock solution in water was not stable at room temperature. Over 15% degradation was observed on bench in a few hours. The IS Cl-ara-ATP was not stable in water but relatively stable in 70% MeOH-tris. Further investigation is needed to define the stable period at room temperature and long term stability in freezer.
Table 4.
Stability of F-ara-ATP (n=3 except for the * marked)
| Conditions | % remained | RSD, % | |
|---|---|---|---|
| Autosampler 24hr, 10 °C | |||
| Low(1.5ng/mL) | 102 | 2.4 | |
| High(40ng/mL) | 102 | 2.2 | |
| PBMC extract 6months, −70 °C | |||
| Low(1.5ng/mL) | 114 | 3.3 | |
| High(40ng/mL) | 102 | 2.8 | |
| PBMC extract 12hr, 22 °C | |||
| Low(1.5ng/mL) | 104 | 5.4 | |
| High(40ng/mL) | 98.6 | 8.1 | |
| Stock in water, 6months, −70 °C | 101 | 0.9 | |
| Stock in water, 1.5hr, 22 °C | 1.1* | ||
| Stock in 70%MeOH-tris, | |||
| 3hr | 94.1 | 1.6 | |
| 22hr | 69.4 | 1.4 | |
| IS in water 3hr, 22 °C | 10.5 | 7.0 | |
| IS in 70% MeOH-tris | |||
| 3hr | 90.6 | 1.4 | |
| 22hr | 70.3 | 3.8 | |
note: F-ara-ATP was degraded in water so quickly that triplicate analysis gave >15% variation.
The assay has been used in an ongoing clinical population PK study using a sparse sampling design: A total of 135 samples were analyzed, and 13 samples were below LLOQ. Two samples were above upper limit of quantification and dilution was performed. During the analysis, 16 QC samples at 3 concentration levels were analyzed along with the samples. The precision of the QC samples was 7.3 at 1.5 ng/mL (n=14), 8.6 at 15 ng/mL (n=13), and 7.0 at 40 ng/mL (n=14). Table 5 summarizes the F-ara-ATP time-concentration data from our clinical study evaluating fludarabine exposure in pediatric patients undergoing stem cell transplantation. Large inter-person variation was observed. At 2-hr after fludarabine infusion, the concentration is varied by 198-fold. At 24-hr after infusion, the variation is 74-fold. On the other hand, the difference of F-ara-ATP median concentration at 2hr and 24hr is only 3-fold, which is significantly small compared to that of plasma fludarabine levels at the same time points (~20-fold, unpublished data), though conversion of fludarabine to F-ara-ATP may present a significant time delay for actual intracellular F-ara-ATP concentration corresponding to the plasma fludarabine level. The intracellular F-ara-ATP accounts for ~1% of plasma fludarabine, assuming 0.4pL is the volume of a PMBC.
Table 5.
Median (range) concentrations of F-ara-ATP in the clinical samples of pediatric patients receiving fludarabine therapy
| Time post start of infusion (hours) |
No. of samples |
Median (range), pmol/million cells |
|---|---|---|
| 2 hours | 70 | 3.19 (0.10–19.75) |
| 24 hours | 65 | 1.03 (0.12–8.89) |
4. Discussion and Conclusion
Two daunting challenges for quantification of intracellular nucleoside triphosphate drugs are sensitivity and selectivity. Low intracellular drug level requires highly sensitive methods. LC-MS/MS is the choice of method for the required sensitivity. Conventional reverse phase columns hardly retain the triphosphate analogs due to the charged phosphate groups, and interference of endogenous nucleotides such as ATP are expected. Consequently, ion pairing agents are commonly added to the mobile phase to achieve better separation [22–26]. However, ion pairing agents also compromise ionization of the analytes in the MS source and thus lower sensitivity of MS detector. Indirect methods (dephosphorylation of the triphosphate drugs and then quantification of the parent drugs) require labor intensive sample preparation including solid phase extraction and dephosphorylation as our laboratory experienced in quantification of zidovudine triphosphate [28–30] and stavudine triphosphate (unpublished data) in PBMC.
In this report, we directly determine fludarabine triphosphate in PBMC extract by using a hypercarb column. With different retention mechanism from conventional reverse phase columns, hypercarb column showed good retention for the highly charged polar molecule F-ara-ATP and good selectivity over structure-similar analogs [31]. Thus, no ion-pairing agent was used in our method, and no interference was observed in the presence of 5000ng/mL ATP. The sample preparation was simple: The PBMC pellets were lysed with 70% MeOH-tris (1.2mM, pH7.4) and the lysed PBMC was directly injected onto the LC-MS/MS system. The LLOQ was 1.52nM for PBMC extract, corresponding to 61 (1.52×50µL×80/100) femtomole F-ara-ATP injected onto the LC-MS/MS system. The LLOQ in this method was about 20-fold lower than a previous method where LLOQ was 30nM [26], enabling us to quantify intracellular F-ara-ATP in PBMC from no more than 4mL blood. The assay has been used in a clinical pediatric population PK study using PBMC isolated from ~4mL blood. Over 90% of samples were above LLOQ. Although 5×106 PBMC cells were planned to be collected, some samples had less cell number. Among the 13 samples below LLOQ, 10 samples had less than 5x106 cells. The initial results suggest the method suitable for the intended pediatric study with a cell amount of ~5×106.
An interesting finding is that there is large inter-person variation on accumulation of F-ara-ATP in PBMC from pediatric patients: At 24hr after infusion of fludarabine, the inter-person difference of F-ara-ATP levels is 74-fold while at 2hr after fludarabine infusion the difference reaches to 198-fold. A previous study reported a 10.5 and 12.5- fold variation in CD4+ and CD8+ cells, respectively, from HCT patients using ex vivo incubation with 5 µM fludarabine for 4hr [20]. The large inter-person variation of F-ara-ATP exposure is attributed to the various clinical responses.
Another finding is the stability of F-ara-ATP in water, 70% MeOH-tris (1.2mM, pH7.4) and PBMC extract was in the order of water<70%MeOH-tris<PBMC extract. It is known that F-ara-ATP is not stable in water. Significant degradation observed in 1–2 hour. Whereas, no significant degradation of F-ara-ATP observed for at least 3 hr in 70% MeOH-tris(1.2mM, pH7.4), the lysis solvent for PBMC, and the best stability observed in PBMC extract. F-ara-ATP was found to be stable for at least 12 hr in PBMC extract.
A major limitation of using a hypercarb column is probably that it is hard to achieve repeatable chromatograms if the column is not washed properly after usage or change of the mobile phase additives. In the early phase of the method development, we tested over 10 different combinations of mobile phase solvents and additives, including 10mM NH4HCO3, 1–5mM NH4OH, 10–100mM NH4AC as mobile phase A and MeOH, acetonitrile, and acetonitrile-isopropanol (1:1, v/v) as mobile phase B. Peak fronting was observed when mobile phase A changed from 100mM NH4AC to 20mM NH4AC, and broad, distorted peak was observed when switching back to 100mM NH4AC from other solvents. It was found a long equilibration time was required when switching solvents. It seems high buffer concentration helps to sharpen the peak and obtain repeatable chromatograms. The column should be washed with 80% acetonitrile overnight and stored in 100% acetonitrile if not in use. The retention mechanism of hypercarb porous graphitic carbon is different from that of reverse phase columns. In addition hydrophobicity, the interaction of polar groups on the analytes with the graphite surface plays a key role in retention of the analytes on the hypercarb column. Probably for the same reason, additives in the mobile phase such as ammonium acetate and trifluoroacetic acid might affect the retention of analytes on the hypercarb column through induced polar-polar interaction and sometimes the additives could permanently change the column if they stick to the column surface.
In summary, the application of hypercarb column and tandem mass spectrometry resulted in a simple, fast and sensitive method for determination for F-ara-ATP in human PBMC to support PK study of F-ara-ATP in pediatric patients. In respect to high polarity of F-ara-ATP, this current method is advantageous over reported methods due to direct analysis without using ion-pairing agents.
Direct analysis of fludarabine triphosphate using hypercarb column
Highly sensitive with an LLOQ at 1.52nM.
The 1st report of a clinical application from pediatric patients.
Provided valuable data for the stability of fludarabine triphosphate.
Acknowledgments
We would like to thank Brian Phillips and Jeannine S McCune at Fred Hutchinson Cancer Research Center, Seattle, WA for providing application information on the hypercarb column. We also thank Dr. Lianxing Liu from the laboratory of Dr. Jay Levy of University of California at San Francisco for preparation of the blank PBMC.
Funding support: This work was supported by Center for AIDS Research (AI027763 and the Thrasher Research Fund. Additionally, support was also provided by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI Grant Number UL1 RR024131 and UCSF-CTSI Grant Number KL2 TR000143. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Abbreviation
- F-ara-ATP
fludarabine triphosphate
- Cl-ara-ATP
2-chloroadenosine triphosphate
- alloHCT
allogeneic hematopoietic cell transplantation
- PK
pharmacokinetic
- PBMC
peripheral blood mononuclear cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grigull L, Beilken A, Schrappe M, Das A, Luecke T, Sander A, Stanulla M, Rehe K, Sauer M, Schmid H, Welte K, Lukacs Z, Gal A, Sykora KW. Transplantation of allogeneic CD34-selected stem cells after fludarabine-based conditioning regimen for children with mucopolysaccharidosis 1H (M. Hurler) Bone Marrow Transplant. 2005;35:265–269. doi: 10.1038/sj.bmt.1704786. [DOI] [PubMed] [Google Scholar]
- 2.Smith FO, King R, Nelson G, Wagner JE, Robertson KA, Sanders JE, Bunin N, Emaunel PD, Davies SM. Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol. 2002;116:716–724. doi: 10.1046/j.0007-1048.2001.03333.x. [DOI] [PubMed] [Google Scholar]
- 3.Gluckman E, Rocha V, Ionescu I, Bierings M, Harris RE, Wagner J, Kurtzberg J, Champagne MA, Bonfim C, Bittencourt M, Darbyshire P, Fernandez MN, Locatelli F, Pasquini R. Results of unrelated cord blood transplant in fanconi anemia patients: risk factor analysis for engraftment and survival. Biol Blood Marrow Transplant. 2007;13:1073–1082. doi: 10.1016/j.bbmt.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JE, Eapen M, MacMillan ML, Harris RE, Pasquini R, Boulad F, Zhang MJ, Auerbach AD. Unrelated donor bone marrow transplantation for the treatment of Fanconi anemia. Blood. 2007;109:2256–2262. doi: 10.1182/blood-2006-07-036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, Chen M, Riddle MJ, Osborn MJ, Lund T, Dolan M, Blazar BR, Tolar J. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629–639. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verneris MR, Eapen M, Duerst R, Carpenter PA, Burke MJ, Afanasyev BV, Cowan MJ, He W, Krance R, Li CK, Tan PL, Wagner JE, Davies SM. Reduced- intensity conditioning regimens for allogeneic transplantation in children with acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2010;16:1237–1244. doi: 10.1016/j.bbmt.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yabe M, Sako M, Yabe H, Osugi Y, Kurosawa H, Nara T, Tokuyama M, Adachi S, Kobayashi C, Yanagimachi M, Ohtsuka Y, Nakazawa Y, Ogawa C, Manabe A, Kojima S, Nakahata T Japanese Childhood MDS Study Group. A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant. 2008;12:862–867. doi: 10.1111/j.1399-3046.2008.00931.x. [DOI] [PubMed] [Google Scholar]
- 8.Awaya T, Kato T, Niwa A, Hiramatsu H, Umeda K, Watanabe K, Shibata M, Yamanaka Y, Maruya E, Saji H, Nakahata T, Adachi S. Successful cord blood transplantation using a reduced-intensity conditioning regimen for advanced childhood- onset cerebral adrenoleukodystrophy. Pediatr Transplant. 2011;15:E116–E120. doi: 10.1111/j.1399-3046.2009.01188.x. [DOI] [PubMed] [Google Scholar]
- 9.Sauer M, Meissner B, Fuchs D, Gruhn B, Kabisch H, Erttmann R, Suttorp M, Beilken A, Luecke T, Welte K, Grigull L, Sykora KW. Allogeneic blood SCT for children with Hurler's syndrome: results from the German multicenter approach MPS-HCT 2005. Bone Marrow Transplant. 2009;43:375–381. doi: 10.1038/bmt.2008.328. [DOI] [PubMed] [Google Scholar]
- 10.Ringden O, Remberger M, Svenberg P, Svahn BM, Dahllof G, Gustafsson B, Hassan Z, Omazic B, Uzunel M, Aschan J, Barkholt L, Winiarski J, Ljungman P, Mattsson J. Fludarabine-based disease-specific conditioning or conventional myeloablative conditioning in hematopoietic stem cell transplantation for treatment of non-malignant diseases. Bone Marrow Transplant. 2007;39:383–388. doi: 10.1038/sj.bmt.1705602. [DOI] [PubMed] [Google Scholar]
- 11.Dow LW, Bell DE, Poulakos L, Fridland A. Differences in metabolism and cytotoxicity between 9-beta-D arabinofuranosyladenine and 9-beta-D-arabinofuranosyl-2-fluoroadenine in human leukemic lymphoblasts. Cancer Res. 1980;40:1405–1410. [PubMed] [Google Scholar]
- 12.Brockman RW, Cheng YC, Schabel FM, Jr, Montgomery JA. Metabolism and chemotherapeutic activity of 9-beta-D-arabinofuranosyl-2-fluoroadenine against murine leukemia L1210 and evidence for its phosphorylation by deoxycytidine kinase. Cancer Res. 1980;40:3610–3615. [PubMed] [Google Scholar]
- 13.Barrett JS, Della Casa Alberighi O, Laer S, Meibohm B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin. Pharmacol. Ther. 2012;92:40–49. doi: 10.1038/clpt.2012.64. [DOI] [PubMed] [Google Scholar]
- 14.Meibohm B, Laer S, Panetta JC, Barrett JS. Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J. 2005;7:E475–E487. doi: 10.1208/aapsj070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plunkett W, Huang P, Gandhi V. Metabolism and action of fludarabine phosphate. Semin Oncol. 1990;17:S3–S17. [PubMed] [Google Scholar]
- 16.Danhauser L, Plunkett W, Liliemark J, Gandhi V, Iacoboni S, Keating M. Comparison between the plasma and intracellular pharmacology of 1-beta-D- arabinofuranosylcytosine and 9-beta-D-arabinofuranosyl-2-fluoroadenine 5'-monophosphate in patients with relapsed leukemia. Leukemia. 1987;1:638–643. [PubMed] [Google Scholar]
- 17.Danhauser L, Plunkett W, Keating M, Cabanillas F. 9-beta-D-arabinofuranosyl-2-fluoroadenine 5'-monophosphate pharmacokinetics in plasma and tumor cells of patients with relapsed leukemia and lymphoma. Cancer Chemother Pharmacol. 1986;18:145–152. doi: 10.1007/BF00262285. [DOI] [PubMed] [Google Scholar]
- 18.Malspeis L, Grever MR, Staubus AE, Young D. Pharmacokinetics of 2-F-ara-A (9-beta-D-arabinofuranosyl-2-fluoroadenine) in cancer patients during the phase I clinical investigation of fludarabine phosphate. Semin Oncol. 1990;17:s18–s32. [PubMed] [Google Scholar]
- 19.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet. 2002;41:93–103. doi: 10.2165/00003088-200241020-00002. [DOI] [PubMed] [Google Scholar]
- 20.Woodahl EL, Wang J, Heimfeld S, Sandmaier BM, O'Donnell PV, Phillips B, Risler L, Blough DK, McCune JS. A novel phenotypic method to determine fludarabine triphosphate accumulation in T-lymphocytes from hematopoietic cell transplantation patients. Cancer Chemother Pharmacol. 2009;63:391–401. doi: 10.1007/s00280-008-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodahl EL, Wang J, Heimfeld S, Sandmaier BM, McCune JS. Intracellular disposition of fludarabine triphosphate in human natural killer cells. Cancer Chemother Pharmacol. 2009;63:959–964. doi: 10.1007/s00280-008-0829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becher F, Schlemmer D, Pruvost A, Nevers MC, Goujard C, Jorajuria S, Guerreiro C, Brossette T, Lebeau L, Créminon C, Grassi J, Benech H. Development of a direct assay for measuring intracellular AZT triphosphate in humans peripheral blood mononuclear cells. Anal Chem. 2002;74:4220–4227. doi: 10.1021/ac020144r. [DOI] [PubMed] [Google Scholar]
- 23.Becher F, Pruvost A, Goujard C, Guerreiro C, Delfraissy JF, Grassi J, Benech H. Improved method for the simultaneous determination of d4T, 3TC and ddl intracellular phosphorylated anabolites in human peripheral-blood mononuclear cells using high-performance liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:555–565. doi: 10.1002/rcm.605. [DOI] [PubMed] [Google Scholar]
- 24.Compain S, Durand-Gasselin L, Grassi J, Benech H. Improved method to quantify intracellular zidovudine mono- and triphosphate in peripheral blood mononuclear cells by liquid chromatography-tandem mass spectrometry. J Mass Spectrom. 2007;42:389–404. doi: 10.1002/jms.1176. [DOI] [PubMed] [Google Scholar]
- 25.Chen P, Liu Z, Liu S, Xie Z, Aimiuwu J, Pang J, Klisovic R, Blum W, Grever MR, Marcucci G, Chan KK. A LC-MS/MS method for the analysis of intracellular nucleoside triphosphate levels. Pharm Res. 2009;26:1504–1515. doi: 10.1007/s11095-009-9863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalhorn TF, Ren AG, Slattery JT, McCune JS, Wang J. A highly sensitive high-performance liquid chromatography-mass spectrometry method for quantification of fludarabine triphosphate in leukemic cells. J Chromatogr B. 2005;820:243–250. doi: 10.1016/j.jchromb.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 27.Bushman LR, Kiser JJ, Rower JE, Klein B, Zheng JH, Ray ML, Anderson PL. Determination of nucleoside analog mono-, di-, and tri-phosphates in cellular matrix by solid phase extraction and ultra-sensitive LC-MS/MS detection. J Pharm Biomed Anal. 2011;56:390–401. doi: 10.1016/j.jpba.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peter K, Lalezari JP, Gambertoglio JG. Quantification of zidovudine and individual zidovudine phosphates in peripheral blood mononuclear cells by a combined isocratic high performance liquid chromatography radioimmunoassay method. J Pharm Biomed Anal. 1996;14:491–499. doi: 10.1016/0731-7085(95)01649-x. [DOI] [PubMed] [Google Scholar]
- 29.Thevanayagam LN, Jayewardene AL, Gambertoglio JG. The stability of intracellular zidovudine and its anabolites in extracts of peripheral blood mononuclear cells (PBMCs) J Pharm Biomed Anal. 2000;22:597–603. doi: 10.1016/s0731-7085(00)00238-7. [DOI] [PubMed] [Google Scholar]
- 30.Aweeka FT, Rosenkranz SL, Segal Y, Coombs RW, Bardeguez A, Thevanayagam L, Lizak P, Aberg J, Watts DH. NIAID AIDS Clinical Trials Group, The impact of sex and contraceptive therapy on the plasma and intracellular pharmacokinetics of zidovudine. AIDS. 2006;20:1833–1441. doi: 10.1097/01.aids.0000244202.18629.36. [DOI] [PubMed] [Google Scholar]
- 31.Thermofisher Scientific Inc. [Last accessed on July 10, 2013];Chromatography Resource Center. http://www.separatedbyexperience.com/applications/app.aspx?id=lc525. [Google Scholar]