

Abstract
Wegener's granulomatosis (WG) is a systemic vasculitis affecting small and medium-sized vessels with granulomatous formation. Though it is known for respiratory tract and kidney involvement, neurologic manifestation has been also reported. Herein we report a patient who suffered pansinusitis with multiple lower cranial nerve palsies but reached remission by immunosuppressant after the diagnosis of WG. A 54-yr-old female visited with headache, hearing difficulty, and progressive bulbar symptoms. She experienced endoscopic sinus surgeries due to refractory sinusitis. Neurologic examination revealed multiple lower cranial nerve palsies. Vasculitic markers showed no abnormality. Nasal biopsy revealed granulomatous inflammation and vasculitis involving small vessels. Given cyclophosphamide and prednisolone, her symptoms were prominently improved. WG should be considered in the patient with multiple cranial nerve palsies, especially those with paranasal sinus disease. Because WG can be lethal if delayed in treatment, prompt immunosuppressant is warranted after the diagnostic tissue biopsy.
Keywords: Wegener Granulomatosis, Cranial Nerve Diseases, Refractory Sinusitis, c-ANCA Negative
INTRODUCTION
Wegener's granulomatosis (WG) is a rare autoimmune disease of localized granulomatous inflammation of the upper and lower respiratory tract and systemic small and medium-sized vasculitis associated with antineurotrophil cytoplasmic antibody (ANCA). The systemic vasculitic form of the disease can be lethal when renal or pulmonary involvement leads to alveolar hemorrhage related respiratory failure or necrotizing glomerulonephritis. Nevertheless, most common manifestation of the disease is the localized form of granulomatous inflammation of upper respiratory tract, especially around head and neck. The upper respiratory tract symptom including rhino-sinusitis or otitis media is the most frequent initial presentation with more than 75% prevalence (1). The neurologic symptom of the disease is mainly of peripheral nervous system by vasculitis, and much infrequently central nervous system (CNS) manifestations of seizure, cerebrovascular event or pachymeningitis have been documented (2, 3). Herein we present a case of biopsy proven ANCA-negative WG patient presented with progressive multiple lower cranial nerve palsies from VII to XII and extensive necrotizing sinu-oto-mastoiditis but have reached sustainable remission state with timely immunosuppressive treatment.
CASE DESCRIPTION
A 54-yr-old female was referred for neurologic consultation due to progressive dysarthria, dysphagia and left facial palsy for past 5 month in January 2011. She had no underlying medical illness such as diabetes mellitus, and had underwent first functional endoscopic sinus surgery (FESS) for bilateral maxillary sinusitis 4 yr ago. She had remained tolerable after the surgery for about 3. 5 yr then the symptoms of headache, hearing difficulty, dysarthria and dysphagia slowly emerged and progressed for about 6 months. Under the diagnosis of recurrent sinusitis involving frontal, ethmoid, sphenoid and maxillary sinuses, second FESS and broad-spectrum antibiotics were treated. Although these surgical and medical treatments were adequately performed, her symptoms worsened as aggravated hearing problem, severe bulbar symptoms and newly developed left facial palsy. The first brain MRI with using contrast enhancement was performed, only to reveal still existing bilateral otomastoiditis and extensive sinusitis (Fig. 1A-D). Cerebrospinal fluid (CSF) had no cell counts and normal protein level. The empiric steroid therapy (methysol 2 mg/kg/day) without definite diagnosis has somewhat stabilized these series of aggravating symptoms. At then, she was referred to our clinic for more detailed evaluation.
Fig. 1.

Brain MRI. (A to D) The first MRI. Initial outside brain MRI was not remarkable, except bilateral mastoiditis and extensive sinusitis. (E to H) The second MRI. Repeated brain MRI after 5 months shows new meningeal thickening with enhancement around the tentorium cerebelli, inferior aspect of frontotemporal lobe adjacent to sinuses and nasopharynx. Also diffuse sinusitis in bilateral frontal, ethmoid, sphenoid, and maxillary sinuses were still existed.
In January 2011 when she was admitted in our clinic, all the vital signs including body temperature were stable. Accompanied by severe bulbar symptoms of dysarthria and dysphagia, she complained of constitutional symptoms such as general weakness, poor oral intake and weight loss of up to 12 kilograms during past 6 months. Neurologic examination revealed various lower cranial nerve palsies of both sides, which were overall more prominent on left side than right side. Demonstrated clinical manifestations and positive neurologic findings are; facial diplegia, progressive hearing difficulty resulting in near-deafness, bilateral tinnitus and hyperacusis, right deviation of uvula, bilateral decreased gag reflexes, flaccid dysarthria with breathy voice and hypernasality, dysphagia especially in liquid food, left vocal cord palsy, left sternocleidomastoid muscle weakness, impaired tongue protrusion and giggling, and fasciculation with slight atrophy on the left side tongue. In contrast to these various lower cranial nerve involvements, there was no afferent pupillary defect, and no limitation in extraocular eye movements. Otolaryngology report was given as extensive sinusitis involving bilateral frontal, sphenoid, and maxillary sinuses with nasal cartilage erosion and bilateral chronic otitis media.
Laboratory studies revealed increased C-reactive protein (CRP) of 29.90 mg/dL (normal range 0-0.3 mg/dL), increased erythrocyte sedimentation rate (ESR) of 67 mm/hr, mild anemia (hemoglobin 10.8 g/dL), but no eosinophila and mild leukocytosis (12,800/µL). Renal function (BUN 15 mg/dL, creatinine 0.6 mg/dL, glomerular filtration rate 111 mL/min) and microscopic urine analysis were normal. Serology test for anti-Ro/SSA, anti-La/SSB, antinuclear, anticardioplin antibody, and ANCA were negative. Neither immunofluorescence method nor direct enzyme-linked immunosorbent assay method for proteinase 3 and myeloperoxidase antigens could detect ANCA. FANA screening and VDRL were also negative, and angiotensin converting enzyme was within normal as 28 U/L (normal range 18-55 U/L). Rheumatoid factor was minimally elevated up to 20.4 IU/mL (normal range 10-18 IU/mL). Serum folate, vitamin B1 and vitamin B12 were measured as normal level. Tumor markers including AFP, CEA, CA19-9, and CA125 were normal.
Chest CT revealed no hilar enlargement or parenchymal lung lesion. Neck CT showed high density lesions in the nasopharynx with enhancement, penetrating the skull base and extensive sinusitis expanding into orbit and retromaxillary fat-pad (Fig. 2). Whole body positron emission tomography (PET) showed hypermetabolic lesion in bilateral tonsil area with multiple lymph nodes in level II/III of neck and also in bilateral frontal, ethmoid, sphenoid, and maxillary sinuses (Fig. 3).
Fig. 2.
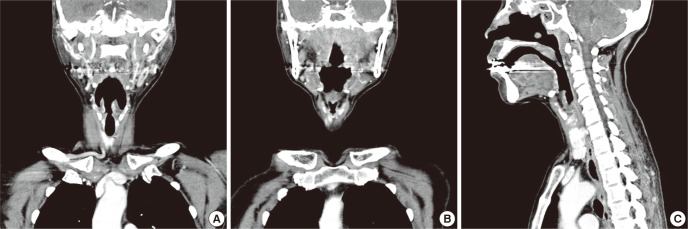
Neck CT. (A to C) Quite extensive sinusitis expanding to orbit and retromaxillary fat pad is observed. Note that massive swollen lesions of nasopharynx with positive enhancement penetrated the skull base.
Fig. 3.

18F-FDG PET. (A to C) Prominent hypermetabolism in bilateral tonsil area of nasopharynx and multiple lymph nodes of neck level II and III were shown. (D-F) Multiple sinusitis were also demonstrated as hypermetabolic lesions. 18F-FDG PET: 18F-fluorodeoxyglucose positron-emission tomography.
In the facial nerve conduction study, the compound muscle action potential (CMAP) amplitude of left facial nerve was decreased to 60% that of right side. Blink reflex test showed delayed ipsilateral R1 and R2, but normal contralateral R2 responses on both supraorbital nerve stimulations. These electrophysilogical findings were indicative of bilateral facial neuropathies, more severe in the left side.
Endoscopic biopsy was performed twice due to failure to obtain pathologic confirmation at first biopsy. The first biopsy specimen was obtained from nasopharyeal wall layer through a punch biopsy under local anesthesia, but the second endoscopic biopsy was performed far deeper layer of retropharyngeal area under general anesthesia. Tissues obtained from the second biopsy showed granulomatous inflammation and vasculitis involving small sized blood vessels with ischemic necrosis and destruction of cartilage (Fig. 4). Period acid-Schiff, Gomori methenamine silver and acid-fast bacilli stains showed no histological evidence of fungal organism or mycobacterium. Immunostains for CD3 and CD56 had no abnormal infiltration of natural killer T cells. Ebstein-Barr virus-encoded RNA in situ hybridization resulted in negative findings.
Fig. 4.
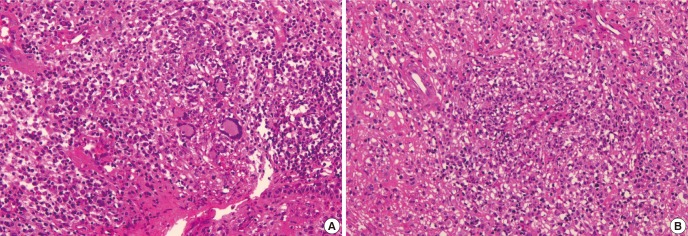
Nasopharygenal biopsy. (A) The nasopharyngeal tissue shows dense polymorphous inflammatory cell infiltrates composed of mature lymphocytes, plasma cells, histiocytes, eosinophils and neutrophils. There are few multinucleated giant cells which represent granulomatous component of the disease. (B) Granulomatous inflammation involving vessel wall and lumen (vasculitis) with necrosis is evident.
All the clinical manifestations, laboratory and imaging studies indicated ANCA negative WG involving otolaryngeal space, upper respiratory tract and multiple lower cranial nerves. She was transferred to rhematology department and began therapy with steroid (methysol 1 mg/kg/day) and cyclophosphamide (2 mg/kg/day). Within the ten days of these treatments, the patient stated that her sufferings of headache and constitutional symptoms were prominently diminished. The hearing difficulty, which compelled her to wear a hearing aid, improved dramatically leaving no more need of the device. After three months of combination therapy of tapering steroid (prednisolone 0.6 mg/kg/day) and steady cyclophosphamide (2 mg/kg/day), the patient restored to almost her normal bulbar function and removed nasogastric tube for oral intake. Further repeated ANCA study was negative. Brain MRI was re-evaluated with a time interval of five months from the first MRI study, and showed persistent sinusitis of both frontal, ethmoid, sphenoid, and maxillary sinuses, in despite of marked improvement of clinical symptoms. Moreover, focal thickening with enhancement of the meninges around the area of tentorium cerebelli, inferior aspect of frontotemporal lobe adjacent to sinuses and nasopharynx were newly observed (Fig. 1E-H). Clinical significance of these imaging findings was somewhat debatable because previous neurologic defects such as dysarthria, dysphagia, and hearing impairment had improved remarkably. However, re-evaluated CRP was still high as 5.75 mg/dL, which implied for further immunotherapy. Ultimately, potent immunotherapy consisting of high dose steroid (methysol 2 mg/kg/day) and cyclophosphamide (3 mg/kg/day) was performed. Three weeks later, the follow up CRP fall to 0.39 mg/dl and ESR normalized as 3 mm/hr. Taking only cyclophosphamide (0.25 mg/kg/day) after tapering steroid, she is still in a remission state for more than a year.
DISCUSSION
The initial clinical symptoms and neurologic signs of the patient have shown bilateral multiple lower cranial neuropathies with chronic, insidious and progressive course, coexisting with chronic sinusitis, rhinitis and otitis media refractory to usual treatments. These symptoms and signs requires careful differential diagnosis within chronic inflammatory disease such as sarcoidosis, systemic vascular conditions including WG and antiphospholid syndrome, infectious disease such as tuberculosis or fungal infection, diffuse infiltrative lymphoma mainly involving in head and neck, and meningitis carcinomatosis secondary to unknown primary malignancy. A series of CSF analysis, laboratory test, brain MRI, neck CT and whole-body PET were strongly suggestive of diffuse infiltrating malignancy or vasculitic disease mainly involving in upper respiratory tract. Eventually, histopathologic study by two endoscopic biopsy of nasopharynx confirmed granulomatous inflammation and vasculitis involving small sized blood vessels. To conclude, WG was the final diagnosis through the course of exclusion of differential diagnosis.
The vasculitis is defined histologically as inflammation within the wall of the blood vessels, which can be granulomatous or associated with fibrinoid necrosis of the vessel wall. Classification of systemic vasculitis is mainly based on histological features and the size of the vessel involved predominantly (4). WG and Churg-Strauss syndrome (CSS) are two main systemic vasculitides, both affecting mainly small-sized vessels and involving the upper respiratory tract. In the case of our patient, WG was far more possible diagnosis rather than CSS since the coexisting refractory sinusitis and otitis media could be more integrally explained under a single disease entity.
Although WG is one of the most common forms of systemic vasculitides, a reported annual incidence is merely 10 cases per one million people (2). Systemic vasculitides are relatively rare disease to encounter, particularly in the neurological field. The diagnostic difficulty is much more serious if clinical manifestations are not typical. The systemic vasculitis, however, is a disease entity worth to be remembered and should be included in the differential diagnosis in the case of multiple cranial neuropathies. The fact that WG is potential life-threatening disease, if the diagnosis is delayed and appropriate treatment is not promptly initiated, intensifies the importance of clinical suspicion.
While WG is generally known for its predilection for upper and lower airway tracts and kidney, quite a number of various systems can be affected. Thus, the involvement of nervous system is possible but not so usual. Especially, neurologic signs as presenting features are by far rare. If the nervous system is affected, the peripheral nervous system is implicated more frequently than the CNS, mainly as single mononeuropathy or mononeuritis multiplex (3). In contrast to other subsets of vasculitis, peripheral neuropathy in WG usually occurs in the late stage of disease, and uniquely painless. Involvement of the CNS occurs in 2%-8% of the patients, most commonly as cranial neuropathy. Cranial nerves II, VI, and VII are the most frequently involved, either solely or in combination (5). Lower cranial nerve palsies are reported as unusual involvement and as occurring in later stage of the disease, with the unilateral distribution or associated with skull-base pachymeningitis (6).
The main three patterns of CNS involvement in WG are; vasculitic changes, direct meningeal involvement due to adjacent granulomatous disease in the skull base, paranasal or orbital region and isolated granulomatous meningocerebral lesions (7-9). Among these three mechanisms, the most possible causes of our patient's multiple cranial nerve palsies could be direct extension of the granulomatous inflammation into the basilar meninges probably from the middle ear or paranasal cavities, resulting in cranial nerve palsies due to mechanical compression. The presence of enhancing high density lesions penetrating the skull base in neck CT and also focal enhancing meningeal thickening in the second brain MRI, support the possibility of this direct spreading mechanism. Moreover, asymmetrity of the symptom, which was more severe in the left side, suggested focal mechanical compression rather than diffuse inflammatory process.
On the contrary to direct spreading pattern, CNS vasculitis affect diffusely in the brain, which lead to various clinical symptoms other than the cranial neuropathy. Headache, loss of consciousness, seizure, transient or permanent ischemic attack, and even subarachnoid or cerebral hemorrhage due to vascular rupture are possible manifestations (10). Unfortunately, vasculitic changes of the cerebral vessels are barely detectable features because the affected vessels in WG are mostly small in size, and thus, are not able to be visualized by conventional cerebral angiography (11). In the present case, conventional MR angiography showed normal findings, and overall clinical symptoms and courses were by far less suggestive of vasculitic processes.
Cranial neuropathy can be the first obvious clinical manifestation in many various vasculitides including WG. Unfortunately, no single diagnostic modality can be reliable in the diagnosis of limited WG of CNS involvement. CSF analysis often shows only a slight rise in protein and a mild lymphocytic pleocytosis. Therefore, the diagnosis of CNS involvement in WG is based upon the combination of neurologic symptoms and signs, brain MRI, and histopathology.
A study of imaging in WG reported about 30% of meningeal thickening and contrast enhancement, 26% meningeal involvement by extracerebral granulomatous disease, 20% cerebral infarcts, and about 50% non-specific white matter lesions (12). Enhancing diffuse or focal dural thickening and multiple non-specific white matter lesions with increased signal on T2-weighted and fluid attenuated inversion recovery (FLAIR) images are relatively common imaging findings (13). Remote brain parenchymal granulomatous lesions of brain MRI are extremely rare.
Nasal biopsy is useful for confirming the diagnosis with a reported sensitivity of about 53% (14, 15). Classically, a triad of microscopic findings of vasculitis, granulomatous inflammation and tissue necrosis is the histologic characteristic of WG (8). However, rarely will all three histologic features be present at the same time, particularly in limited WG. One study reported that only 16% of biopsies in head and neck WG have all three defining criteria and in most cases only one criterion is found (16). In spite of these limitations, the biopsy remains as a crucial component of the diagnostic workup in suspected WG.
Anti-neutrophilic cytoplasmic antibodies with a cytoplasmic staining pattern (c-ANCA) is an established diagnostic tool of WG more easily used than biopsy. ANCA is known as a pathogenic factor in inflammatory processes that underlies necrotizing vasculitis. The sensitivity of c-ANCA in active Wegener's disease is up to 91% and a specificity of 99% (17). However, its titer depends on the extent and phase of the disease. In complete remission status, c-ANCA is not detectable. In the limited form, when WG is localized to the respiratory tract or the adjacent CNS, c-ANCA confers about 40 to 50% specificity. On the other hand, it has the specificity is 85%-100% in the generalized form (18, 19). Our patient corresponded as the limited form of WG confined to the upper respiratory tract and CNS, without the evidences of renal or lung involvement. Thus our laboratory result of negative c-ANCA could be explained well by the previous reports that a detection rate of c-ANCA is low in the limited WG than generalized. Despite the attraction of using c-ANCA as a diagnostic test due to its ease, in almost every case it should not be placed in for biopsy to diagnose WG.
Current management of WG is based upon the regimen of oral prednisolone 1 mg/kg/day in combination with oral cyclophosphamide 2 mg/kg/day for remission induction. Daily prednisolone is continued for four weeks and tailed down gradually over 1 to 2 months before converting to an alternate day regimen. Then the dose is further tapered down till patient is solely on cyclophosphamide. Long-term experience with cyclophosphamide provided many evidences for its efficacy of 80% survival rate, with 91% of patients having significant improvement and 75% achieving complete remission (20). In severe disease, however, cyclophosphamide is started at higher dose of 3-5 mg/kg/day, and prednisolone is also given higher from 2 to 15 mg/kg/day. A few case studies of limited WG confined to CNS showed relatively favorable clinical response to standard protocol consisting of steroid and cyclophosphamide. Though some case reported that there was only halting of further progression without definite neurological improvement after standard immunotherapy (21), and others reported that some clinical improvement was followed by rapid deterioration leading to fatal outcome (9, 22). The case reports of dramatic response or near full recovery after a standard therapy was not uncommon (6, 10, 23, 24). For few progressive cases in spite of the combination therapy of cyclophosphamide and prednisolone, other agents including methotrexate, leflunomide, etanercept, mycophenolate, tumor necrosis factor, and anti-lymphocyte directed therapy have been tried (25, 26). Eventhough, no specific regimen has been established for those refractory cases or focally localized cases of WG (4), several studies reported of successful treatments of focal meningeal involved WG by using infliximab or rituximab (27-29).
In summary, we described a limited WG presenting with refractory sinusitis and various cranial neuropathies having slowly progressive feature. WG should be considered in patients with multiple cranial nerve palsies, especially those with underlying chronic paranasal sinus disease, with or without systemic involvement. Since WG could be lethal if untreated, prompt treatment with potent immunosuppressants including cyclophosphamide is necessary promptly after the tissue biopsy to confirm the diagnosis.
Footnotes
This study was supported by a grant of the Korea Healthcare technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI10C2020).
References
- 1.Martinez Del Pero M, Sivasothy P. Vasculitis of the upper and lower airway. Best Pract Res Clin Rheumatol. 2009;23:403–417. doi: 10.1016/j.berh.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Rodrigues CE, Callado MR, Nobre CA, Moura FE, Vieira RM, de Albuquerque LA, Vieira WP. Wegener's granulomatosis: prevalence of the initial clinical manifestations: report of six cases and review of the literature. Rev Bras Reumatol. 2010;50:150–164. [PubMed] [Google Scholar]
- 3.Cattaneo L, Chierici E, Pavone L, Grasselli C, Manganelli P, Buzio C, Pavesi G. Peripheral neuropathy in Wegener's granulomatosis, Churg-Strauss syndrome and microscopic polyangiitis. J Neurol Neurosurg Psychiatry. 2007;78:1119–1123. doi: 10.1136/jnnp.2006.111013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagnoux C, Wolter NE. Vasculitis of the upper airways. Swiss Med Wkly. 2012;142:w13541. doi: 10.4414/smw.2012.13541. [DOI] [PubMed] [Google Scholar]
- 5.Nishino H, Rubino FA, DeRemee RA, Swanson JW, Parisi JE. Neurological involvement in Wegener's granulomatosis: an analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol. 1993;33:4–9. doi: 10.1002/ana.410330103. [DOI] [PubMed] [Google Scholar]
- 6.Armani M, Spinazzi M, Andrigo C, Fassina A, Mantovan M, Tavolato B. Severe dysphagia in lower cranial nerve involvement as the initial symptom of Wegener's granulomatosis. J Neurol Sci. 2007;263:187–190. doi: 10.1016/j.jns.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 7.Murphy JM, Gomez-Anson B, Gillard JH, Antoun NM, Cross J, Elliott JD, Lockwood M. Wegener granulomatosis: MR imaging findings in brain and meninges. Radiology. 1999;213:794–799. doi: 10.1148/radiology.213.3.r99dc11794. [DOI] [PubMed] [Google Scholar]
- 8.Kodama S, Nomi N, Suzuki M. Wegener's granulomatosis with extensive bone abnormalities mimicking fungal sinusitis. Case Rep Otolaryngol. 2012;2012:103403. doi: 10.1155/2012/103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kashiyama T, Suzuki A, Mizuguchi K. Wegener's granulomatosis with multiple cranial nerve involvements as the initial clinical manifestations. Intern Med. 1995;34:1110–1113. doi: 10.2169/internalmedicine.34.1110. [DOI] [PubMed] [Google Scholar]
- 10.Spísek R, Kolouchová E, Jensovský J, Rusina R, Fendrych P, Plas J, Bartůnková J. Combined CNS and pituitary involvement as a primary manifestation of Wegener granulomatosis. Clin Rheumatol. 2006;25:739–742. doi: 10.1007/s10067-005-0065-5. [DOI] [PubMed] [Google Scholar]
- 11.Akahoshi M, Yoshimoto G, Nakashima H, Miyake K, Inoue Y, Tanaka Y, Tsukamoto H, Horiuchi T, Otsuka T, Harada M. MPO-ANCA-positive Wegener's granulomatosis presenting with hypertrophic cranial pachymeningitis: case report and review of the literature. Mod Rheumatol. 2004;14:179–183. doi: 10.1007/s10165-004-0288-3. [DOI] [PubMed] [Google Scholar]
- 12.Warnatz K, Peter HH, Schumacher M, Wiese L, Prasse A, Petschner F, Vaith P, Volk B, Weiner SM. Infectious CNS disease as a differential diagnosis in systemic rheumatic diseases: three case reports and a review of the literature. Ann Rheum Dis. 2003;62:50–57. doi: 10.1136/ard.62.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen SD, Harvey CJ. Imaging of Wegener's granulomatosis. Br J Radiol. 2007;80:757–765. doi: 10.1259/bjr/34705892. [DOI] [PubMed] [Google Scholar]
- 14.Del Buono EA, Flint A. Diagnostic usefulness of nasal biopsy in Wegener's granulomatosis. Hum Pathol. 1991;22:107–110. doi: 10.1016/0046-8177(91)90030-s. [DOI] [PubMed] [Google Scholar]
- 15.Madhira S, Hamid QA, Prayaga SM, Kolloju S. Limited Wegener's granulomatosis with predominant otological presentation. Indian J Otolaryngol Head Neck Surg. 2011;63:4–5. doi: 10.1007/s12070-011-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller S, Eljack S, DelGaudio JM. Clinical pathologic conference case 1: Wegener's granulomatosis. Head Neck Pathol. 2011;5:268–272. doi: 10.1007/s12105-011-0291-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal PK, Gogia A. Limited Wegener's granulomatosis with p-ANCA positivity. J Indian Acad Clin Med. 2004;5:348–350. [Google Scholar]
- 18.Said MS. Upper respiratory tract symptoms, renal involvement and vasculitis: a case report and review of wegener granulomatosis. J Clin Med Res. 2010;2:189–193. doi: 10.4021/jocmr412w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swain S, Ray R. Wegener's granulomatosis of nose: a case report. Indian J Otolaryngol Head Neck Surg. 2011;63:402–404. doi: 10.1007/s12070-011-0282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langford CA. Cyclophosphamide as induction therapy for Wegener's granulomatosis and microscopic polyangiitis. Clin Exp Immunol. 2011;164:31–34. doi: 10.1111/j.1365-2249.2011.04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daderian AD, Chayasirisobhon S. An unusual case of multiple cranial nerve palsies in Wegener's granulomatosis. J Natl Med Assoc. 2000;92:455–457. [PMC free article] [PubMed] [Google Scholar]
- 22.Nordmark G, Boquist L, Rönnblom L. Limited Wegener's granulomatosis with central nervous system involvement and fatal outcome. J Intern Med. 1997;242:433–436. doi: 10.1046/j.1365-2796.1997.00215.x. [DOI] [PubMed] [Google Scholar]
- 23.Nowack R, Wachtler P, Kunz J, Rasmussen N. Cranial nerve palsy in Wegener's granulomatosis: lessons from clinical cases. J Neurol. 2009;256:299–304. doi: 10.1007/s00415-009-0121-1. [DOI] [PubMed] [Google Scholar]
- 24.Loke YK, Tan MH. An unusual case of Wegener's granulomatosis. Med J Malaysia. 1998;53:107–109. [PubMed] [Google Scholar]
- 25.Langford CA, Talar-Williams C, Barron KS, Sneller MC. A staged approach to the treatment of Wegener's granulomatosis: induction of remission with glucocorticoids and daily cyclophosphamide switching to methotrexate for remission maintenance. Arthritis Rheum. 1999;42:2666–2673. doi: 10.1002/1529-0131(199912)42:12<2666::AID-ANR24>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 26.Langford CA. Wegener's granulomatosis: current and upcoming therapies. Arthritis Res Ther. 2003;5:180–191. doi: 10.1186/ar771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK, et al. RAVE-ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermann J, Reittner P, Scarpatetti M, Graninger W. Successful treatment of meningeal involvement in Wegener's granulomatosis with infliximab. Ann Rheum Dis. 2006;65:691–692. doi: 10.1136/ard.2005.043885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Just SA, Knudsen JB, Nielsen MK, Junker P. Wegener's granulomatosis presenting with pachymeningitis: clinical and imaging remission by rituximab. ISRN Rheumatol. 2011;2011:608942. doi: 10.5402/2011/608942. [DOI] [PMC free article] [PubMed] [Google Scholar]