

Abstract
Rett syndrome and MECP2 duplication syndrome are neurodevelopmental disorders that arise from loss-of-function and gain-of-function alterations in methyl-CpG binding protein 2 (MeCP2) expression, respectively. Although there have been studies examining MeCP2 loss of function in animal models, there is limited information on MeCP2 overexpression in animal models. Here, we characterize a mouse line with MeCP2 overexpression restricted to neurons (Tau–Mecp2). This MeCP2 overexpression line shows motor coordination deficits, heightened anxiety, and impairments in learning and memory that are accompanied by deficits in long-term potentiation and short-term synaptic plasticity. Whole-cell voltage-clamp recordings of cultured hippocampal neurons from Tau–Mecp2 mice reveal augmented frequency of miniature EPSCs with no change in miniature IPSCs, indicating that overexpression of MeCP2 selectively impacts excitatory synapse function. Moreover, we show that alterations in transcriptional repression mechanisms underlie the synaptic phenotypes in hippocampal neurons from the Tau–Mecp2 mice. These results demonstrate that the Tau–Mecp2 mouse line recapitulates many key phenotypes of MECP2 duplication syndrome and support the use of these mice to further study this devastating disorder.
Introduction
Methyl-CpG binding protein 2 (MeCP2) is a transcriptional regulator important for controlling gene expression. MeCP2 functions as a transcriptional repressor by binding directly to methylated CpG dinucleotides and recruiting corepressor proteins, such as histone deacetylases (HDACs) and Sin3a to the promoters of target genes (Jones et al., 1998; Nan et al., 1998). Recent evidence suggests that MeCP2 may also function as an activator of gene transcription, particularly when bound to lightly methylated DNA (Yasui et al., 2007; Chahrour et al., 2008; Ben-Shachar et al., 2009). Although there are many genes that have been suggested as putative MeCP2 target genes, brain-derived neurotrophic factor (BDNF), known to play critical roles in various neuronal functions, is one of the best characterized (Chen et al., 2003; Martinowich et al., 2003).
MECP2 duplication syndrome is the result of increased MECP2 copy number (Van Esch et al., 2005). Clinical evidence suggests that males with MECP2 duplication syndrome display autistic features, heightened anxiety, mental retardation, epilepsy, motor dysfunction, and progressive neurological symptoms similar to those seen in females with Rett syndrome (Meins et al., 2005; Ramocki et al., 2009, 2010). Although there have been many studies examining loss of MeCP2 function in animal models, there are limited data regarding overexpression of MeCP2 in animals. A recent report showed that transgenic mice in which MECP2 was expressed using a large genomic clone of the human MECP2 locus (Mecp2Tg1) display enhanced cerebellar and hippocampal learning and decreased anxiety, along with enhanced long-term synaptic plasticity and excitatory synaptic activity in hippocampal neurons (Collins et al., 2004). These findings were unexpected given the clinical phenotype observed in MECP2 duplication syndrome individuals, in particular the learning and memory deficits and the heightened anxiety commonly observed in these afflicted individuals.
Here, we examine an alternative MeCP2 overexpression mouse line to determine whether it recapitulates key phenotypes observed in MECP2 duplication individuals. This transgenic MeCP2 overexpression line was generated by targeting Mecp2 into the locus of the neuron-specific gene Tau (Tau–Mecp2). These mice were shown previously to have decreased body weight and motor dysfunction compared with control littermates, although additional behavioral analysis was not performed (Luikenhuis et al., 2004). Here, we show that Tau–Mecp2 mice have impairments in motor coordination and heightened anxiety-like behavior, similar to phenotypes observed in MECP2 duplication individuals. We also find that Tau–Mecp2 mice have impairments in learning and memory as well as long-term potentiation (LTP) and paired-pulse facilitation (PPF). To assess synaptic function, we examined the impact of MeCP2 overexpression in hippocampal neurons and find a selective increase in excitatory spontaneous neurotransmission that can be rescued by an inhibitor of HDACs, demonstrating that MeCP2 acts as a transcriptional repressor to mediate this synaptic phenotype. Together, these results indicate that Tau–Mecp2 mice recapitulate key features of MECP2 duplication syndrome, as well as have specific alterations in measurements of synaptic plasticity and excitatory neurotransmission, and present a valuable animal model to study this disorder.
Materials and Methods
Tau–Mecp2 mice.
The transgenic Tau–Mecp2 line was generated by Dr. Rudolf Jaenisch (Whitehead Institute, Cambridge, MA) and colleagues as described previously (Luikenhuis et al., 2004). This line was backcrossed for >10 generations to C57BL/6 mice. Genomic DNA isolated from tails was used to confirm genotype of the mice. The Tau–Mecp2 mice were genotyped by PCR using the following primer sequences, Tau138 (5′-CTG GCA GAT CTT CCC GTC TA-3′), Tau1078 (5′-TGC CTG ACA GAG TCC AGA TG-3′), and Neo1323 (5′AGG GGA TCC GTC CTG TAA GT-3′), which amplified a 941 bp fragment from the wild-type (WT) allele and a 796 bp fragment from the overexpressing allele. The Tau–Mecp2 and WT littermate controls were generated by crossing heterozygous female Tau–Mecp2 with male C57BL/6 mice. All behavioral experiments used only male Tau–Mecp2 mice or WT littermate controls aged 10–16 weeks of age. Mice were maintained on a 12 h light/dark cycle with ad libitum access to food and water. Before all behavioral testing, mice were allowed to habituate in the behavior room for 1 hour. Mice were tested in a panel of behavioral tests as follows: locomotor activity, dark/light (DL), elevated plus maze (EPM), rotarod, and fear conditioning. Different sets of mice were used for cue fear conditioning, extinction, and novel object recognition (NOR) experiments. All data were analyzed and scored by an observer blind to the genotype. All animal procedures were approved by the Institutional Animal Care and Use Committee at University of Texas Southwestern Medical Center.
Western blot.
The specific brain regions were dissected and flash frozen in dry ice before being homogenized. Protein was isolated using RIPA buffer (5 mm EDTA, 150 mm NaCl, 1% NP40, and 50 mm HEPES). To probe for BDNF, 10 μg of protein was loaded and separated on an 8% SDS gel before being transferred to a nitrocellulose membrane. Membranes were probed with anti-MeCP2 and anti-actin (1:2000, Affinity Bioreagents PA1–888; and 1:2500, Sigma-Aldrich A5060, respectively, overnight at 4°C) proteins. Membranes were incubated with fluorescent secondary antibody (1:10,000, 827-11081, anti-rabbit LI-COR IRDye) for 30 min at room temperature (RT). Blots were scanned using Odyssey Imager (LI-COR Biosciences). To probe for BDNF, 100 μg of protein was loaded and separated on a 15% SDS gel before being transferred to a PVDF membrane. Membranes were blocked at 10 and 3% milk for BDNF and actin, respectively, and then probed with anti-BDNF and anti-actin antibodies (1:200, Santa Cruz Biotechnology N-20, sc-546; and 1:100,000, Sigma-Aldrich A5060, respectively, overnight at 4°C). Membranes were incubated with secondary antibody (1:10,000 for BDNF and 1:50,000 for actin, anti-rabbit, Vector Laboratories PI-1000) for 1 h at RT. Bands were visualized using chemiluminescence (GE Healthcare ECL Plus). Optical density was calculated for either MeCP2 or BDNF and normalized to actin.
Immunohistochemistry.
Immunohistochemistry to detect MeCP2 in brain was similar to a previously published protocol with slight modifications (Gemelli et al., 2006). Mice were transcardially perfused with ice-cold 0.1 m PBS before perfusion with 4% paraformaldehyde (PFA). Whole brains were dissected and postfixed in 4% PFA overnight and then cryoprotected in 20% glycerol. Coronal sections (30 μm) were prepared on a freezing microtome. Sections from hippocampus were mounted on Superfrost plus slides and dried overnight. Slide-mounted sections were blocked and permeabilized in 3% normal goat serum in 0.3% Triton X-100/0.1 m PBS for 30 min at RT and then incubated overnight with MeCP2 antibody (1:1000) at RT. Sections were rinsed with 0.1 m PBS and incubated with Cy3-conjugated anti-rabbit secondary (1:200) for 2 h at RT. After secondary antibody treatment, sections were dehydrated in an ethanol series and coverslipped in DPX mounting medium.
Locomotor activity.
Mice were placed in a standard mouse cage with fresh bedding. Locomotor activity was assessed for 20 min under red light by photocell beams linked to computer data acquisition software (San Diego Instruments).
Rotarod.
Mice were given eight rotarod sessions across 2 d (four trials per day). Each mouse was placed on a rotating rod (IITC Life Science), the speed of which gradually increased over 5 min. The test session ended when either the animal fell off the rod or after 5 min. The amount of time spent on the rotarod was determined by IITC Life Science software.
DL test.
The DL test consisted of a box partitioned into two compartments, a dark side and a light side. Mice were placed into the dark side for 2 min before a gate that separates the two compartments is lifted to allow mice to explore either the dark or light side for 10 min. The amount of time spent in the light side and dark side was quantified through computer data acquisition software (Med Associates).
EPM.
Mice were placed in the center of an EPM measuring 33 × 5 cm for each platform and located 1 m above the floor for 5 min. Time spent in the open arm, closed arms, and center (where the open and closed arms are crossed) was quantified by Ethovision tracking software (Noldus Information Technology).
Fear conditioning.
Training and testing were conducted under red light as described previously with slight modifications (Monteggia et al., 2004; Barbosa et al., 2008). Mice were placed in individual chambers and habituated for 2 min, during which time baseline freezing behaviors were recorded. Freezing behavior was defined as the absence of movement except for respiration (Fanselow, 1980). White noise (90 dB) was played for 30 s, which coterminated with a 2 s footshock (0.5 mA). Mice were given an interstimulus interval of 1 min before a second presentation of a tone paired with shock. Mice remained in conditioning chambers for 1 min after the second tone and shock presentation before being returned to their home cages. Chambers were cleaned using NPD Cleaner Disinfectant (Steris Corporation) between each trial. Context- and cue-dependent fear conditioning were tested the following day. To test context-dependent fear conditioning, 24 h after training, mice were placed in the same chamber without tone or shock, and freezing behavior was scored for 5 min. Four hours after the context-dependent fear conditioning test, cue-dependent fear conditioning tests were performed. Mice were placed in the fear conditioning chambers with a novel environment scented with vanilla extract and lit with fluorescent lights for 6 min. Animals were allowed to adapt for 3 min before presentation of tone for 3 min, and freezing behavior was scored.
Extinction training.
Extinction training was initiated 24 h after the cue-dependent fear conditioning test. Mice were placed in the fear conditioning chambers with a novel environment as described above, and baseline freezing (3 min) and freezing during presentation of tone (3 min) were assessed once per day for 9 d. Mice were given once daily extinction training sessions that were terminated once WT controls reach baseline levels of freezing during tone presentation.
NOR.
On day 1, mice were habituated to a white rectangular box (88 × 17 cm) for 10 min. Four hours later during the familiarization phase, mice were again reintroduced to the rectangular box now containing two identical objects (A and A) placed 35 cm apart for 10 min. The following day, mice were placed back in the rectangular box with two objects present, the object to which the mice were exposed during the familiarization phase (A) and a novel object (B). Mice were allowed to explore the objects for 10 min. A mouse was scored exploring an object whenever its front paws made contact with the object or if the mouse's nose was oriented toward the object. The difference score was calculated by subtracting the time spent with the familiar object from the time spent with the novel object (Bevins and Besheer, 2006).
Hippocampal slice electrophysiology.
Mice that were 8–12 weeks of age were anesthetized with Euthasol (30 mg/ml, 0.2 ml, i.p.) before decapitation. Brains were removed and immersed for 2–3 min in ice-cold artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4 (continuously bubbled with 95% O2 and 5% CO2). The hippocampi were dissected out and cut with a vibratome into 350-μm-thick transverse sections in ice-cold ACSF. Sections recovered in oxygenated ACSF for at least 1 h at 32°C. Hippocampal slices were transferred into a recording chamber and superfused with ACSF at a constant rate of 2.5 ml/min at 30°C. Field EPSPs (fEPSPs) were recorded with glass recording electrodes filled with ACSF (Sutter Instruments; resistance, 1–2 MΩ). Extracellular stimuli were delivered by placing a bipolar platinum–tungsten stimulating electrode at the region of interest (A-M Systems Isolated Pulse Stimulator model 2100). The stimulating electrode was inserted to stimulate fibers of the Schaffer collateral pathway, and the recording electrode was inserted into the CA1 just beneath the molecular layer. The stimulating and recording electrodes were separated by a distance of 300–350 μm. Electrical signals were amplified (A-M Systems AC amplifier model 1800), digitized, and stored on a personal computer for subsequent analysis using Labview 8.6 software (National Instruments). Input/output (I/O) relationship was determined by providing an ascending series of stimulus input intensities (range, 40 to ∼400 μA) until the maximum amplitude response was reached. An input stimulus intensity that induces 40–60% of the maximum response was used for measuring PPF and LTP. PPF was measured by giving two pulses at decreasing interstimulus intervals (500, 400, 200, 100, 50, 30, and 20 ms) and analyzed by dividing the fEPSP slope of pulse 2 by pulse 1. After 20 min of stable baseline, LTP was induced by high-frequency stimulation (HFS) using an input stimulus intensity that produces the maximum response (two 100 Hz trains with 100 pulses with an interburst interval of 20 s).
Single-cell recordings.
Dissociated hippocampal cultures were prepared based on previously published protocols (Nelson et al., 2006, 2008; Akhtar et al., 2009). Hippocampi were dissected from Tau–Mecp2 and WT mice on postnatal days 0–3. Tissue was trypsinized for 10 min at 37°C, mechanically dissociated with siliconized glass pipettes, and plated onto Matrigel-coated coverslips. A concentration of 4 μm cytosine arabinoside (Sigma) was added at 1 d in vitro (DIV). Single-cell recording experiments were performed on 14–21 DIV pyramidal hippocampal neurons in culture using a whole-cell voltage-clamp technique. Data were acquired using an Axopatch 200B amplifier and Clampex 9.0 software (Molecular Devices). Recordings were filtered at 2 kHz and sampled at 200 μs. A modified Tyrode's solution containing 150 mm NaCl, 4 mm KCl, 2 mm MgCl2, 2 mm CaCl2, 10 mm glucose, and 10 mm HEPES, pH 7.4, was used as an external bath solution. Picrotoxin (50 μm) was used to isolate miniature EPSCs (mEPSCs). The drug 6-nitro-7-sulfamoylbenzo-(f)quinoxaline-2,3-dione (10 μm) was used to isolate miniature IPSCs (mIPSCs). The pipette internal solution contained the following (in mm): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na3GTP, pH 7.35, and 10 QX-314 [N- (2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide] (with osmolarity at 300 mOsm/L). Hippocampal cultures were treated for 24 h with trichostatin A (TSA; 1 μm; Sigma) or dimethylsulfoxide (1:1000) as a control.
Statistics.
Data are presented as mean ± SEM. Total locomotor activity, DL, EPM, context- and cue-dependent fear conditioning, basal synaptic transmission, and Western blot data were all analyzed using (Student's two-tailed) independent t tests. Rotarod, extinction training, and LTP data were analyzed using repeated-measures ANOVA. Post hoc analyses (i.e., Fisher's LSD) were conducted after significant interaction effects were found. A p value of <0.05 was required for statistical significance.
Results
Increased expression of MeCP2 protein in brain regions of Tau–Mecp2 mice
Previous work had shown increased MeCP2 overexpression in whole brain of Tau–MeCP2 mice (Luikenhuis et al., 2004) To assess the level of MeCP2 expression in specific brain regions of Tau–Mecp2 mice, we dissected tissue from the caudate–putamen, frontal cortex, hippocampus, and cerebellum and performed Western blot analysis. An increase in MeCP2 protein, as assessed by Western blot analysis, was seen in the caudate–putamen, frontal cortex, hippocampus, and cerebellum of Tau–Mecp2 mice compared with WT controls (Fig. 1C). Using immunohistochemistry, we also visualized MeCP2 protein within specific subregions of the hippocampus and observed increased MeCP2 expression in Tau–Mecp2 mice compared with WT controls (Fig. 1A,B).
Figure 1.

Increased expression of MeCP2 in Tau–Mecp2 mice compared with WT control littermates. A, B, Immunofluorescence images of the CA1 and dentate gyrus (DG) areas of the hippocampus. Left column shows MeCP2 protein in WT control littermates. Right column shows MeCP2 in Tau–Mecp2 mice. Top row is at a 10× magnification, and the bottom row is at a 40× magnification. C, Western blot data demonstrating an increase in MeCP2 protein expression in Tau–Mecp2 mice in different areas of the brain. FC, Frontal cortex; CPu, caudate–putamen; HC, hippocampus; CBL, cerebellum. WT mice, n = 4; Tau–Mecp2 mice, n = 4. *p < 0.05.
Tau–Mecp2 mice have decreased body weight and impaired motor coordination
In agreement with a previous report (Luikenhuis et al., 2004), Tau–Mecp2 mice weighed significantly less than WT littermate controls (data not shown), and this phenotype persisted as mice aged. We assessed locomotor activity in these mice by recording the number of beam breaks while animals were allowed to freely explore in a home-cage environment for 20 min. Total locomotor activity was indistinguishable between Tau–Mecp2 and WT mice (Fig. 2A, inset). To more closely examine locomotor activity, we plotted the data in 5 min epochs that did not reveal significant differences between Tau–Mecp2 and WT mice at any time point (Fig. 2A). We next assessed motor coordination using the rotarod test. Tau–Mecp2 mice spent significantly less time on the rotarod compared with WT mice at most trials (Fig. 2B), indicating motor coordination deficits in these mice.
Figure 2.

Behavioral phenotypes observed in mice that overexpress Mecp2. A, Locomotor activity is not significantly different between WT and Tau–Mecp2 mice. B, Tau–Mecp2 mice spend significantly less time on the rotarod on trials 2–4, 6, and 7 compared with WT mice, demonstrating motor coordination deficits. Significant main effects of time (F(7,315) = 19.5, p < 0.05) and genotype (F(7,315) = 8.2, p < 0.05). Post hoc Fisher's LSD tests revealed significant differences between Tau–Mecp2 and WT mice at trials 2–4, 6, and 7. C, DL test demonstrating an increased anxiety-like phenotype in mice that overexpress Mecp2 compared with WT mice (t(21) = 4.99, p < 0.05). Tau–Mecp2 mice spend significantly less time in the light side than WT mice. D, EPM data showing increased anxiety-like behavior in Tau–Mecp2 mice, which spend less time in the open arms and more time in the closed arms compared with WT controls (t(21) = 3.30, p < 0.05). WT mice, n = 11;Tau–Mecp2 mice, n = 12. *p < 0.05 between Tau–Mecp2 and WT mice.
Tau–Mecp2 mice display heightened anxiety-like behavior
We examined anxiety-related behavior in Tau–Mecp2 mice using the EPM and DL tests, behavioral paradigms that incorporate aspects of human anxiety and have predictive validity for anxiolytic drugs (Holmes et al., 2001; Karlsson et al., 2008). In the DL test, the Tau–Mecp2 mice spent significantly less time in the light side and significantly more time in the dark side, suggesting a heightened anxiety-like phenotype (Fig. 2C). Consistent with these findings, in the EPM, the Tau–Mecp2 mice spent significantly less time in the center, more time in the closed arms, and less time in the open arms compared with WT littermate controls (Fig. 2D).
Tau–Mecp2 mice display impairments in learning and memory
We assessed learning and memory in the Tau–Mecp2 mice by using the fear-conditioning paradigm. In this test, mice learn to associate a context (novel environment) or cue (auditory tone) with a footshock. Baseline freezing behavior between Tau–Mecp2 and WT mice was indistinguishable in the absence of a paired stimulus (data not shown). Twenty-four hours after training, Tau–Mecp2 mice displayed a significant increase in freezing behavior in response to context (Fig. 3A). We also found a significant increase in freezing behavior in response to cue-dependent fear conditioning in Tau–Mecp2 mice compared with those in WT littermate mice (Fig. 3A).
Figure 3.
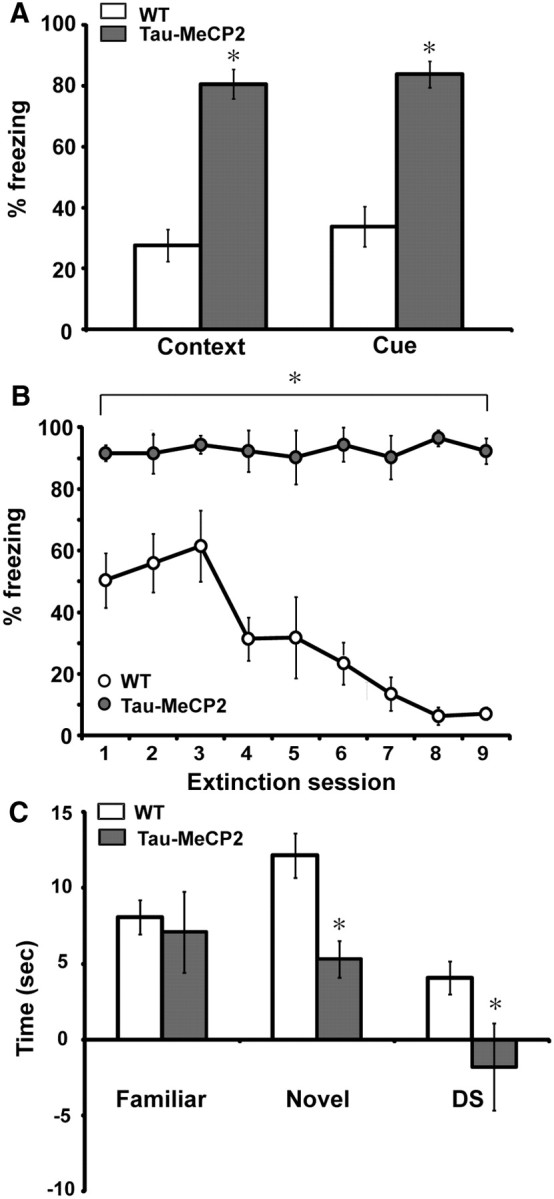
Learning and memory deficits in Tau–Mecp2 mice. A, Tau–Mecp2 mice (n = 12) show increased freezing responses in both context and cue fear conditioning compared with WT (n = 11) littermates (t(20) = 7.4, t(20) = 6.3, p < 0.05, respectively). B, Tau–Mecp2 mice do not learn to extinguish freezing behavior to repeated presentations of cue after nine extinction sessions, whereas WT control littermates extinguish freezing responses by the fourth extinction session, indicating learning and memory deficits in Tau–Mecp2 mice (WT, n = 7; Tau–Mecp2, n = 6). A repeated-measures ANOVA revealed a significant interaction effect between trials and genotype (F(8,125) = 4.8, p < 0.05), and a Fisher's LSD analysis revealed significant differences between all extinction trials. C, Mice that overexpress Mecp2 are not able to distinguish between a novel object and a familiar one. Tau–Mecp2 mice spend less time exploring the novel object than WT mice (t(22) = 3.4, p < 0.05) and also spend less time with the novel object versus the familiar object as demonstrated by their significant negative difference score (DS) (t(22) = 2.1, p < 0.05). There is no difference, however, in the amount of time spent with the familiar object between Tau–Mecp2 and WT mice. *p < 0.05 between WT mice and Tau–Mecp2 mice.
To examine whether this increase in freezing behavior reflects enhanced associative learning, we assessed the animal's ability to extinguish conditioned responses to the cue during extinction training. Extinction is considered an active form of learning (Robleto et al., 2004); thus, to test whether Tau–Mecp2 mice can learn new contingencies, mice were trained to extinguish freezing behavior to cue. Control WT mice were able to extinguish freezing responses by the fourth extinction session (t(6) = 2.74, p = 0.03) (Fig. 3B). In contrast, Tau–Mecp2 mice displayed consistent freezing levels across all nine extinction trials, indicating impairments in extinction learning in these mice.
We also tested Tau–Mecp2 mice in the NOR task, which tests an animal's ability to recognize a novel object (Bevins and Besheer, 2006). During familiarization, there were no significant differences in the amount of time spent with the objects (data not shown). However, during the NOR test on the following day, Tau–Mecp2 mice spent significantly less time with the novel object than WT mice and showed less preference for the novel object over the familiar, as demonstrated by the difference score (Fig. 3C).
Tau–Mecp2 mice have alterations in hippocampal synaptic plasticity
LTP is a form of synaptic plasticity considered to be a putative cellular mechanism for learning and memory (Maren, 2005). To examine LTP in hippocampal slices from Tau–Mecp2 mice, we stimulated the Schaffer-collateral pathway using HFS (two trains of enhanced pulses at 100 Hz), which has been shown previously to induce LTP in a separate MeCP2 overexpression line (Collins et al., 2004). We found that HFS-induced LTP was significantly attenuated in hippocampal slices from Tau–Mecp2 mice compared with WT controls for the length of the 120 min recording (Fig. 4A).
Figure 4.

LTP induced by HFS is impaired in Tau–Mecp2 hippocampal slices. A, Tau–Mecp2 mice show attenuated LTP responses induced by HFS compared with WT controls (WT, n = 16; Tau–Mecp2, n = 11). A significant time × genotype interaction effect was seen (F(271,7343) = 3.0, p < 0.05). Post hoc comparisons using Fisher's LSD revealed differences at almost all time points after HFS except for 15 time points of the 78 time points analyzed. Representative traces are shown in the inset (black, before stimulation; red, after LTP stimulation). B, There were no significant differences in the slope of the I/O relationship between Tau–Mecp2 and WT mice (WT, n = 8; Tau–Mecp2, n = 10). C, Tau–Mecp2 hippocampal slices show impairments in PPF. PPF was significantly augmented by MeCP2 overexpression, with Tau–Mecp2 mice showing increased PPF at interstimulus intervals 30 ms (t(16) = 2.2, p < 0.05) and 50 ms (t(16) = 3.1, p < 0.05) compared with WT controls, indicating a decreased probability of neurotransmitter release (WT, n = 8; Tau–Mecp2, n = 10). *p < 0.05 between WT mice and Tau–Mecp2 mice.
We next examined whether MeCP2 overexpression had effects on baseline transmission and short-term plasticity as assessed by I/O relationships and PPF. Stimulating electrodes were placed in the Schafffer collaterals and field potentials recorded in the CA1 region of the hippocampus to generate I/O curves. I/O slopes of presynaptic volley amplitude to the fEPSP slope recorded from Tau–Mecp2 and WT hippocampal slices were not significantly different (Fig. 4B). We also assessed PPF, an index of presynaptic release probability, by recording the response to two pulses separated by varying interstimulus intervals and taking the ratio of the second response to first. PPF was significantly augmented in the Tau–Mecp2 mice at interstimulus intervals of 30 and 50 ms (Fig. 4C).
Tau–Mecp2 hippocampal neurons display enhanced miniature excitatory neurotransmission
To further assess the impact of MeCP2 overexpression on basal synaptic function, we made primary dissociated hippocampal cultures from newborn Tau–Mecp2 and WT mice and then performed whole-cell voltage-clamp recordings of spontaneous synaptic currents in pyramidal neurons at 14–21 DIV. Previous work has demonstrated that the maturation state of individual synaptic terminals and their functional properties were relatively constant in this time range (Mozhayeva et al., 2002). To take into account potential changes in synapse numbers, all experiments were conducted in a pairwise manner on sister coverslips at the same day. In neurons cultured from Tau–Mecp2 mice, we found a significant increase in mEPSC frequency with no change in mEPSC amplitudes compared with WT controls (Fig. 5A–C). We also found no significant differences in mIPSC frequency or amplitudes in Tau–Mecp2 cultured neurons compared with WT (Fig. 5D–F).
Figure 5.

Increased excitatory neurotransmission in mice that overexpress Mecp2. A, Representative traces of mEPSC frequency in WT and Tau–Mecp2 hippocampal neuronal cultures. B, mEPSC frequency was significantly increased in hippocampal Tau–Mecp2 cultures compared with WT cultures (*p < 0.05). C, There were no significant differences in the cumulative distribution of mEPSC amplitudes. D, Representative traces of mIPSC frequency in WT and Tau–Mecp2 hippocampal neurons. E, There were no significant differences in mIPSC frequency. F, Cumulative distribution of mIPSC amplitudes was not significantly altered in Tau–Mecp2 hippocampal neurons. The number of cells recorded in mEPSC and mIPSC experiments are shown in the respective bar graph.
Alterations in synaptic transmission of Tau–Mecp2 hippocampal neurons is attributable to transcriptional repression
To explore mechanisms underlying MeCP2 overexpression on synaptic transmission, we examined the level of the MeCP2 gene target BDNF, which if altered, could explain the synaptic deficiencies we observed. In previous work, depolarization of cultured neurons by potassium chloride was shown to reduce MeCP2 binding with the BDNF promoter and increase BDNF transcription (Chen et al., 2003; Martinowich et al., 2003). In separate work, activity-dependent phosphorylation of MeCP2 at serine 421 was shown to increase BDNF transcription (Zhou et al., 2006). However, the link between MeCP2 and BDNF is complex because BDNF protein is reduced in Mecp2 null mice, and overexpressing BDNF in the brain of Mecp2 null mice slows disease progression (Chang et al., 2006). Moreover, previous work has also shown that MeCP2 overexpression increases BDNF mRNA based on its role as a transcriptional activator (Chahrour et al., 2008). We found that BDNF protein levels were unaltered in the hippocampus of Tau–Mecp2 compared with WT mice (Fig. 6A). To examine whether the transcriptional repressor function of MeCP2 is mediating the synaptic phenotype, we treated primary dissociated hippocampal cultures from Tau–Mecp2 mice with the HDAC inhibitor TSA for 24 h and then recorded effects on excitatory synaptic transmission. This manipulation is expected to alter MeCP2 function because MeCP2 acts in conjunction with HDACs to regulate gene expression. In neurons from Tau–Mecp2 mice treated with TSA, we found a significant decrease in mEPSC frequency that rescued excitatory synaptic transmission to a level comparable with vehicle-treated cultures (Fig. 6B).
Figure 6.

Inhibition of transcriptional repressor processes rescue the synaptic deficits in hippocampal neurons from Tau–Mecp2 mice. A, BDNF protein expression was quantified relative to actin in Tau–Mecp2 and WT mice. There were no significant differences in relative BDNF protein expression in Tau–Mecp2 compared with WT mice (n = 4 for each group). B, Left, Representative traces of mEPSC frequency in WT and Tau–Mecp2 hippocampal neuronal cultures treated with TSA. B, The significant increase in mEPSC frequency seen in Tau–Mecp2 hippocampal neurons is reduced back to WT levels with 24 h TSA treatment (WT, n = 9; Tau–Mecp2, n = 8; Tau + TSA, n = 8; *p < 0.05).
Discussion
Here, we demonstrate that Tau–Mecp2 mice recapitulate key features of MECP2 duplication syndrome, namely motor dysfunction, heightened anxiety, and learning and memory deficits. The impairments in learning and memory as well as LTP observed in the Tau–Mecp2 mice are consistent with the mental retardation phenotype found in individuals with MECP2 duplication syndrome. Interestingly, we observed an increase in freezing behavior in context and cue-dependent fear conditioning 24 h after training, suggestive of an enhancement in associative learning. Because an enhancement in learning and memory seemed counterintuitive when compared with the phenotypes observed in individuals with MECP2 duplication syndrome, we performed subsequent experiments with the Tau–Mecp2 mice and found that these animals are not capable of extinguishing their conditioned response (e.g., freezing) to cue when cue is presented alone (i.e., without shock), indicating severe impairments in extinction learning and/or associative learning. The Tau–Mecp2 mice do not show enhanced freezing behavior in the absence of a paired stimulus (baseline freezing) or before the tone in cued fear conditioning, suggesting that these mice in fact learn to associate the cue with the shock. Extinction training is considered a new learning process in that animals learn to adapt their behavior to new contingencies (Santini et al., 2008; Whittle et al., 2010). Although it is possible that the Tau–Mecp2 mice have a strong memory of the initial training, this explanation is unlikely given the fact that the persistent freezing response suggests that these mice are inflexible learners (i.e., new contingencies are not readily acquired by these mice). Other strains of mice also show this same inability to acquire extinction learning (Hefner et al., 2008; Soliman et al., 2010). This inflexibility in the Tau–Mecp2 mice is maladaptive behavior especially considering that extinction learning does not require that the animal forget the original contingency. Studies have shown that animals adapt their behavior to new contingencies (i.e., diminished freezing response) during extinction training while still maintaining the original memory (Rescorla and Heth, 1975; Quirk, 2002; Chang et al., 2009; Stafford and Lattal, 2011). Thus, based on our data, Tau–Mecp2 mice are incapable of suppressing the expression of fear memories, or alternatively are not capable of forming new associations.
We also assessed learning and memory using the NOR test, a paradigm that induces little stress to avoid any confounds of the heightened anxiety phenotype of the Tau–Mecp2 mice. The Tau–Mecp2 mice show significant impairments in NOR compared with WT littermate controls. Given that NOR is generally considered a test of episodic memory, these data suggest that these mice may be impaired at forming episodic memories (Ennaceur and Delacour, 1988; Bevins and Besheer, 2006; Davis et al., 2010). Deficits in the NOR paradigm have also been reported with male hemizygous Mecp2 mice and female heterozygous Mecp2 mutant mice suggesting that this behavioral task is sensitive to fluctuations in MeCP2 expression levels (Stearns et al., 2007). Collectively, the fear conditioning and NOR data suggest impairments in both hippocampal-dependent and -independent learning and memory in the Tau–Mecp2 mice.
In agreement with the learning and memory deficits, the Tau–Mecp2 mice had significant impairments in hippocampal LTP. The Tau–Mecp2 mice displayed normal I/O curves, suggesting that Schaffer-collateral input into CA1 neurons is intact. Interestingly, we found enhanced PPF, a form of short-term synaptic plasticity, from hippocampal slices of Tau–Mecp2 mice. Previous work with constitutive Mecp2 null and Mecp2308/y mice has demonstrated impairments in LTP in the CA1 region of the hippocampus accompanied by normal I/O curves but decreased PPF (Asaka et al., 2006; Moretti and Zoghbi, 2006; Weng et al., 2011). These data suggest significant susceptibility of several synaptic measures to alterations in MeCP2 expression.
To examine the effect of MeCP2 overexpression on spontaneous neurotransmission, we recorded from Tau–Mecp2 cultured hippocampal neurons and found enhanced mEPSCs indicative of increased resting excitatory drive with no effect on mIPSC properties. These data suggest that MeCP2 overexpression selectively impacts excitatory neurotransmission and are in agreement with recent findings demonstrating an increase in mEPSC frequency in autaptic cultures expressing a twofold increase in MeCP2 levels (Chao et al., 2007). Collectively, we find that Tau–Mecp2 mice have increased PPRs of evoked excitatory neurotransmission, indicating a decrease in evoked release probability but an increase in spontaneous release. These results are intriguing based on previous findings using Mecp2 knock-out (KO) mice that report decreased PPRs of evoked excitatory neurotransmission, suggesting an increase in neurotransmitter release probability (Asaka et al., 2006; Moretti and Zoghbi, 2006; Nelson et al., 2006, 2011), coupled with a decrease in spontaneous excitatory neurotransmission (Dani et al., 2005; Asaka et al., 2006; Moretti and Zoghbi, 2006; Nelson et al., 2006). Together, the impact of MeCP2 on evoked and spontaneous neurotransmission appears to be bidirectional, suggesting a direct relationship between MeCP2 expression levels and presynaptic function. However, evoked and spontaneous forms of neurotransmission are affected in the opposite direction in both loss-of-function and overexpression models, consistent with the premise that the two forms of neurotransmission constitute independent pathways (Ramirez and Kavalali, 2011).
To gain mechanistic insight into the impact of MeCP2 overexpression on neuronal function, we examined levels of BDNF, a putative MeCP2 target gene that impacts learning and memory as well as synaptic plasticity and neurotransmission. Our findings that hippocampal BDNF levels are not altered in Tau–Mecp2 mice suggest that this gene is not responsible for the behavioral and synaptic deficits in these mice. Although we cannot rule out alterations in BDNF mRNA levels in the Tau–Mecp2 mice, the lack of a protein change makes it difficult to attribute the phenotypic differences to BDNF.
Although we do not have a selective gene target to explain the phenotypes observed in the Tau–Mecp2 mice, we examined whether MeCP2 may be mediating the effects via its activity as a transcriptional repressor. We have shown previously that treatment of WT neurons with the HDAC inhibitor TSA produces a significant reduction in mEPSC frequency (∼50%) that was occluded in the absence of MeCP2 (Nelson et al., 2006; Akhtar et al., 2009), demonstrating that loss of MeCP2 impacts synaptic transmission through dysregulation of transcriptional repression. To investigate whether a similar mechanism mediates the effects of MeCP2 overexpression, we treated Tau–Mecp2 cultures with TSA and found that the increase in mEPSC frequency recorded from Tau–Mecp2 neurons was rescued by TSA. The TSA treatment of Tau–Mecp2 neurons resulted in an equivalent 50% reduction in mEPSC frequency to that observed after TSA treatment of WT neurons, suggesting that the overexpressed MeCP2 pool is indistinguishable from the endogenous MeCP2 and functions in a similar manner to regulate mEPSC frequency. Treatment with TSA increases gene expression by inhibiting transcriptional repression; thus, the rescue of the synaptic phenotype in the Tau–Mecp2 suggests that MeCP2 overexpression is downregulating gene expression to produce the synaptic deficits. Here, we cannot fully exclude that TSA inhibition of mEPSC frequency may be partly independent of MeCP2 function. However, combined with our previous finding that the TSA effect is occluded in the MeCP2 KO neurons, we consider this as unlikely.
The deficits in motor coordination and increased anxiety in the Tau–Mecp2 mice recapitulates features of MECP2 duplication syndrome, in contrast to a previous study with the Mecp2Tg1 line that reported enhanced motor coordination and decreased anxiety (Collins et al., 2004). The Mecp2Tg1 mice also show premature death, which is not observed in the Tau–Mecp2 mice. The phenotypic differences between these two MeCP2 overexpression lines may be attributable to variations in generation methods of each mouse line. The Tau–Mecp2 mice were generated by expressing the mouse Mecp2 gene under control of the Tau promoter (Luikenhuis et al., 2004), whereas the Mecp2Tg1 mice were generated by using a P1-derived artificial chromosome that contains the human MECP2 gene (Collins et al., 2004). Although concerns have been raised that targeting the Tau locus may impact neuronal function, recent data suggest this is not the case (Harada et al., 1994; Brandt, 1996). Another possibility is that human MECP2 may not be under the same transcriptional control in mice. Indeed, although the level of increased MeCP2 expression in the Mecp2Tg1 mice is twice that of WT, a cross between Mecp2Tg1 and Mecp2308/y KO mice did not result in WT levels of MeCP2 protein (Collins et al., 2004). MeCP2 expression in the Tau–Mecp2 mice is also not under endogenous control because only Mecp2 cDNA was inserted into the Tau locus; thus, additional experiments will be necessary to resolve the differences observed between the Tau–Mecp2 and Mecp2Tg1 lines.
In summary, the present study demonstrates that neuron-specific MeCP2 overexpression results in cognitive impairments, motor coordination deficits, a pronounced anxiety-like phenotype, and specific alterations in excitatory synaptic transmission. Moreover, we show that the certain synaptic deficits in Tau–Mecp2 neurons can be rescued by an HDAC inhibitor consistent with alterations in transcriptional repression mechanisms underlying synaptic phenotypes in Tau–Mecp2 mice. These data demonstrate that the Tau–Mecp2 mice recapitulate many of the core symptoms of human MEPC2 duplication syndrome, thus providing a viable animal model for the study of this devastating neurodevelopmental disorder.
Footnotes
This work was supported by National Institute of Health Grant MH081060 (L.M.M.), National Institutes of Mental Health Training Fellowship T32 MH076690 (E.S.N.), and a NARSAD Independent Investigator Award (E.S.N.). We thank Dr. Rudolf Jaensich for generously providing the Tau–Mecp2 mice. We acknowledge Drs. Michael Morris, Elena Nosyreva, and Caroline Smith for technical assistance with hippocampal field recordings. We also thank Aroon Karra and Heena Pranav for assistance with breeding of the Tau–Mecp2 line, Dr. Waseem Akhtar for assistance with cell culture, and Dr. Shari Birnbaum and Ami Pettersen for assistance with the behavioral experiments.
References
- Akhtar MW, Raingo J, Nelson ED, Montgomery RL, Olson EN, Kavalali ET, Monteggia LM. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J Neurosci. 2009;29:8288–8297. doi: 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, Olson EN. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A. 2008;105:9391–9396. doi: 10.1073/pnas.0802679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet. 2009;18:2431–2442. doi: 10.1093/hmg/ddp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.”. Nat Protoc. 2006;1:1306–1311. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Brandt R. The tau proteins in neuronal growth and development. Front Biosci. 1996;1:d118–d130. doi: 10.2741/a120. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Knapska E, Orsini CA, Rabinak CA, Zimmerman JM, Maren S. Fear extinction in rodents. Curr Protoc Neurosci Chapter. 2009;8 doi: 10.1002/0471142301.ns0823s47. Unit8.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, Sweatt JD, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Renaudineau S, Poirier R, Poucet B, Save E, Laroche S. The formation and stability of recognition memory: what happens upon recall? Front Behav Neurosci. 2010;4:177. doi: 10.3389/fnbeh.2010.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1. Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, Bussey TJ, Singewald N, Holmes A. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Iles JP, Mayell SJ, Rodgers RJ. Prior test experience compromises the anxiolytic efficacy of chlordiazepoxide in the mouse light/dark exploration test. Behav Brain Res. 2001;122:159–167. doi: 10.1016/s0166-4328(01)00184-x. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Karlsson RM, Choe JS, Cameron HA, Thorsell A, Crawley JN, Holmes A, Heilig M. The neuropeptide Y Y1 receptor subtype is necessary for the anxiolytic-like effects of neuropeptide Y, but not the antidepressant-like effects of fluoxetine, in mice. Psychopharmacology (Berl) 2008;195:547–557. doi: 10.1007/s00213-007-0945-2. [DOI] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Bal M, Kavalali ET, Monteggia LM. Selective impact of MeCP2 and associated histone deacetylases on the dynamics of evoked excitatory neurotransmission. J Neurophysiol. 2011;106:193–201. doi: 10.1152/jn.00751.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ. Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn Mem. 2002;9:402–407. doi: 10.1101/lm.49602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET. Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol. 2011;21:275–282. doi: 10.1016/j.conb.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, Zoghbi HY. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66:771–782. doi: 10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- Robleto K, Poulos AM, Thompson RF. Brain mechanisms of extinction of the classically conditioned eyeblink response. Learn Mem. 2004;11:517–524. doi: 10.1101/lm.80004. [DOI] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville LH, Voss HU, Glover G, Ballon DJ, Liston C, Teslovich T, Van Kempen T, Lee FS, Casey BJ. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lattal KM. Is an epigenetic switch the key to persistent extinction? Neurobiol Learn Mem. 2011;96:35–40. doi: 10.1016/j.nlm.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 2007;146:907–921. doi: 10.1016/j.neuroscience.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng SM, McLeod F, Bailey ME, Cobb SR. Synaptic plasticity deficits in an experimental model of rett syndrome: long-term potentiation saturation and its pharmacological reversal. Neuroscience. 2011;180:314–321. doi: 10.1016/j.neuroscience.2011.01.061. [DOI] [PubMed] [Google Scholar]
- Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J Neurosci. 2010;30:13586–13596. doi: 10.1523/JNEUROSCI.0849-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]