

Abstract
The chlorosulfolipid mytilipin A has been synthesized in racemic form in only seven steps and in enantioenriched form in eight steps. Key transformations include a highly diastereoselective bromoallylation of a sensitive α,β-dichloroaldehyde, a kinetic resolution of a complex vinyl epoxide, a convergent and highly Z-selective alkene cross metathesis, and a chemoselective and diastereoselective dichlorination of a complex diene.
Keywords: chlorosulfolipids, alkene cross metathesis, total synthesis, substrate control, kinetic resolution
The chlorosulfolipids are a most unusual group of lipids known since the independent reports of chlorinated C22 lipids isolated from the alga Ochromonas danica by the Vagelos and Haines groups in 1969.[1,2] These first compounds were characterized as bis-sulfates of 1,14-docosanediol, with varying levels of chlorination up to the hexachloride, now known as danicalipin A (1, Figure 1). In the decades since, this family has grown to include ill-characterized C24 analogs (also from O. danica),[3] the unusual chlorovinyl sulfate-containing lipid malhamensilipin A (2) that was isolated from the related alga Poterioochromonas malhamensis by Slate and Gerwick,[4] and the mytilipins, a small group of lipids isolated in very small quantities from toxic Adriatic mussels and reported by Fattorusso and co-workers.[5,6] These last compounds include the C15 lipid mytilipin A (3), which has some structural resemblance to danicalipin A and malhamensilipin A, and mytilipins B and C (4 and 5), two C24 lipids with an astounding level of stereochemical complexity that includes 11 chlorine-bearing centers. More recently, Okino has uncovered more natural congeners in the danicalipin series in a careful study of O. danica,[7] and the Sheu group isolated analogs of mytilipin A from an octocoral from the Strait of Taiwan.[8] Given the diversity of sources and structures, it is reasonable to expect that more members will be added to the chlorosulfolipid family in the years to come.[9]
Figure 1.

Representative chlorosulfolipids
Over the past four years, several groups, including our own, have reported syntheses of members of the chlorosulfolipid family. The Carreira group registered the first synthesis when they disclosed an elegant route to racemic mytilipin A.[10] Shortly thereafter, our group reported the stereochemical elucidation of danicalipin A and its synthesis in racemic form,[11] followed by the structural revision and enantioselective synthesis of malhamensilipin A.[4b,12] These three syntheses featured similar overall strategies with introduction of the polar substituents via alkene oxidation reactions.[13] The Yoshimitsu group developed a substantially different approach, featuring their method for stereospecific deoxydichlorination of enantioenriched epoxides,[14] which culminated in clever asymmetric syntheses of both mytilipin A and danicalipin A,[15] the latter of which was contemporaneous with a third very different, creative approach from the Matsuda group.[16] Finally, the Carreira group recently reported the synthesis of the proposed structure of mytilipin B as shown in Figure 1, and came to the conclusion that this chlorosulfolipid requires stereochemical revision.[17]
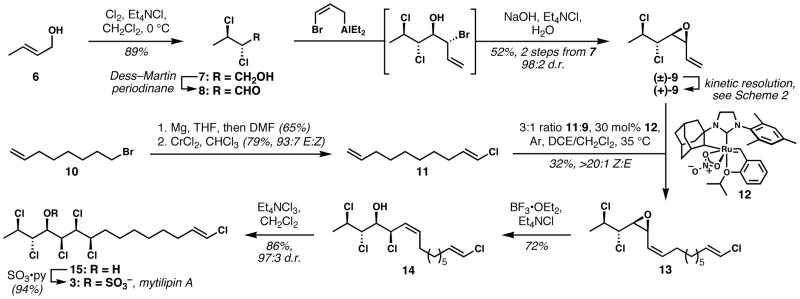
We now report a new synthesis of mytilipin A via a longest linear sequence of only seven steps for racemic material, eight for enantioenriched chlorosulfolipid, with several key features: (1) a highly diastereoselective haloallylation of a sensitive α,β-dichloroaldehyde; (2) a kinetic resolution of a complex vinyl epoxide; (3) a convergent olefination via Z-selective alkene cross metathesis; and (4) excellent levels of stereocontrol throughout.
Crotyl alcohol was treated with molecular chlorine in the presence of Et4NCl to afford the anti-dichloride 7; presumably this procedure generates Mioskowski’s reagent[18] (Et4NCl3) in situ. Oxidation with the Dess–Martin periodinane followed by a careful workup afforded the sensitive and volatile aldehyde 8 in crude form, which was immediately converted to vinyl epoxide 9. The bromoallylaluminum reagent[19] shown added with high diastereocontrol consistent with both the Felkin–Anh and Cornforth models,[20,21] and the resulting bromohydrin was converted to the epoxide upon treatment with aqueous base. The moderate yield in this case might be attributed to volatility of the intermediate aldehyde and the product vinyl epoxide; in related systems with longer alkyl chains, yields of about 75% have been obtained. Although strategically attractive in the context of the chlorosulfolipids, carbonyl addition reactions to α,β-dichloroaldehydes can be plagued by facile elimination reactions, and only Yoshimitsu has previously embraced this type of C–C bond forming reaction en route to these targets.[15] In our studies, we have found that a variety of allylation reagents that presumably react via closed transition states provide products with high levels of diastereoselectivity and without destruction of the sensitive substrate.
The alkene partner 11 required for the convergent step was obtained in two steps from 8-bromo-1-octene. Formylation of the Grignard reagent derived from 10 followed by Takai chloroolefination[22] afforded 11 in multigram scale.
The key convergent bond-formation—Z-selective alkene cross metathesis[23] using the Grubbs cycloadamantyl catalyst 12[24]—generated Z-vinyl epoxide 13 with complete control of alkene geometry. We attribute the relatively low yield in this particular case to catalyst deactivation by the vinyl epoxide; however, this step compares favorably overall with previous syntheses of this type of epoxyalkene via poorly selective Wittig reactions that require several extra steps.[10–12] Unfortunately, we were unable to achieve effectively more than a single turnover with 12;[25] however, this reaction is notable for the ability to directly introduce the vinyl chloride, which eliminates at least three post-convergence steps.[10,15a] The success of the cross metathesis in the face of possible RCM pathways can be attributed to: 1. the intrinsically slow reactivity of vinyl chlorides; 2. the slow kinetics of cyclooctene formation; and 3. the high kinetic selectivity of catalyst 12 for Z-alkenes, which presumably prevents reaction with the E-vinyl chloride in either RCM or cross metathesis events. Vinyl epoxide chlorinolysis with inversion of configuration[11,12] proceeded smoothly under the conditions shown, providing diene 14. Uncertain about the reactivity of the isolated vinyl chloride relative to the particularly electron-deficient allylic chloride in 14, we were pleased to observe a completely chemoselective, highly diastereoselective (93:7 d.r. of crude product, purified to 97:3), and efficient chlorination using Mioskowski’s reagent. The generation of hexachloride product 15 constitutes a formal synthesis of mytilipin A, and sulfation of the secondary alcohol according to Carreira’s conditions[10] completes the seven step (longest linear sequence) synthesis of racemic chlorosulfolipid. This route has enabled the preparation of over 100 mg of the target molecule to date, with a nearly 9% yield over the entire sequence. Although the final steps of the synthesis are related to those previously reported by us and the Carreira group, our rapid access to 13 takes advantage of a completely new strategy designed for general access to several of the chlorosulfolipids.
The convergent Z-selective alkene cross metathesis to form 13 is noteworthy for its complete diastereoselectivity if not for its efficiency. To see if the extremely high selectivity we observed was general for vinyl epoxides, as well as to investigate the low catalytic activity of 12 with respect to the specific case in Scheme 1, we tested the reactivity of cis-3,4-epoxy-1-undecene (18) with 1-decene (eq 1). With 10 mol% catalyst 12, complete conversion to Z-vinyl epoxide 19 was observed (83% isolated yield, >20:1 Z:E). With 1 mol% catalyst, the product was isolated in 43% yield (incomplete conversion), with equal selectivity. Therefore, it appears that vinyl epoxides are subject to highly Z-selective cross metathesis with catalyst 12, and that the poor efficiency observed in the convergent step to form 13 is likely specific to chlorinated substrates of type 9.
Scheme 1.
Synthesis of mytilipin A via a seven-step longest linear sequence
 |
(1) |
In principle, adaptation of our route to the enantioselective preparation of mytilipin A simply requires access to enantioenriched dichloroalcohol 7. However, methods for the asymmetric dichlorination of alkenes, including the allylic alcohol substrates pertinent to our synthesis, are not yet at a level of sophistication appropriate for the first step in complex molecule synthesis.[26] We contemplated accessing enantioenriched dichloroalcohols using either the stereospecific deoxydichlorination of an optically active epoxyalcohol derivative according to the method of Yoshimitsu,[14] or by resolution. Because it would add only one step to the sequence, we opted to investigate resolution strategies. Unfortunately, attempts at kinetic resolution of dichloroalcohol 7 (and dichloroaldehyde 8) using a variety of approaches were unsuccessful; however racemic vinyl epoxide 9 could be resolved by adapting the meso-epoxide desymmetrizing chlorinolysis of Denmark and co-workers (Scheme 2).[27] The strong preference for ring-opening of the vinyl epoxide at the allylic position (as opposed to the position proximal to the electron-withdrawing chloride residues) is presumably key to success; a selectivity factor of 13 was realized, allowing for material of 93.5:6.5 e.r. to be obtained at 57% conversion (by NMR analysis) and in 43% isolated yield.[28] On the basis of this result, we have defined an eight-step enantioselective synthesis of mytilipin A.[29,30] More details about this interesting resolution will be forthcoming in a full account.
Scheme 2.

Kinetic resolution affords enantioenriched vinyl epoxide 9
A few short years ago, the highly chlorinated lipids shown in Figure 1 might have appeared to be intractable problems for chemical synthesis, because of the dearth of previous work on stereochemically complex polychlorinated compounds. Now, with the recent advances made by several groups,[10–16] and with the design and execution of our new direct synthesis described here, it has become clear that the chlorosulfolipids can be assembled in a relatively straightforward fashion. The synthesis of racemic mytilipin A that we report requires only seven linear steps from commercially available starting materials and features only productive chemical transformations. Using kinetic resolution of a vinyl epoxide, enantioenriched chlorosulfolipid can be obtained with only one additional operation. Furthermore, our synthesis demonstrates that α,β-dichloroaldehydes can participate in efficient and highly stereoselective carbonyl addition reactions, and that Z-selective alkene cross metathesis is a powerful method for convergent olefination en route to the chlorosulfolipids. The successful application of these catalysts in this context further documents the ever-expanding reach of metathesis processes for stereocontrolled natural product synthesis.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 GM086483). C.D.V. is grateful for additional funding from an AstraZeneca Excellence in Chemistry Award, an Eli Lilly Grantee Award, and an A. P. Sloan Foundation Fellowship. J.S.C. is supported by a National Science Foundation Graduate Research Fellowship. D.K.B. was supported by a UC Irvine–Eli Lilly Graduate Fellowship and a UC Irvine Physical Sciences Dissertation Fellowship. We thank Materia for generous donation of metathesis catalyst 12, and Dr. Martin Schnermann, Dr. Paresma Patel, Dr. Vanessa Marx, and Prof. Robert Grubbs for invaluable assistance with the convergent metathesis step.
Footnotes
Supporting information for this article is available on the WWW at http://www.angewandte.org or from the author.
Dedicated to Professor Larry Overman on the occasion of his 70th birthday.
References
- 1.Elovson J, Vagelos PR. Proc Natl Acad Sci USA. 1969;62:957–963. doi: 10.1073/pnas.62.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haines TH, Pousada M, Stern B, Mayers GL. Biochem J. 1969;113:565–566. doi: 10.1042/bj1130565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haines TH. Prog Chem Fats Lipids. 1971;11:297–345. [Google Scholar]
- 4.(a) Chen JL, Proteau PJ, Roberts MA, Gerwick WH, Slate DL, Lee RH. J Nat Prod. 1994;57:524–527. doi: 10.1021/np50106a015. [DOI] [PubMed] [Google Scholar]; (b) Pereira AR, Byrum T, Shibuya GM, Vanderwal CD, Gerwick WH. J Nat Prod. 2010;73:279–283. doi: 10.1021/np900672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Ciminiello P, Fattorusso E, Forino M, Magno S, Di Rosa M, Ianaro A, Poletti R. J Org Chem. 2001;66:578–582. doi: 10.1021/jo001437s. [DOI] [PubMed] [Google Scholar]; (b) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Di Rosa M, Ianaro A, Poletti R. J Am Chem Soc. 2002;124:13114–13120. doi: 10.1021/ja0207347. [DOI] [PubMed] [Google Scholar]; (c) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Magno S, Di Meglio P, Ianaro A, Poletti R. Tetrahedron. 2004;60:7093–7098. [Google Scholar]
- 6.The mytilipins initially went unnamed, and then were given the descriptive names hexachlorosulfolipid (for 3) and undecachlorosulfolipids A and B (for 4 and 5, respectively). Recently, the mytilipin name has been adopted in light of the origin of these compounds from the mussel Mytilus galloprovincialis.
- 7.Kawahara T, Kumaki Y, Kamada T, Ishii T, Okino T. J Org Chem. 2009;74:6016–6024. doi: 10.1021/jo900860e. [DOI] [PubMed] [Google Scholar]
- 8.Chao CH, Huang HC, Wang GH, Wen ZH, Wang WH, Chen IM, Sheu JH. Chem Pharm Bull. 2010;58:944–946. doi: 10.1248/cpb.58.944. [DOI] [PubMed] [Google Scholar]
- 9.For reviews on the chlorosulfolipids, see: Bedke DK, Vanderwal CD. Nat Prod Rep. 2011;28:15–25. doi: 10.1039/c0np00044b.Nilewski C, Carreira EM. Eur J Org Chem. 2012:1685–1698.
- 10.Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]
- 11.Bedke DK, Shibuya GM, Pereira A, Gerwick WH, Haines TH, Vanderwal CD. J Am Chem Soc. 2009;131:7570–7572. doi: 10.1021/ja902138w. [DOI] [PubMed] [Google Scholar]
- 12.Bedke DK, Shibuya GM, Pereira AR, Gerwick WH, Vanderwal CD. J Am Chem Soc. 2010;132:2542–2543. doi: 10.1021/ja910809c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibuya GM, Kanady JS, Vanderwal CD. J Am Chem Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimitsu T, Fujumoto N, Tanaka T. J Org Chem. 2009;74:696–702. doi: 10.1021/jo802093d. [DOI] [PubMed] [Google Scholar]
- 15.(a) Yoshimitsu T, Fujumoto N, Nakatani R, Kojima N, Tanaka T. J Org Chem. 2010;75:5425–5437. doi: 10.1021/jo100534d. [DOI] [PubMed] [Google Scholar]; (b) Yoshimitsu T, Nakatani R, Kobayashi A, Tanaka T. Org Lett. 2011;13:908–911. doi: 10.1021/ol1029518. [DOI] [PubMed] [Google Scholar]
- 16.Umezawa T, Shibata M, Kaneko K, Okino T, Matsuda F. Org Lett. 2011;13:904–907. doi: 10.1021/ol102882a. [DOI] [PubMed] [Google Scholar]
- 17.Nilewski C, Deprez NR, Fessard TC, Li DB, Geisser RW, Carreira EM. Angew Chem Int Ed. 2011;50:7940–7943. doi: 10.1002/anie.201102521. [DOI] [PubMed] [Google Scholar]
- 18.Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Angew Chem Int Ed. 1997;36:2341–2344. [Google Scholar]
- 19.The corresponding chloroallylaluminum reagents have been previously reported: Hosomi A, Kohra S, Tominaga Y, Ando M, Sakurai H. Chem Pharm Bull. 1987;35:3058–3061.
- 20.The addition of organometallic nucleophiles to α-chloroaldehydes generally proceeds to afford the anti product, consistent with the polar Felkin–Anh model (hyperconjugative stabilization of the transition state/steric control), though the Cornforth model (dipole minimization/ steric control) also accounts for the stereochemistry of the products. For an excellent discussion and lead reference, see: Cee VJ, Cramer CJ, Evans DA. J Am Chem Soc. 2006;128:2920–2930. doi: 10.1021/ja0555670.
- 21.Additions to simple α-chloroaldehydes are often highly selective. See: Britton R, Kang B. Nat Prod Rep. 2013;30:227–236. doi: 10.1039/c2np20108a.
- 22.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408–7410. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 23.For the first application of Z-selective alkene cross-metathesis in natural product synthesis, see: Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461–466. doi: 10.1038/nature09957.
- 24.(a) Endo K, Grubbs RH. J Am Chem Soc. 2011;133:8525–8527. doi: 10.1021/ja202818v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keitz BK, Endo K, Patel PR, Herbert MB, Grubbs RH. J Am Chem Soc. 2012;134:693–699. doi: 10.1021/ja210225e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Herbert MB, Marx VM, Pederson RL, Grubbs RH. Angew Chem Int Ed. 2012;52:310–314. doi: 10.1002/anie.201206079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rosebrugh LE, Herbert MB, Marx VM, Keitz BK, Grubbs RH. J Am Chem Soc. 2013;135:1276–1279. doi: 10.1021/ja311916m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Under otherwise identical conditions, we obtained 10% yield of 13 with 10 mol% 12, 34% of 13 with 50 mol% 12, and only 39% of 13 with 100 mol% 12. However, we note that we have seen examples where more than one turnover is observed, and do not believe that there is a mechanistic explanation for the limitation in terms of turnover; rather, catalyst decomposition appears to be competitive with the desired transformation.
- 26.For the most advanced system to date for catalytic asymmetric dichlorination of allylic alcohols, and an excellent lead reference, see: Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. J Am Chem Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m.For a stoichiometric reagent system that enabled enantioselective dichlorination in the context of a complex natural product synthesis, see: Snyder SA, Tang ZY, Gupta R. J Am Chem Soc. 2009;131:5744–5745. doi: 10.1021/ja9014716.
- 27.(a) Denmark SE, Barsanti PA, Wong KT, Stavenger RA. J Org Chem. 1998;63:2428–2429. doi: 10.1021/jo9801420. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Barsanti PA, Beutner GL, Wilson TW. Adv Synth Catal. 2007;349:567–582. [Google Scholar]
- 28.Surprisingly, the monomeric catalyst described in ref 27a was less selective than 16. That contrasts with the report in ref 27b that dimeric catalysts of type 16 were less effective at meso-epoxide desymmetrizations than the corresponding monomeric catalysts.
- 29.The stereochemical outcome of the resolution of vinyl epoxide (±)-9 was opposite to what we expected on the basis of Denmark’s epoxide desymmetrization; ultimately, this unanticipated outcome resulted in the synthesis of the unnatural enantiomer of mytilipin A.
- 30.The Carreira group used a Sharpless epoxidation of racemic anti-4,5-dichloro-2-buten-1-ol followed by diastereomer separation to render their synthesis of mytilipin A enantioselective: Geisser RW. D Sc Dissertation. ETH Zürich; 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.