

Abstract
Scope
The flavanol (-)-epicatechin (Epi), a component of cacao, has cardiac protective benefits in humans. Our previous study demonstrated Epi has δ-opioid receptor (DOR) binding activity and promotes cardiac protection. Here we examined the effects of 10 days of Epi treatment on: cardiac mitochondrial respiration, ROS production, calcium swelling, and mitochondrial membrane fluidity.
Methods & Results
Mice were randomized into four groups: (1) Control (Saline), (2) Naltrindole (Nalt; DOR antagonist), (3) Epi, and (4) Epi+Nalt and received 1 mg kg−1 Epi or water via oral gavage. Nalt groups received 5 mg kg−1 ip per day for 10 days. Significant increases in mitochondrial respiration and enhanced free radical production during state 3 respiration were observed with Epi. Additionally, we observed significant increases in rigidity of mitochondrial membranes and resistance to calcium induced mitochondrial swelling with Epi treatment. Blocking the DOR with Nalt resulted in decreases in all of the observed parameters by Epi treatment.
Conclusion
These findings indicate that Epi induces an integrated response that includes metabolic and structural changes in cardiac mitochondria resulting in greater functional capacity via DOR. Mitochondrial targeted effects of epicatechin may explain the physiologic benefit observed on cardiac protection and support epicatechin’s potential clinical application as a cardiac protective mimetic.
Keywords: epicatechin, heart, superoxide, opioids
1. Introduction
It is of great interest to identify cardiac protective natural products and nutritional supplements to improve cardiac performance and possibly attenuate the pathological progression of cardiac disease. Evidence suggests that a common theme among cardiovascular diseases is increased mitochondrial damage and dysfunction [1, 2]. Flavonoids can protect cells from insults that lead to mitochondria mediated cell death [3, 4]. (-)-Epicatechin (Epi), the predominant flavonoid present in dark chocolate is capable of inducing cardiac protection. Studies have linked the consumption of small amounts of dark chocolate (a product of cacao) with notable reductions in the risk of cardiovascular diseases [5]. Interestingly, epicatechin plays a role in preserving mitochondrial integrity in the setting of disease and enhances mitogenesis [6], and has been shown to regulate mitochondrial volume and cristae abundance in rodent myocardium [7, 8].
Our group has focused on cardiovascular effects of epicatechin and has demonstrated that at low doses there is reduced myocardial injury and enhanced cardiac protective signaling induced by epicatechin that is δ-opioid receptor (DOR) dependent [9]. Opioid receptors have been implicated in protection against various stresses in several organs, including the heart. Recent studies suggest that activation of opioid receptors contributes to the initiation of cardiac protective signaling pathways [10, 11]. Some evidence of opioid-mitochondrial interactions exists where DOR activate mitochondrial ATP-sensitive K channels (mKATP) in cardiac myocytes [12-14]. Whether the downstream changes induced by epicatechin on mitochondrial function and structure are also opioid receptor dependent is unknown. In the current study we tested the hypothesis that epicatechin enhances mitochondrial function and structure by activating DOR. We examined the effects of low dose epicatechin administration (10 days) in the presence and absence of DOR blockade on mitochondrial 1) respiration, 2) ROS production, 3) calcium-induced swelling, and 4) mitochondrial membrane fluidity.
2. Materials and Methods
Animals
All animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals, and animal use protocols were approved by the Veterans Affairs San Diego Healthcare System Institutional Animal Care and Use Committee (San Diego, CA). C57Bl/6 male mice (aged 8–10 wk, and 24–26 g body wt) were purchased from Jackson Laboratories (Bar Harbor, ME). The animals were kept on a 12-h:12-h light-dark cycle in a temperature- and humidity-controlled room.
Experimental design
Mice were randomly assigned to the following 4 groups: control, naltrindole (Nalt), Epi, and Epi+Nalt. Control animals were given 0.3 ml of saline (vehicle) by oral gavage for 10 days. Epi was administered by oral gavage daily (1 mg/kg body wt, dissolved in 0.3 ml of saline) for 10 days. Nalt was administered by intraperitoneal injection daily (5 mg/kg body wt, dissolved in 0.3 ml of saline) for 10 days.
Mitochondrial isolation
Mice were sacrificed and hearts removed. Ventricles were placed in ice-cold mitochondrial isolation media (MIM: 0.3M Sucrose, 10mM HEPES, 250μM EDTA), minced, and then homogenized with a Tissuemiser. Homogenates were rinsed in MIM. Samples were centrifuged at 600g to clear nuclear/membrane debris. The resulting supernatant was spun at 8,000g for 15min. The resulting pellet was resuspended in MIM in the presence of 1mM BSA followed by another 8,000g spin for 15min. The resulting pellet was resuspended in isolation buffer with BSA and spun again at 8,000g. Metabolically active mitochondria were then suspended in 150μL MIM for functional studies.
To isolate pure mitochondria for membrane fluidity analysis, the washing steps were repeated with MIM in a final 2 mL resuspension of the pellet in mitochondrial re-suspension buffer (MRB: 500μM EDTA, 250mM mannitol, 5mM HEPES). The mitochondria were layered on top of a 30% Percoll/70% MRB solution. The Percoll gradient was spun at 95,000g for 30min. The mitochondrial band was removed from the gradient, and volume was increased 10-fold with MRB to remove the Percoll by an 8,000g centrifugation for 15min. The mitochondrial pellet was re-suspended in 50-150μl of MRB and subjected to further analysis.
Mitochondrial respiration by Oxygraph analysis
Mitochondrial respiratory function was studied according to published protocols [15]. Oxygen consumption was measured using a Clark-type oxygen electrode (Oxygraph∣, Hansatech, Norfolk, UK) during the sequential additions of substrates and inhibitors to purified mitochondria. Purified mitochondria (~100-200 μg protein) were added to the oximetry chamber in a 300 ml solution containing 100 mM KCl, 75 mM mannitol, 25 mM sucrose, 5 mM H3PO4, 0.05 mM EDTA and 10 mM Tris-HCl, pH =7.2 at 37 °C. After 2 minutes of equilibration, 5 mM pyruvate and 5 mM malate were added and oxygen consumption followed for ~1-2 minutes (State 4). ADP (250 μM) was added to measure State 3 (phosphorylating) respiration. To switch from NAD+- to FAD+- linked respiration, we first eliminated complex I through the inhibition of the back electron transfer using 0.5 μM rotenone and triggered complex II activity by the addition of 10 mM succinate. Next, we inhibited complex III by the addition of 5 μM antimycin A. Complex IV activity was measured in the presence of 0.5 mM TMPD + 2 mM ascorbate. Oxygen utilization traces and rate determinations were obtained using Oxygraph software and normalized to protein concentrations.
ROS measurement by electron paramagnetic resonance (EPR)
For EPR studies, immediately after mixing mitochondria (0.1–0.2 mg of protein) with 70 mM 5-(diisopropoxyphosphoryl)-5-ethyl-1-pyrroline-N-oxide (DEPMPO) and appropriate combinations of the substrates, the mixture was loaded into 50 μl glass capillary tubes and introduced into the EPR cavity of a Magnettech MiniScope MS300 Benchtop spectrometer. We confirmed that the detected EPR signals are substrate specific, and not due to redox cycling in the studied mixtures, by lack of signals when DEPMPO was mixed with combinations of substrates and inhibitors in the absence of mitochondria. Assignment of the observed signals from mitochondria was confirmed through computer-assisted spectral simulation using the WinSim software (http://epr.niehs.nih.gov/pest.html). In most cases a mixture of signals due to DEPMPO-OOH and DEPMPO-OH adducts, with occasional contribution from a carbon-centered radical, was detected but the complete removal of these signals upon the inclusion of SOD confirmed that superoxide radical was the exclusive source of the observed EPR-active species. Signals were quantified by measuring the peak amplitudes of the observed spectra and normalized by mitochondrial protein concentrations.
EPR for membrane fluidity
Hydrocarbon chain mobility was measured using fatty acid spin labeling EPR analysis using 5-nitroxyl stearate (5-DSA) as a spin probe [16, 17]. The number designation indicates the relative position of the nitroxide on the stearic acid relative to the polar carboxylic group. In the case of 5-DSA, the spin probe is firmly held in place by the head groups of the lipids, which is reflected in broad EPR lines. Purified mitochondrial membranes were incubated for 15 minutes with 5-DSA (1 mM final concentration) at 25°C. The mixture was then loaded into a 50 μl-glass capillary tube and inserted into the EPR cavity of a MiniScope MS300 Benchtop spectrometer (Magnettech, Berlin, Germany), maintained at 37°C, where the EPR spectra registered. EPR conditions were the following: microwave power, 5 mW; modulation amplitude, 2 G; modulation frequency, 100 kHz; sweep width, 150 G centered at 3349.0 G; scan rate, 7.5 G/s, with each spectrum representing the average of 5 scans. The fluidity parameters T∥ and T⊥ were used to calculate the order parameter as previously described [16].
Calcium-induced mitochondrial swelling assay
Calcium-induced mitochondrial swelling was measured on an Infinite M200 plate reader at 540nm over the span of 20 minutes. Crude mitochondria (0.5μg/uL) in the absence of calcium were loaded onto a clear flat bottom 96 well plate and challenged with 250μM calcium with absorbance measured every 10 seconds. Change of absorbance at 540nm was compared between samples.
Statistics
All data are presented as mean ± SEM. GraphPad Prism 6 software (GraphPad Software, Inc., San Diego, CA, USA) was used for all statistical analysis. Statistical analyses were performed by one-way ANOVA followed by a Bonferroni’s post-hoc test.
3. Results
DOR inhibition attenuates epicatechin mediated increases in mitochondrial respiration
Mitochondria from the hearts of epicatechin treated mice have greater respiration rates during state 3 conditions in response to the addition of malate, pyruvate and ADP (Figure 1A). Upon the administration of Nalt, Epi treated hearts showed attenuated state 3 respiration. No changes were observed in state 4 respiration (Figure 1B), FAD+-linked respiration through complex II in the presence of succinate (Figure 1C), or altered complex IV activity in response to 2,2,4-trimethyl-1,3-pentanediol (TMPD) and ascorbate (Figure 1D). These results indicate that DOR activation by Epi enhances cardiac mitochondrial respiratory function, likely through alteration of complex I activity while effects on all other components of the electron transport chain are equivocal.
Figure 1. Epicatechin alters mitochondrial respiration rates dependent on opioid receptor stimulation.

Mitochondria isolated from epicatechin treated animals show increased respiratory rates during state 3 as triggered by malate + pyruvate + ADP (complex 1 substrates) and this effect was blocked by naltrindole administration in conjunction with epicatechin (A). No changes were observed in state 4 respiraiton with complex 1 substrates (B) or with complex II substrates under state 3 (C) and 4 (D) respiraiton. Respiration was calculated as nanomoles of oxygen consumed per minute per milligram of protein. The values are expressed as mean±SEM of 4 to 6 mice. p<0.05 vs Ctrl (*) or vs epicatechin (+).
Epicatechin treated mice have enhanced mitochondrial ROS production under state 3 conditions
The role of ROS remains controversial in whole organ and whole animal models. There is increasing evidence that low concentrations of ROS may play a subtle role in cellular signaling and cardiac protection [18]. ROS have been shown to activate various pro-survival kinases in cardiac myocytes [19]. Mitochondria isolated from epicatechin treated hearts produced greater superoxide signals during state 3 respiration whereas blocking the DOR attenuated the epicatechin-induced increase in ROS production (Figure 2A). ROS production under state 3 respitation with complex II substrates resulted in elevation only with epicatechin with naltrindole group and not with any other group. ROS production under state 4 respiration with complex I and II substrates were not altered in any group relative to control. This result implies that complex I is the likely source of mitochondrial ROS by epicatechin that may have signaling potential, though under certain conditions it is possible that opioid antagonist may target mitochondrial ROS as well.
Figure 2. ROS production is enhanced by epicatechin dependent on opioid receptor stimulation.

Electron paramagentic resonanse (EPR) spin trapping spectroscopy was used to trace DEPMPO-hydroxyl radical adduct originated from superoxide radical trapping. EPR spectra were acquired after mixing isolated cardiac mitochondria from hearts of drug treated animals with DEPMPO, substrates (malate/pyruvate, succinate) with (state 3 respiration) or without ADP (state 4 respiration). Cardiac mitochondria form epicatechin treated animals showed increased superoxide production during state 3 respiration with complex 1 substrates and this superoxide increase was blocked with naltrindole administration (A). No changes were observed in superoxide generation under state 4 conditions with complex I substrates (B). In isolated mitochondrial from naltrindole treatment alone there was a trend towards increased superoxide which was significantly increased with combination of epicatechin and naltrindole under state 3 conditions with complex II substrates; no changes were observed with epicatechin alone under these conditions (C). No changes was observed in superoxide signal under state 4 conditions with complex 2 substrates (D). Representative EPR spectra are included above the summary data for each conditon and treatment. The values are expressed as mean±SEM of 4 to 6 mice. p<0.05 vs Ctrl (*) or vs epicatechin (+).
Epicatechin alters mitochondrial membrane structure
Doxyl stearic acid spin labels can be used to reflect fluidity closer to the membrane surface (5-DSA) by EPR. By analyzing the spectrum of 5-DSA an order parameter can be calculated from the outer and inner hyperfine splitting 2T⁄⁄ and 2T⊥ (Figure 3A). Order parameter reveals lipid peroxidation and permeability variations due to oxidative insults or redox changes. Epi treatment increased the mitochondrial membrane order parameter. Furthermore, Epi treatment caused disproportionate variations in lipid composition by increasing rigid membrane regions vs. fluid microenvironments as revealed by increased rigid/fluid ratio (Figure 3B). This effect of epicatechin was blocked by naltrindole (Figure 3B).
Figure 3. Mitochondrial membrane fluidity/rigidity is altered by epicatechin in an opioid receptor dependent manner.
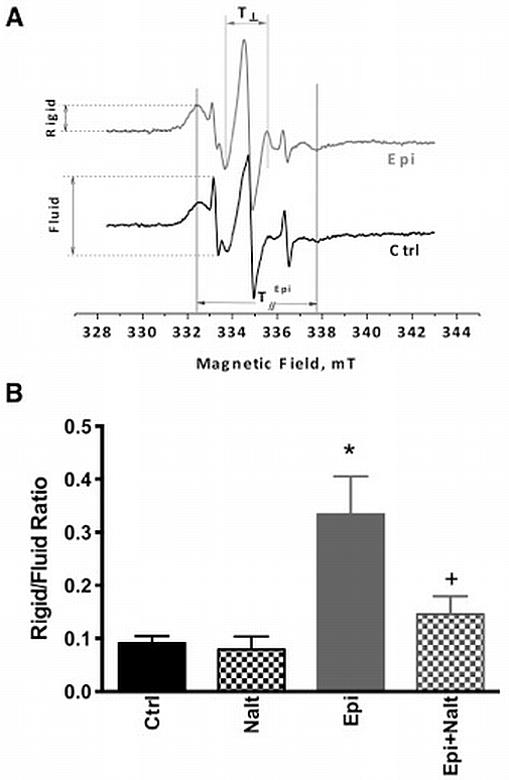
Isolated mitochondria from heart tissue were treated with the spin label 5-DSA followed by EPR spectroscopy to estimate the fluidity of the membrane (rigid/fluid ratio). Two component spectra were registered when 5-DSA was used as a probe reflecting rigid as well as fluid membrane microenvironments, T⁄⁄ and T⊥ refer to outer and inner hyperfine splitting (A). Cardiac mitochondria from epicatechin treated animals showed significant increases in membrane rigididity which was blocked with naltrindole (B). The values are expressed as mean±SEM of 3 to 5 mice. p<0.05 vs Ctrl (*) or vs epicatechin (+).
Improved Ca2+ tolerance of mitochondria by epicatechin is mediated by DOR
The mitochondrial membrane structural change is reflected in increased resistance to calcium (250 μm) load in isolated mitochondria (Figure 4). Mitochondria from epicatechin treated hearts are more resistant to calcium-induced mitochondrial swelling than control and this effect is blocked by naltrindole. To confirm opioid specific changes to mitochondrial calcium loading we added two additional groups with SNC-121 (a DOR selective agonist, IP, 5mg/kg day for 10 days) with and without naltrindole treatment. SNC-121 resulted in similar protection of mitochondria from calcium swelling as that observed with epicatechin and this protective effect was blocked by naltrindole (Figure 4).
Figure 4. Altered calcium induced mitochondrial swelling by epicatechin is opioid receptor dependent.
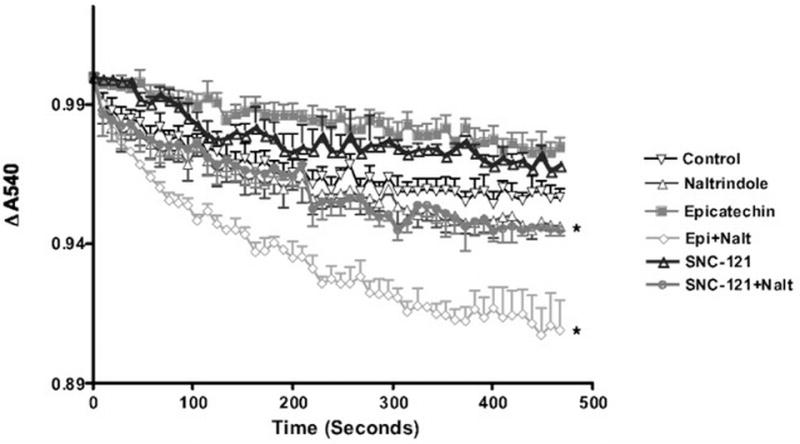
Mitochondria isolated from cardiac tissue of all control and treatment groups were challenged with calcium and decreases in absorbance at 540nm were observed over time. Epicatechin and SNC-121 treated mice were resistant to calcium triggered mitochondrial swelling. This resistance to mitochondrial swelling was blocked in each group by naltrindole. The values are expressed as mean±SEM of 3 to 5 mice. p<0.05 vs Ctrl (*).
4. Discussion
The present data show that epicatechin can modulate mitochondrial function and structure via δ-opioid receptor stimulation in mouse myocardium. To our knowledge, this is the first time a flavonoid has been shown to produce a significant and sustained effect on mitochondrial energetics and structure through a receptor mediated mechanism. The results of the present study further expand evidence to support the concept that the DOR dependent effects of epicatechin treatment over an extended period of time directly alter mitochondrial function and structure.
Stimulation of DOR triggers many mechanisms at multiple levels in the cell to increase cardiac myocyte survival including preservation of Ca2+ homeostasis and increases in pro-survival signaling and antioxidant capacity [11, 20, 21]. Previous studies with DOR agonists have demonstrated an interaction of opioid receptors and KATP channels in the heart and other organ systems. We have previously shown that epicatechin-induced cardiac protection via DOR was blocked by 5-HD, a mKATP channel blocker, suggesting involvement of mitochondrial signaling in the protection [9]. McPherson et al. [22], found that 5-HD and BNTX (KATP channel blocker and selective DOR antagonist, respectively) antagonized the cardiac protective effects of several DOR specific agonists such as morphine and BW373U86, which indicates some selectivity of DOR in transducing these effects via KATP channels and downstream signaling pathways. Opening of the mitochondrial KATP channel potentially may alter the mitochondrial membrane potential and subsequently uncouple the electron transport chain (ETC). These data raise the possibility that activation of the KATP channel is important in initiating the DOR mediated mitochondrial effect of Epi. Dorta et al. [23] have previously demonstrated the effect of flavonoids on mitochondria energetics and possibly mitochondrial structure. They suggested that the chemical makeup of flavonols, including catechin, makes them ideal for integration into the mitochondria and altering liquid ordered states. However, their data in liver mitochondria showing that mitochondrial function is dysregulated suggest a possible apoptosis inducing effect. We show the opposite effect for epicatechin in our study. The differences in our findings may be explained by the differences in the chemical structure of (-)-epicatechin compared to the agents (i.e., quercetin, taxifolin, catechin and galangin) they tested and our use of a low dose (-)-epicatechin administration to the whole animal over many days vs. direct treatment of isolated mitochondria. It is possible that cardiac mitochondria in our treatment regimen were exposed to a much lower effective concentration of epicatechin that produced a signaling/protective effect rather than apoptosis. Recent data to support this contention have shown both in rodents and humans that low dose epicatechin is able to alter mitochondrial energetics and structure [6-8].
Reactive oxygen species traditionally are thought to damage DNA, proteins, and lipids to disrupt cell function [24-26]. With the discovery that reactive species such as nitric oxide have signaling potential this dogma has been challenged and the thought that low levels of ROS are beneficial to the cell have been advanced [27, 28]. (-)-Epicatechin and other flavonols at high doses are thought to work as antioxidants and low dose effects appear to be independent of the antioxidant potential [29]. Our data suggest that low dose Epi during state 3 respiration with complex I substrates produces a spike in superoxide generation. It is possible that this increase in ROS generation by Epi is linked to the cardiac protective effect. Low dose opioid treatment has been shown to trigger a cardiac protective signaling cascade in a ROS dependent manner where the protection is attenuated by application of free radical scavengers prior to agonist treatment [30, 31]. The possibility exists that Epi utilized opioid receptors via a similar mechanism of ROS generation to trigger cardiac protection.
Our data suggests that the modulation of mitochondrial function is mechanistically tied to a receptor mediated effect dependent on a selective molecular singling cascade. It is unclear how such signaling is transduced specifically to mitochondria. It is possible that other potential mediators of epicatechin induce mitochondrial changes. Lagoa et al. [32], showed that epicatechin stoichiometrically reduces purified cytochrome C. Alternatively, Epi may mediate mitochondrial responses to modulation of classic protective pathways involving mKATP channels. KATP is an important downstream effector in mediating the mitochondrial effect of opioids in the intact heart [14]. It is possible, alterations in mitochondrial permeability transition, membrane depolarization and mitochondrial volume associated with KATP opening may be involved in the protective effect of Epi in the heart that are reflective of the changes in mitochondrial function and structure we observed. We did not specifically test this possibility. Future studies employing a proteomic approach may be able to model changes in specific cellular compartments including mitochondria that potentially are induced by epicatechin and shed further light onto the molecular mechanism of epicatechin action.
We also observed that the opioid antagonist, naltrindole, has some ability to modulate mitochondrial function as with complex II substrates under state 3 conditions we observed increased generation of ROS when combined with epicatechin. Under this conditions no changes were observed with epicatechin. Such data are perplexing and suggest that both agonist and antagonist of opioidergic signaling may modulate mitochondrial function. It is possible that such an antagonist may possess mixed agonist/antagonist properties that may manifest under certain conditions as modification of naltrindole possess such mixed effects [33]. Further studies are needed to tease out this particular observation.
In summary, the results of the present study demonstrate that the effects of epicatechin on mitochondrial structure and function in the heart are dependent on opioid receptor stimulation. However, the precise molecular mechanism involved in mediating the mitochondrial effects of epicatechin through DOR remain unknown and deserve further investigation. Given the ability of epicatechin to regulate mitochondrial structure and function, it may be possible to advance a therapeutic benefit of epicatechin treatment in a variety of disease processes including but not limited to diabetes, aging in heart and brain, and neurodegenerative disorders that have pathophysiologies that involve mitochondrial defects.
Acknowledgments
This work was supported by grants from National Institutes of Health HL091071 (HHP) and HL107200 (HHP).
Abbreviations
- 5-DSA
5-nitroxyl stearate
- DEPMPO
5-(diisopropoxyphosphoryl)-5-ethyl-1-pyrroline-N-oxide
- DOR
delta opioid receptor
- Epi
epicatechin
- EPR
electron paramagnetic resonance
- Nalt
naltrindole
- MIM
mitochondrial isolation media
- ROS
reactive oxygen species
References
- 1.Rosenberg P. Mitochondrial dysfunction and heart disease. Mitochondrion. 2004;4:621–8. doi: 10.1016/j.mito.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Knowlton AA. Mitochondrial dynamics in heart failure. Congest Heart Fail. 2011;17:257–61. doi: 10.1111/j.1751-7133.2011.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang WT, Li J, Haung HH, Liu H, et al. Baicalein protects against doxorubicin-induced cardiotoxicity by attenuation of mitochondrial oxidant injury and JNK activation. J Cell Biochem. 2011;112:2873–81. doi: 10.1002/jcb.23201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang EJ, Min JS, Ku HY, Choi HS, et al. Isoliquiritigenin isolated from Glycyrrhiza uralensis protects neuronal cells against glutamate-induced mitochondrial dysfunction. Biochem Biophys Res Commun. 2012;421:658–64. doi: 10.1016/j.bbrc.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 5.Engler MB, Engler MM. The emerging role of flavonoid-rich cocoa and chocolate in cardiovascular health and disease. Nutr Rev. 2006;64:109–18. doi: 10.1111/j.1753-4887.2006.tb00194.x. [DOI] [PubMed] [Google Scholar]
- 6.Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Perkins G, et al. Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: effects of epicatechin rich cocoa. Clin Transl Sci. 2012;5:43–7. doi: 10.1111/j.1752-8062.2011.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huttemann M, Lee I, Malek MH. (-)-Epicatechin maintains endurance training adaptation in mice after 14 days of detraining. FASEB J. 2012;26:1413–22. doi: 10.1096/fj.11-196154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nogueira L, Ramirez-Sanchez I, Perkins GA, Murphy A, et al. (-)-Epicatechin enhances fatigue resistance and oxidative capacity in mouse muscle. J Physiol. 2011;589:4615–31. doi: 10.1113/jphysiol.2011.209924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panneerselvam M, Tsutsumi YM, Bonds JA, Horikawa YT, et al. Dark chocolate receptors: epicatechin-induced cardiac protection is dependent on delta-opioid receptor stimulation. Am J Physiol Heart Circ Physiol. 2010;299:H1604–9. doi: 10.1152/ajpheart.00073.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Zhang H, Li T, Zhang B. Involvement of adenosine monophosphate-activated protein kinase in morphine-induced cardioprotection. J Surg Res. 2011;169:179–87. doi: 10.1016/j.jss.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Peart JN, Gross GJ. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am J Physiol Heart Circ Physiol. 2006;291:H1746–53. doi: 10.1152/ajpheart.00233.2006. [DOI] [PubMed] [Google Scholar]
- 12.Cao Z, Liu L, Van Winkle DM. Activation of delta-and kappa-opioid receptors by opioid peptides protects cardiomyocytes via KATP channels. Am J Physiol Heart Circ Physiol. 2003;285:H1032–9. doi: 10.1152/ajpheart.01004.2002. [DOI] [PubMed] [Google Scholar]
- 13.Fang X, Tang W, Sun S, Weil MH. delta-Opioid-induced pharmacologic myocardial hibernation during cardiopulmonary resuscitation. Crit Care Med. 2006;34:S486–9. doi: 10.1097/01.CCM.0000246015.05214.5A. [DOI] [PubMed] [Google Scholar]
- 14.Schultz JE, Gross GJ. Opioids and cardioprotection. Pharmacol Ther. 2001;89:123–37. doi: 10.1016/s0163-7258(00)00106-6. [DOI] [PubMed] [Google Scholar]
- 15.Ali SS, Marcondes MC, Bajova H, Dugan LL, et al. Metabolic depression and increased reactive oxygen species production by isolated mitochondria at moderately lower temperatures. J Biol Chem. 2010;285:32522–8. doi: 10.1074/jbc.M110.155432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabbita SP, Butterfield DA, Hensley K, Shaw W, et al. Aging and caloric restriction affect mitochondrial respiration and lipid membrane status: an electron paramagnetic resonance investigation. Free Radic Biol Med. 1997;23:191–201. doi: 10.1016/s0891-5849(97)00043-9. [DOI] [PubMed] [Google Scholar]
- 17.Gabbita SP, Subramaniam R, Allouch F, Carney JM, et al. Effects of mitochondrial respiratory stimulation on membrane lipids and proteins: an electron paramagnetic resonance investigation. Biochim Biophys Acta. 1998;1372:163–73. doi: 10.1016/s0005-2736(98)00040-6. [DOI] [PubMed] [Google Scholar]
- 18.Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1433–40. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- 19.Rosc-Schluter BI, Hauselmann SP, Lorenz V, Mochizuki M, et al. NOX2-derived reactive oxygen species are crucial for CD29-induced pro-survival signalling in cardiomyocytes. Cardiovasc Res. 2012;93:454–62. doi: 10.1093/cvr/cvr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventura C, Spurgeon H, Lakatta EG, Guarnieri C, et al. Kappa and delta opioid receptor stimulation affects cardiac myocyte function and Ca2+ release from an intracellular pool in myocytes and neurons. Circ Res. 1992;70:66–81. doi: 10.1161/01.res.70.1.66. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Xia X, Zhang Y, Wang Q, et al. delta-Opioid receptor activation attenuates oxidative injury in the ischemic rat brain. BMC Biol. 2009;7:55. doi: 10.1186/1741-7007-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPherson BC, Yao Z. Morphine mimics preconditioning via free radical signals and mitochondrial K(ATP) channels in myocytes. Circulation. 2001;103:290–5. doi: 10.1161/01.cir.103.2.290. [DOI] [PubMed] [Google Scholar]
- 23.Dorta DJ, Pigoso AA, Mingatto FE, Rodrigues T, et al. The interaction of flavonoids with mitochondria: effects on energetic processes. Chem Biol Interact. 2005;152:67–78. doi: 10.1016/j.cbi.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Wagner BA, Buettner GR, Burnes CP. Free radical-mediated lipid peroxidation in cells: oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry. 1994;33:4449–4453. doi: 10.1021/bi00181a003. [DOI] [PubMed] [Google Scholar]
- 25.Dean RT, Fu S, Stocker R, Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem Journal. 1997;324:1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schraufstatter IU, Hyslop PA, Jackson J, Cochrane CC. Oxidant injury of cells. Int J Tis React. 1987;9:317–324. [PubMed] [Google Scholar]
- 27.VandenHoek TL, Becker LB, Shao Z, Li C, et al. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 28.Moncada S, Higgs EA. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995;9:1319–1330. [PubMed] [Google Scholar]
- 29.Xu JZ, Yeung SY, Chang Q, Huang Y, et al. Comparison of antioxidant activity and bioavailability of tea epicatechins with their epimers. Br J Nutr. 2004;91:873–81. doi: 10.1079/BJN20041132. [DOI] [PubMed] [Google Scholar]
- 30.Patel HH, Hsu A, Gross GJ. Delayed cardioprotection is mediated via a non-peptide delta opioid agonist, SNC-121, independent of opioid receptor stimulation. Bas Res Cardiol. 2004;99:38–45. doi: 10.1007/s00395-003-0438-3. [DOI] [PubMed] [Google Scholar]
- 31.Patel HH, Hsu A, Moore J, Gross GJ. BW373U86, a delta opioid agonist, partially mediates delayed cardioprotection via a free radical mechanism that is independent of opioid receptor stimulation. J Mol Cell Cardiol. 2001;33:1455–65. doi: 10.1006/jmcc.2001.1408. [DOI] [PubMed] [Google Scholar]
- 32.Lagoa R, Graziani I, Lopez-Sanchez C, Garcia-Martinez V, et al. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochim Biophys Acta. 2011;1807:1562–72. doi: 10.1016/j.bbabio.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 33.Ananthan S, Johnson CA, Carter RL, Clayton SD, et al. Synthesis, opioid receptor binding, and bioassay of naltrindole analogues substituted in the indolic benzene moiety. J Med Chem. 1998;41:2872–81. doi: 10.1021/jm980083i. [DOI] [PubMed] [Google Scholar]