

Abstract
It has been recognized that changes in mitochondrial structure plays a key role in development of cardiac dysfunction, and autophagy has been shown to exert maintenance of mitochondrial homeostasis effects. Our previous study found that anti-β1-adrenergic receptor autoantibodies (β1-AABs) could lead to cardiac dysfunction along with abnormalities in mitochondrial structure. The present study tested the hypothesis that β1-AABs may induce the decline in mitochondrial membrane potential (ΔΨm) by suppression of cardiac autophagy, which contributed to cardiac dysfunction. Male adult rats were randomized to receive a vehicle or peptide corresponding to the second extracellular loop of the β1 adrenergic receptor (β1-AAB group, 0.4 μg/g every two weeks for 12 weeks) and treated with rapamycin (RAPA, an autophagy agonist) at 5 mg/kg/day for two days before detection. At the 4th week, 8th week and 12th week of active immunization, the rats were sacrificed and cardiac function and the levels of cardiac LC3 and Beclin-1 were detected. ΔΨm in cardiac myocytes was determined by myocardial radionuclide imaging technology and JC-1 staining. In the present study, β1-AABs caused cardiac dysfunction, reduced ΔΨm and decreased cardiac autophagy. Treatment with RAPA markedly attenuated β1-AABs-induced cardiac injury evidenced by recovered ΔΨm. Taken together, these results suggested that β1-AABs exerted significant decreased ΔΨm, which may contribute to cardiac dysfunction, most likely by decreasing cardiac autophagy in vivo. Moreover, myocardial radionuclide imaging technology may be needed to assess the risk in developing cardiac dysfunction for the people who have β1-AABs in their blood.
Introduction
Cardiac dysfunction is the end-stage syndrome of various cardiovascular diseases. In recent years, the prevalence of cardiovascular diseases in China has increased from 31.4% in 1993 to 50.0% in 2003 [1]. Cardiovascular diseases have become the leading causes of death among Chinese adults [2] and myocardial dysfunction accounted for roughly 40% of all cardiovascular disease mortality [3]. Cardiovascular diseases, especially cardiac function deterioration, have become the main factors affecting human health in China. Despite advances in cardiac dysfunction treatment, mortality remains high, with 1-year mortality rates of nearly 38% in China [4]. There are still many unknown factors involved in the development of depressed cardiac function. Therefore, further exploration of its etiology is the key to reducing the mortality of cardiac dysfunction.
Accumulating evidence indicates that the changes in cardiac energy metabolism play an important role in the pathogenesis of cardiac function deterioration [5]. The regulation of cardiac energy metabolism is expected to become a new strategy for treatment of cardiac dysfunction [6]. As the energy source of myocardial cells, mitochondria structure and function play a central role in the maintenance of energy metabolism. Studies have confirmed that mitochondrial ATP production decreases in the failing heart and is linked with mitochondrial structural abnormalities [7] and a reduction in mitochondrial respiration [8]. Such mitochondrial dysfunction is always accompanied by alterations in cardiac cellular homeostasis and may lead to various heart diseases [9]. The mitochondrial membrane potential (ΔΨm) is currently considered an important parameter of mitochondrial function [10]. If ΔΨm is improved and energy supply is increased, it is possible that heart function may be improved. However, the cause of cardiac mitochondrial dysfunction is still elusive and thus some of the strategies to improve mitochondrial dysfunction have still not achieved the desired results [11].
In recent years, anti-β1-adrenergic receptor autoantibodies (β1-AABs) have been shown to be prevalently distributed in the sera of patients with dilated cardiomyopathy (DCM) [12], chronic Chagas heart disease [13] and heart failure caused by ischemic cardiomyopathy [14]. Binding of the specific autoantibody with the second extracellular loop of β1-adrenergic receptor (β1-AR-ECII) resulted in agonist-like effects, such as increasing the beating frequency of neonatal rat cardiomyocytes [15], enhancing atrial contractility [16], augmenting intracellular cyclic adenosine monophosphate (cAMP) accumulation [17] and activating the Ca2+ channels [18]. Ours [19] and others [12] researches have demonstrated that the long-term presence of β1-AABs resulted in myocardial remodeling in rats as well as impaired cardiac function. Moreover, the morphologic hallmarks of mitochondrial dysfunction that include mitochondria swelling and condensation were revealed by electron microscopy with the existence of β1-AABs [19]. It was not clear, however, whether the changes in ΔΨm (an indicator of mitochondrial function) were caused by β1-AABs. Yet another study has shown that the β-adrenergic receptor agonist isoproterenol can reduce autophagy, which is required for the maintenance of cellular homeostasis [20]. However, whether autophagy is changed with the long-term existence of β1-AABs remains largely unknown. If it does, what is the contribution of autophagy to the changes in ΔΨm?
Therefore, the aims of the present study were to identify the effects of β1-AABs on ΔΨm and to determine whether autophagy was involved in ΔΨm changes.
Materials and Methods
Ethics Statement
All the animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996), approved by the Institutional Animal Care and Use Committee of Shanxi Medical University, and the Guide for the Care and Use of Laboratory Animals according to the regulation in the People's Republic of China. The Wistar rats used in the present study were obtained from the Animal Center of Shanxi Medical University, P.R.China.
β1-AAB-positive rat model and rapamycin (RAPA) treatment
Animals were randomly assigned to two experimental groups: Vehicle group and β1-AAB group. A synthetic peptide of (197~223, H-W-W-R-A-E-S-D-E-A-R-R-C-Y-N-D-P-K-C-C-D-F-V-T-N-R-A, rat homology 100%, synthesized by GL Biochemical Co., LTD, Shanghai, purity was greater than 95%) was dissolved in Na2CO3 solution (concentration of 1 mg/ml), and emulsified with the same volume of complete Freund's adjuvant (CFA) (Sigma, F5881). Then, the rats in the β1-AAB group were injected with this antigen emulsified in CFA (0.4 μg/g, subcutaneous injection) posteriorly along the back at multiple sites for the first time. Afterwards, a booster immunization was given posteriorly along the back every two weeks with a mixture of equal volume of peptide solution and incomplete Freund's adjuvant (Sigma, F5506). The rats in the Vehicle group were injected with 1 ml of normal saline (1 mg/ml) mixed with 1 ml of incomplete Freund's adjuvant in the same manner as described above. Blood samples were collected 1 day before booster injection to test the generation of β1-AABs after immunization. A subset of β1-AAB animals were treated with RAPA for two days at the end of the study (β1-AAB+RAPA group). RAPA (Sigma, R0395) was dissolved in dimethyl sulfoxide (DMSO) (25 mg/ml) and further diluted with phosphate-buffered saline (PBS) before intraperitoneally (i.p) injection. The final dose was 5 mg/kg/day of RAPA.
Enzyme-linked immunosorbent assay (ELISA) and preparation of IgG (immunoglobulin G)
Peptides corresponding to the sequence of the second extracellular loops of human β1-adrenoceptors were synthesized, and ELISA was performed as previously described [21]. In brief, fifty microliters of the peptide (50 µg/ml) in 100 mM Na2CO3 solution (pH 11.0) was coated on NUNC (Kastrup, Denmark) microtitre plates overnight at 4 °C. The wells were then saturated with PMT [PBS supplemented with 3% skimmed milk (W/V), 0.1% Tween 20 (V/V) and 0.01% thimerosal (W/V)] for 1 h at 37 °C. After washing the wells three times with PMT, 5 μl sera were added to 95 μl PMT solution and was incubated for 1 h at 37 °C. After washing again three times with PMT, an affinity-purified biotinylated sheep anti-rat IgG (H+L) antibody (1:2000 dilution, Beijing Zhongshan Golden Bridge Biotechnology, ZB-2040) was added and allowed to react for 1 h at 37 °C. After three times washing, the bound biotinylated antibody was detected by incubating the plates for 1 h with horseradish peroxidase streptavidin (1:3000 dilution, Vector, SA-5004). Then the plate was washed with PBS three times and the substrate [2.5 mM H2O2, 2mM 2, 2'-azinodi (ethylbenzthiazoline) sulfuric acid (ABTS, Bio Basic Inc., AD0002)] was added. Optical density was read after 30 min at 405 nm by using a microplate reader (Molecular Devices Corp., USA). The positive reaction of the sera against the peptides was confirmed as reported by Liu et al [22]. Immunoglobulin G (IgG) was affinity purified from β1-AAB-positive serum by MAbTrap™ Kit (GE Healthcare, 17-1128-01) and the total purified IgG concentration (mg/ml) was determined by the Bicinchoninic Acid (BCA) Protein Assay (thermo scientific, #23225).
Measurement of cardiac function in vivo
The left ventricular function of rats in the β1-AAB group and the Vehicle group were determined at the 4th, 8th and 12th week after immunization as described previously [22]. Briefly, after anesthesia, a cannula was inserted into the left ventricle via the right carotid artery to measure the following primary and derived variables, including the left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), and maximal positive and negative values of the instantaneous first derivative of left ventricular pressure (+dP/dtmax and −dP/dtmax).
Myocardial radionuclide imaging
Myocardial radionuclide imaging is a technique used to reflect the ΔΨm [23]. Myocardial uptake of 99mTc-methoxyisobutylisonitrile (99mTc-MIBI) was measured as H/M count ratio, by use of regions of interest positioned over the heart (H) and upper mediastinum (M) [24]. The decline in cardiac 99mTc-MIBI uptake means decreased ΔΨm.
A 18.5-MBq dose of 99mTc-MIBI was injected slowly through the rat tail vein. The planar and single photon emission computed tomography views were obtained approximately 30 min after injection. For image processing, we used a conventional gamma scintillation camera (Mobile Radioisotope Camera, Model BHP6602, Hamamatsu, Japan), equipped with a low-energy, high-resolution collimator and selected a 20% energy window encompassing the 140 keV photopeak. Imaging was performed by projection reconstruction with the chest open and collected in a 256 × 256 matrix format.
Myocyte isolation and culture
At the 4th week after immunization, rats from the Vehicle group, β1-AAB group and β1-AAB+RAPA group were heparinized (1000 U/ kg) and anaesthetized. The hearts were excised and the aorta was cannulated and immediately perfused on a Langendorff apparatus. Hearts were initially perfused for a 5 min period with Ca2+-free Tyrode's solution, followed by a 25 min period with Ca2+-free Tyrode’s solution containing collagenase B and collagenase D (Roche Chemical Co.). The ventricles were then excised, rinsed and cut into small pieces in Ca2+-free Tyrode's solution supplemented with 0.1mM CaCl2 and 1% BSA). The cells were collected using 50 mesh screen filter and the supernatant was centrifuged at 50g for 1min at room temperature and then resuspended for a three-step Ca2+ restoration procedure (i.e. 0.1mM, 0.5mM, 1mM). The freshly isolated cardiomyocytes were then suspended in M199 medium (Hyclone, SH30253.01B) and incubated at 37 °C in a 5% CO2 atmosphere.
Cell culture and RAPA treatment
Rat cardiomyocyte-derived cell line H9c2 was purchased from Cell Bank of China Science Academy (Shanghai, China). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Hyclone, SH30022.01B) containing 10% fetal bovine serum (FBS) (Sijiqing, W0001), 100 U/mL penicillin and 100 μg/mL streptomycin (Solarbio, P1400-100) and incubated at 37 °C in a 5% CO2 atmosphere. H9c2 Cells were treated with β1-AABs (1μmol/L) for 24 hours or pretreated with 10 ng/mL RAPA for 30 minutes and then treated with β1-AABs (1μmol/L) for 24 hours in the continued presence of RAPA. The vehicle to administer RAPA was DMSO (0.1% total volume).
JC-1 staining
JC-1 (Beyotime Biotech, C2006), a sensitive fluorescent probe for ΔΨm, was employed to measure the ΔΨm of cardiomyocytes. In healthy cells with high mitochondrial ΔΨm, JC-1 spontaneously forms complexes known as JC-1 aggregates with intense red fluorescence. On the other hand, in apoptotic or unhealthy cells with low ΔΨm, JC-1 remains in the monomeric form, which shows only green fluorescence. According to the manufacturer's directions, after indicated treatments, cells were incubated with JC-1 staining solution (5 μg/mL) for 20min at 37 °C and then rinsed twice with JC-1 staining buffer. The images were viewed and scanned under laser confocal microscopy (OLYMPUS, FV1000, Japan) at 488 nm excitation and 530 nm emission for green, and at 543 nm excitation and 590 nm emission for red. All the parameters used in confocal microscopy were kept constant in each sample, such as laser power, pinhole, gain, scan speed and offset. Four repeated measurements were performed. In addition, to quantify JC-1 data, 100 μl of cell sample was used to read the fluorescence in 96-well clear-bottom black plate (Corning, #3603) at Ex488/Em530 and Ex543/Em590 as previously described [25] in SpectraMax® M2e Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). The ratio of red to green fluorescence was calculated as ΔΨm. Mitochondrial depolarization is indicated by an decrease in the red/green fluorescence intensity ratio. In addition, the percentage of low Δψm H9c2 cells were assessed by flow cytometry (BD FACSCanto, NJ, USA). The X-axis (FL-1 channel) of flow cytometry results indicated the green fluorescence intensity (JC-1 monomers) and the Y-axis (FL-2 channel) was used to detect the red fluorescence (JC-1 aggregates). The R2 (region 2) encloses the low ΔΨm cell population.
Western blot
The protein levels of Beclin-1 and LC3 were detected by western blot. For western immunoblotting studies, cardiac tissue (70 mg) was lysed in RIPA buffer (Beyotime Biotech, P0013B). Protein concentration was determined by BCA Protein Assay Kit (Thermo scientific, 23225) and 40 μg of protein was resolved on a 12% SDS-PAGE gel, electrophoresed (80 V for 30 min followed by 120 V for 90 min), and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, IPVH00010). The membrane was blocked for 2 h at room temperature in Tris-buffered saline (TBS) pH 7.4/Tween 0.1% containing 0.3% gelatin (Sigma-aldrich, A9418), and then incubated using the primary antibodies at 4°C overnight followed by detection with the second antibodies. The following primary antibodies were used: anti-Beclin-1 monoclonal antibody (dilution 1:1000, Santa Cruz, sc-48341) or anti-LC3B polyclonal antibody (dilution 1:1000, Cell Signaling, 2775) or anti-β-actin monoclonal antibody (dilution 1:1000, Sigma-aldrich, A1978). Binding of specific antibody was detected with a horseradish peroxidase-conjugated anti-mouse IgG or peroxidase-conjugated anti-rabbit IgG at a dilution of 1:3000 for 1 h (Beijing Zhongshan Golden Bridge Biotechnology, ZB-2305, ZB-2301). Specific antibody binding was detected using electrochemiluminescence. The density of the scanned protein bands was measured by image analysis software and the results were presented as a percentage change of the loading control.
Real-time PCR
mRNA expressions of Beclin-1 and LC3 were detected by Real-time PCR with SYBR Premix Ex TaqTM II (TaKaRa, DRR820A) detection in the Stratagene MX 3005P real-time PCR system. Total RNA was isolated from the left ventricle for analysis using the RNAiso plus (TaKaRa, D9108B). 3μg of total RNA was reversely transcribed into cDNA. The thermal profile for SYBR Green PCR was 95°C for 30s, followed by 40 cycles of denaturation at 95°C for 5s and annealing/extension at 60°C for 20s. The primer sequences were as follows: LC3, sense: 5’-CATGCCGTCCGAG-AAGACCT-3’ and antisense: 5’-GATGAGCCGGACATCTTCCACT-3’ (GenBank TM accession number, NM022867.2); Beclin-1, sense: 5’-TTGGCCAATAAGATGG-GTCTGAA-3’ and antisense: 5’-TGTCAGGGACTCCAGATACGAGTG-3’ (GenBankTM accession number, NM001034117.1). Samples were normalized against GAPDH expression to ensure equal loading. The specificity of the amplified product was monitored by its dissociation curve. The results, expressed as the fold difference in the number of LC3 or Beclin-1 copies relative to the number of GAPDH gene copies, were determined by the relative quantitative 2-ΔΔCt method. ΔΔCt = ΔCt (target gene)-ΔCt (GAPDH) and ΔCt (target gene) = Ct (experimental-target)-Ct (control-target) and ΔCt (GAPDH) = Ct (experimental-GAPDH)-Ct (control- GAPDH).
Immunofluorescence
Beclin-1 and LC3 were detected by immunofluorescence. Myocardial tissue samples were embedded in Tissue-Tek OCT compound (Sakura Finetechnical Co., 4583) and were sectioned at 10 μm thickness with cryostat (CM3050 S, Leica, Deerfield, IL), air-dried for 60 min, fixed with acetone for 15 min at 4°C and stored at -20°C until used. These frozen sections were incubated with antibodies against LC3B (1:400; Cell Signaling, 2775) and Beclin-1 (1:50, Santa Cruz Biotechnology, sc-48341) in a humidified container at 4°C overnight. After washing with PBS, the frozen sections were incubated with tetramethylrhodamine isothiocyanate (TRITC)-conjugated second antibodies. TRITC-labeled second anti-rabbit IgG (1:50) and TRITC-labeled second anti-mouse IgG (1:50) were from Beijing Zhongshan Golden Bridge Biotechnology (ZF-0316, ZF-0313). After washing three times with PBS, 2-(4-Amidino-phenyl)-6-Indolecarba-midine dihydrochloride (DAPI, Beyotime Biotech, C1005) solution was added to stain the cell nucleus for 3 min. Sections were then washed in PBS and sealed with a coverslip. The slides were analyzed with laser confocal microscopy (OLYMPUS, FV1000, USA).
Statistical analysis
Data were presented as mean ± standard deviation (SD). Statistical analysis was performed with the SPSS 15.0 program. T-tests were used to compare two independent sample means, and one-way ANOVA with Bonferroni post hoc tests were performed for comparing means of more than two samples. Statistical significance was defined as p < 0.05.
Results
β1-AAB-positive rat models were established
In both experimental groups, the OD value of β1-AABs in the sera before treatment were at a very low level. However, these were markedly increased in the β1-AAB group after two weeks of immunization. Moreover, the serum levels remained relatively high until the end of the experiment (Figure 1).
Figure 1. The generation of β1-AABs after active immunization against β1-AR-ECII.
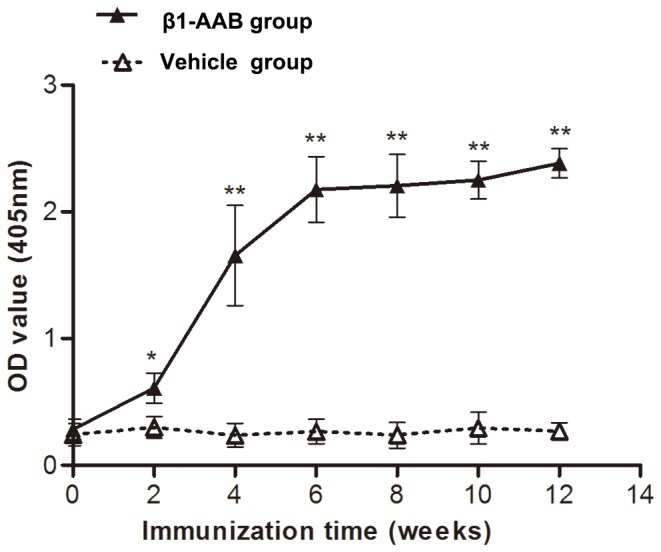
Adult rats were actively immunized with β1-AR-ECII. Antibody titer is defined by OD value. Data are expressed as Mean ± SD (n=12 per group). *P < 0.05; **P < 0.01.
Cardiac function was decreased with the existence of β1-AABs
At the 4th week and 8th week, no obvious change has been found on the left ventricular function parameters. However, at the 12th week after immunization, the left ventricular systolic and diastolic functions, expressed by LVSP, +dP/dtmax and LVEDP, -dP/dtmax, decreased significantly in the β1-AAB group compared with the Vehicle group (Figure 2A-D). These results indicated that cardiac function was declined as a result of immunization.
Figure 2. Assessment of cardiac function at different time points (4w, 8w and 12w) after active immunization.

Comparison of LVSP (A), LVEDP (B), +dp/dtmax (C) and −dp/dtmax (D) between the rats in the Vehicle group and β1-AAB group. Data are expressed as Mean ± SD (n=8 per group). *P < 0.05; **P < 0.01.
The ΔΨm in rat cardiac myocytes declined with the existence of β1-AABs
The ΔΨm is an important parameter of mitochondrial function [10]. In this study, myocardial radionuclide imaging technology and JC-1 staining were executed to evaluate the alteration in ΔΨm.
Myocardium uptake of 99mTc-MIBI, expressed by the H/M ratio [regions of interest positioned over the heart (H) and upper mediastinum (M)] [24], can be used to reflect the ΔΨm in cardiac myocytes. As shown in Figure 3A, the H/M ratio declined at the 4th week after immunization and this trend continued until the end of the study in β1-AAB group versus the Vehicle group (Results are not reported). Meanwhile, JC-1 staining was used to detect the ΔΨm. When the ΔΨm is high, JC-1 accumulates in the mitochondrial matrix to form JC-1 aggregates that produce red fluorescence. Alternatively, green fluorescence is generated by the JC-1 monomers when JC-1 cannot assemble in the mitochondrial matrix. In the present study, at the 4th week after immunization, ventricular cardiomyocytes were isolated and stained with JC-1. After adding JC-1, the intensity of the red fluorescence of JC-1 aggregates was significantly weaker and the green fluorescence of JC-1 monomers was stronger in the β1-AAB group compared with the Vehicle group (Figure 3B, C). To further confirm these results, H9c2 cells, derived from embryonic rat heart, were treated with β1-AABs (1μmol/L) for 24 hours and stained with JC-1, followed by flow cytometry. The data showed that the ΔΨm rapidly decreased, as shown by increased cell population in R2 (Figure 4). These results demonstrated that the ΔΨm was declined with the existence of β1-AABs.
Figure 3. Assessment of ΔΨm at the 4th week after active immunization.

(A) Representative images of cardiac radionuclide imaging. Cardiac 99mTc-MIBI uptake was measured as H/M count ratio, by use of regions of interest positioned over the heart (H) and upper mediastinum (M). Data are expressed as Mean ± SD (n=6 per group). *P < 0.05. (B) Representative images of JC-1 staining. The adult rat ventricular cardiomyocytes were isolated, stained with JC-1 and then the stained cells were imaged using confocal microscopy. Scale bar: B = 50 μm. (C) Quantitative analysis of the shift of mitochondrial red fluorescence to green fluorescence among groups. The fluorescence was measured using a SpectraMax M2e microplate reader and the ratio of red to green fluorescence was calculated as ΔΨm. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05.
Figure 4. Changes in ΔΨm of cardiomyocytes (H9c2 Cells) with the existence of β1-AABs and RAPA.

Representative images of JC-1 fluorescence with flow cytometry and quantitative analysis of ΔΨm. After β1-AAB treatment, decreased ΔΨm was indicated by increased cell population in R2. Induction of autophagy by RAPA lead to the restoration of red fluorescence indicated by decreased cell population in R2. Data are expressed as Mean ± SD (n=6 per group). **P < 0.01 vs. Vehicle; #P < 0.05 vs. β1-AAB group.
Decreased myocardial autophagy contributes to the decline in ΔΨm with the existence of β1-AABs
To determine the extent of autophagy are influenced by β1-AABs, LC3 and Beclin-1, which have been used as molecular markers of autophagic activity, were measured in the present study. Both protein (Figure 5A) and mRNA levels (Figure 5B) of LC3 and Beclin-1 were significantly decreased at the 4th week in the β1-AAB group compared with the Vehicle group. Moreover, compared with the Vehicle group, the Beclin-1 and LC3 punctate dots were significantly reduced in the β1-AAB group by immunofluorescence staining (Figure 5C). Meanwhile, β1-AR-ECII treatment did not affect the level of autophagy and cell survival in H9c2 cardiac cells (File S1, Figure S1 and Figure S2). To further evaluate the role of decreased autophagy in ΔΨm, RAPA, an mTOR inhibitor, was used to enhance autophagy [26]. β1-AAB animals were treated with RAPA (β1-AAB+RAPA group) at 5 mg/kg/day for two days before detection. As shown in Figure 6A, pretreatment with RAPA significantly increased the myocardium uptake of 99mTc-MIBI. Meanwhile, cultured cardiac myocytes were isolated from the rats in β1-AAB+RAPA group and stained with JC-1 showing the restoration of ΔΨm, as indicated by the increase of the red : green fluorescence ratio (Figure 6B, C). To further confirm the result, cardiomyocytes (H9c2 Cells) were pretreated with 10 ng/mL RAPA or DMSO vehicle (0.1% total volume) for 30 minutes and then treated with β1-AABs (1μmol/L) for 24 hours in the continued presence of RAPA or DMSO. As shown in Figures S3 (A-C) and S4, pretreatment with RAPA increased the level of autophagy whereas DMSO vehicle alone did not affect the level of autophagy in β1-AAB-treated H9c2 cardiomyocytes. In addition, β1-AAB+RAPA-treated H9c2 cells were stained with JC-1 (Figure 4). The data showed an increase in ΔΨm as indicated by decreased cell population in R2. Taken together, these results demonstrated that the induction of autophagy could improve ΔΨm in cardiac myocytes with declined ΔΨm due to the existence of β1-AABs.
Figure 5. The level of autophagy decreased with the existence of β1-AABs at the 4th week.

(A and B) The levels of LC3 and Beclin-1 protein and mRNA expression. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05. (C) Beclin-1 and LC3 immunoreactivity were present as punctate pot (red) and the nucleus were stained with DAPI (blue). Scale bar: C=10 μm.
Figure 6. Induction of autophagy by RAPA can improve the ΔΨm in cardiomyocytes.

(A) Induction of autophagy by RAPA results in enhanced myocardium uptake of 99mTc-MIBI. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05. (B) Induction of autophagy can increase red fluorescence intensity of JC-1 aggregates. Scale bar: B=50 μm. (C) Induction of autophagy increase the ratio of red to green fluorescence. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05.
Discussion
In the present study, we demonstrated that the long-term existence of β1-AABs in rat resulted in decreased autophagy and the decline in ΔΨm that was earlier than the emergence of abnormal cardiac function. Additionally, activation of autophagy by RAPA can effectively improve the ΔΨm in cardiac myocytes.
Since the 1990s, β1-AABs were frequently detected in patients suffering from dilated cardiomyopathy (DCM) [17] and Chagas' heart disease [27]. In a previous study, we detected these autoantibodies in a rat model of heart failure [22] and found that they could influence heart function [19]. In a previous paper [19], when the immunization time was once a month, we demonstrated that the left ventricular systolic and diastolic functions decreased significantly in β1-AAB group at the 18th month after immunization. In the current study, the immunization time we chose is once every two weeks. Because of such a high frequency of immunization, the OD value of β1-AABs in the sera were markedly increased and the heart function aggravated progressively in the β1-AAB group at the 12th week after immunization.
+dp/dtmax, which was used as an index of myocardial contractility, started to decrease at 12 weeks after immunization showing that the existence of β1-AABs could reduce cardiac contractility. Among many factors affecting myocardial contractility, mitochondria regulate cardiac cell contractility by providing ATP for cellular ATPases and by participating in Ca2+ homeostasis [28]. Mitochondria dysfunction results in bioenergetic defect and cardiac function deterioration.
The ΔΨm, which is the electrochemical gradient that is present across the inner mitochondrial membrane, is critical for maintaining the physiological function of the respiratory chain to generate ATP [29], so the ΔΨm reflects the integrity of mitochondrial function and is a key indicator of mitochondrial function [10]. In the present study, the ΔΨm was measured through use of the cardiac radionuclide imaging and the lipophilic mitochondrial probe JC-1.
Cardiac radionuclide imaging uses an intravenous radiopharmaceutical to generate scintigraphic images of the myocardium and the images are acquired using a gamma camera. 99mTc-MIBI, which is a lipophilic cationic myocardial perfusion imaging agent, can move across the cytoplasmic membrane and the mitochondrial membrane by passive diffusion in response to transmembrane potential. Approximately 90% of myocardial 99mTc-MIBI is localized within the mitochondrial fraction [30]. Most of the accumulated 99mTc-MIBI is related to mitochondrial uptake. Myocardial uptake of 99mTc-MIBI is dependent on ΔΨm . When the ΔΨm is depolarized, the myocardial uptake of 99mTc-MIBI will be inhibited [23], so the myocardial uptake of 99mTc-MIBI is used to reflect the ΔΨm and mitochondrial function. The advantage of 99mTc-MIBI scanning is that it is a noninvasive inspection and can be better used in clinical practice. In the present study, at the 4th week after immunization, the myocardium uptake of 99mTc-MIBI declined significantly. The existence of β1-AABs caused the disruption of the ΔΨm and may contribute to cardiac dysfunction. Meanwhile, JC-1 staining was used to detect the ΔΨm in cardiac myocytes. JC-1 has been reported to be a more reliable indicator of ΔΨm than other dyes described herein [31,32] and can selectively enter into mitochondria and reversibly change color from red to green as the membrane potential decreases. The ratio of red to green fluorescence of JC-1 depends only on the ΔΨm [33] and can therefore be used as an indicator of ΔΨm [25,34-37]. Our results also found that ΔΨm was decreased, expressed by the increase in green fluorescence and the concomitant attenuation of red fluorescence in the β1-AAB group compared with the Vehicle group. Flow cytometric analysis of cardiomyocytes (H9c2 Cells) showed similar results. As can be seen from the above results, the loss of ΔΨm induced by β1-AABs that emerged in the 4th week was earlier than the declined cardiac function that started to emerge in the 12th week. It has been reported that β1-AABs can be detected in the serum of approximately 10% of healthy human populations [21]. If the functional state of the mitochondria can be evaluated by noninvasive radionuclide scan for those with normal heart function that detected by electrocardiogram, myocardial enzymes, echocardiography and X-ray but β1-AAB-positive, the potentially dangerous conditions that can cause cardiac dysfunction may be prevented earlier.
Mitochondrial dysfunction often leads to diverse cellular responses, including autophagy. The autophagic response, in return, plays a pivotal role in mitochondrial degradation [38]. Decreased ΔΨm could promote mitochondrial dysfunction [39] and induce activation of autophagy [40]. More importantly, autophagy can selectively remove damaged mitochondria to protect cells [39-41]. Impaired autophagy causes the accumulation of dysfunctional organelles such as mitochondria within the cell [42]. Overexpression of LC3, a well-known marker for autophagy, resulted in an improved ΔΨm and enhanced ATP production [43]. Autophagy, a lysosomal degradation pathway that is essential for survival, plays an important role in maintaining cellular homeostasis [44]. Microtubule-associated protein 1 light chain 3 (MAP1-LC3, LC3) is an essential component of the autophagic vacuoles, forming a reliable marker of autophagic activity [45]. There are three isoforms of LC3 in mammalian cells, LC3A, LC3B and LC3C, but only LC3B-II is associated with autophagic vesicle numbers, and therefore the anti-LC3B antibodies were recommended to use for analysis [46]. That is why anti-LC3B antibodies were used for autophagy assays in this study. Upon induction of autophagy, LC3-I, a cytosolic form of LC3, is converted to LC3-II (a lipidated form of LC3). LC3-II is localized in autophagosome membranes and the increase in LC3-II indicates the accumulation of autophagosomes, so LC3-II has been deemed a marker of autophagy [47]. As part of a class III PI3K complex [48], Beclin-1 is thought to be important in mediating the localization of other autophagy proteins to pre-autophagosomal structures [49]. Therefore, it is commonly used during autophagy detection.
Many studies have shown that the level of autophagy was altered in several cardiovascular diseases [50]. Moreover, autophagy was activated by propranolol that is a non-selective β-adrenergic receptor blocking agent [51] and was inhibited by the β-adrenergic agonist isoproterenol [20]. Thus, we speculate that the alteration of the β-adrenergic receptor may result in the change of autophagy. However, whether autophagy alteration is accompanied by ΔΨm and decreased cardiac function caused by β1-AABs remains unknown. If it does, we aimed to understand the contribution of autophagy to the changes in ΔΨm.
In the present study, the expression of LC3 and Beclin-1, markers of the autophagic process, declined with the existence of β1-AABs. Interestingly, induction of autophagy by RAPA results in the restoration of ΔΨm, expressed by the increase of myocardium uptake of 99mTc-MIBI and the repression in green fluorescence accompanied by the restoration in red fluorescence. Flow cytometric analysis of cardiomyocytes (H9c2 Cells) showed similar results. These results suggested that decreased autophagy was involved in the loss of ΔΨm.
In the present study, rat models with β1-AABs were established by active immunization which is the induction of immunity after exposure to a synthetic peptide corresponding to β1-AR-ECII. However, we cannot rule out the possibility that antigenic peptides themselves may influence the heart. Thus, the H9c2 cell line, derived from the embryonic rat heart, was treated with β1-AR-ECII for 24 hours and the data indicated that the levels of autophagy and cell survival did not change significantly.
We concede that there are some limitations to our study and further research is required. Firstly, we only observed the phenomenon that decreased ΔΨm was caused by β1-AABs. However, whether the changes in ΔΨm induced by β1-AABs were associated with mitochondrial dysfunction, mitochondrial damage or disorders of energy metabolism need to be further validated. We will do further research to confirm our results. Secondly, we found that the induction of autophagy contributed to improving the ΔΨm, but whether this restoration induced an improvement of the left ventricular systolic and diastolic functions requires further study. Even so, it can also be concluded that the decreased ΔΨm was induced by β1-AABs in which autophagy plays an important regulatory role. Our preliminary observations may open new insights into the pathogenesis and prevention of β1-AAB-positive heart dysfunction.
Supporting Information
The autophagy did not change with β1-AR-ECII in the rat cardiomyocyte-derived cell line H9c2. H9c2 cells were incubated with β1-AABs and β1-AR-ECII for 24 hours and then lysised, and mRNA levels of LC3 (A) and Beclin-1 (B) were detected with Real-time PCR. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05.
(TIF)
β1-AR-ECII did not significantly alter survival of H9c2 cells. After being stimulated for 24 hours by β1-AABs, the level of H9c2 cell survival declined significantly and had no change in the absence of β1-AR-ECII. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05.
(TIF)
RAPA can induce autophagy in β1-AAB-treated rats. (A and B) The differences in LC3 and Beclin-1 protein and mRNA expression after treatment with RAPA. (n=6 per group) (C) Confocal images of Beclin-1 and LC3. The red punctate pots recovered by RAPA. *P < 0.05 vs. Vehicle; #P < 0.05 vs. β1-AAB group. Scale bar: C=10 μm.
(TIF)
Pretreatment with RAPA increased the level of autophagy in β1-AAB-treated H9c2 cardiomyocytes. The differences in LC3 and Beclin-1 protein expression after treatment with RAPA. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05 vs. Vehicle; #P < 0.05 vs. β1-AAB group.
(TIF)
Supplemental materials and methods.
(DOC)
Acknowledgments
We are grateful to Lindsey Devillier for correcting the English spelling and grammar.
Funding Statement
This study was supported by Natural Sciences Foundation of China (grant number 81070263) and Science Foundation for Youths of Shanxi Medical University (grant number 02201004). In addition, the work during the revision process was supported by Scientific and Technologial Innovation Programs of Higher Education Institutions in Shanxi, STIP. number 2010010. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wang Y, Mi J, Shan XY, Wang QJ, Ge KY (2007) Is China facing an obesity epidemic and the consequences? The trends in obesity and chronic disease in China. Int J Obes (Lond) 31: 177-188. doi: 10.1038/sj.ijo.0803354. [DOI] [PubMed] [Google Scholar]
- 2. He J, Gu D, Wu X, Reynolds K, Duan X et al. (2005) Major causes of death among men and women in China. N Engl J Med 353: 1124-1134. doi: 10.1056/NEJMsa050467. PubMed: 16162883. [DOI] [PubMed] [Google Scholar]
- 3. Jiang H, Ge J (2009) Epidemiology and clinical management of cardiomyopathies and heart failure in China. Heart 95: 1727-1731. doi: 10.1136/hrt.2008.150177. PubMed: 19318343. [DOI] [PubMed] [Google Scholar]
- 4. Kaul P, McAlister FA, Ezekowitz JA, Grover VK, Quan H (2011) Ethnic differences in 1-year mortality among patients hospitalised with heart failure. Heart 97: 1048-1053. doi: 10.1136/hrt.2010.217869. PubMed: 21508417. [DOI] [PubMed] [Google Scholar]
- 5. Neubauer S (2007) The failing heart--an engine out of fuel. N Engl J Med 356: 1140-1151. doi: 10.1056/NEJMra063052. PubMed: 17360992. [DOI] [PubMed] [Google Scholar]
- 6. Taegtmeyer H (2004) Cardiac metabolism as a target for the treatment of heart failure. Circulation 110: 894-896. doi: 10.1161/01.CIR.0000139340.88769.D5. PubMed: 15326079. [DOI] [PubMed] [Google Scholar]
- 7. Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N et al. (2001) Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88: 529-535. doi: 10.1161/01.RES.88.5.529. PubMed: 11249877. [DOI] [PubMed] [Google Scholar]
- 8. Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP et al. (2008) Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res 80: 30-39. doi: 10.1093/cvr/cvn184. PubMed: 18710878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marin-Garcia J, Goldenthal MJ, Moe GW (2001) Mitochondrial pathology in cardiac failure. Cardiovasc Res 49: 17-26. doi: 10.1016/S0008-6363(00)00241-8. PubMed: 11121792. [DOI] [PubMed] [Google Scholar]
- 10. Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA (2011) Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50: 98-115. doi: 10.2144/000113610. PubMed: 21486251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murray AJ, Edwards LM, Clarke K (2007) Mitochondria and heart failure. Curr Opin Clin Nutr Metab Care 10: 704-711. doi: 10.1097/MCO.0b013e3282f0ecbe. PubMed: 18089951. [DOI] [PubMed] [Google Scholar]
- 12. Jahns R, Boivin V, Hein L, Triebel S, Angermann CE et al. (2004) Direct evidence for a beta 1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest 113: 1419-1429. doi: 10.1172/JCI20149. PubMed: 15146239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Labovsky V, Smulski CR, Gómez K, Levy G, Levin MJ (2007) Anti-beta1-adrenergic receptor autoantibodies in patients with chronic Chagas heart disease. Clin Exp Immunol 148: 440-449. doi: 10.1111/j.1365-2249.2007.03381.x. PubMed: 17419712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang L, Hu D, Shi X, Li J, Zeng W et al. (2001) Autoantibodies against the myocardium beta 1-adrenergic and M2-muscarinic receptors in patients with heart failure. Zhonghua Nei Ke Za Zhi 40: 445-447. PubMed: 11798611. [PubMed] [Google Scholar]
- 15. Krause EG, Bartel S, Beyerdörfer I, Wallukat G (1996) Activation of cyclic AMP-dependent protein kinase in cardiomyocytes by anti-beta 1-adrenoceptor autoantibodies from patients with idiopathic dilated cardiomyopathy. Blood Press Suppl 3: 37-40. PubMed: 8973767. [PubMed] [Google Scholar]
- 16. Sterin-Borda L, Gorelik G, Postan M, Gonzalez Cappa S, Borda E (1999) Alterations in cardiac beta-adrenergic receptors in chagasic mice and their association with circulating beta-adrenoceptor-related autoantibodies. Cardiovasc Res 41: 116-125. doi: 10.1016/S0008-6363(98)00225-9. PubMed: 10325959. [DOI] [PubMed] [Google Scholar]
- 17. Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J (1994) Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the beta 1-adrenoceptor with positive chronotropic effect. Circulation 89: 2760-2767. doi: 10.1161/01.CIR.89.6.2760. PubMed: 8205690. [DOI] [PubMed] [Google Scholar]
- 18. Mijares A, Verdot L, Peineau N, Vray B, Hoebeke J et al. (1996) Antibodies from Trypanosoma cruzi infected mice recognize the second extracellular loop of the beta 1-adrenergic and M2-muscarinic receptors and regulate calcium channels in isolated cardiomyocytes. Mol Cell Biochem 163-164: 107-112. doi: 10.1007/BF00408646. PubMed: 8974045. [DOI] [PubMed] [Google Scholar]
- 19. Zuo L, Bao H, Tian J, Wang X, Zhang S et al. (2011) Long-term active immunization with a synthetic peptide corresponding to the second extracellular loop of beta1-adrenoceptor induces both morphological and functional cardiomyopathic changes in rats. Int J Cardiol 149: 89-94. doi: 10.1016/j.ijcard.2009.12.023. PubMed: 20096470. [DOI] [PubMed] [Google Scholar]
- 20. Pfeifer U, Föhr J, Wilhelm W, Dämmrich J (1987) Short-term inhibition of cardiac cellular autophagy by isoproterenol. J Mol Cell Cardiol 19: 1179-1184. doi: 10.1016/S0022-2828(87)80528-X. PubMed: 3443985. [DOI] [PubMed] [Google Scholar]
- 21. Liu HR, Zhao RR, Zhi JM, Wu BW, Fu ML (1999) Screening of serum autoantibodies to cardiac beta1-adrenoceptors and M2-muscarinic acetylcholine receptors in 408 healthy subjects of varying ages. Autoimmunity 29: 43-51. doi: 10.3109/08916939908995971. PubMed: 10052684. [DOI] [PubMed] [Google Scholar]
- 22. Liu HR, Zhao RR, Jiao XY, Wang YY, Fu M (2002) Relationship of myocardial remodeling to the genesis of serum autoantibodies to cardiac beta(1)-adrenoceptors and muscarinic type 2 acetylcholine receptors in rats. J Am Coll Cardiol 39: 1866-1873. doi: 10.1016/S0735-1097(02)01865-X. PubMed: 12039504. [DOI] [PubMed] [Google Scholar]
- 23. Piwnica-Worms D, Kronauge JF, Chiu ML (1990) Uptake and retention of hexakis (2-methoxyisobutyl isonitrile) technetium(I) in cultured chick myocardial cells. Mitochondrial and plasma membrane potential dependence. Circulation 82: 1826-1838. doi: 10.1161/01.CIR.82.5.1826. PubMed: 2225379. [DOI] [PubMed] [Google Scholar]
- 24. Yamada T, Shimonagata T, Fukunami M, Kumagai K, Ogita H et al. (2003) Comparison of the prognostic value of cardiac iodine-123 metaiodobenzylguanidine imaging and heart rate variability in patients with chronic heart failure: a prospective study. J Am Coll Cardiol 41: 231-238. doi: 10.1016/S0735-1097(03)81660-1. PubMed: 12535815. [DOI] [PubMed] [Google Scholar]
- 25. Rizvi F, Heimann T, Herrnreiter A, O'Brien WJ (2011) Mitochondrial dysfunction links ceramide activated HRK expression and cell death. PLOS ONE 6: e18137. doi: 10.1371/journal.pone.0018137. PubMed: 21483866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meijer AJ, Codogno P (2004) Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 36: 2445-2462. doi: 10.1016/j.biocel.2004.02.002. PubMed: 15325584. [DOI] [PubMed] [Google Scholar]
- 27. Rosenbaum MB, Chiale PA, Schejtman D, Levin M, Elizari MV (1994) Antibodies to beta-adrenergic receptors disclosing agonist-like properties in idiopathic dilated cardiomyopathy and Chagas' heart disease. J Cardiovasc Electrophysiol 5: 367-375. doi: 10.1111/j.1540-8167.1994.tb01174.x. PubMed: 8019712. [DOI] [PubMed] [Google Scholar]
- 28. Kaasik A, Joubert F, Ventura-Clapier R, Veksler V (2004) A novel mechanism of regulation of cardiac contractility by mitochondrial functional state. FASEB J 18: 1219-1227. doi: 10.1096/fj.04-1508com. PubMed: 15284222. [DOI] [PubMed] [Google Scholar]
- 29. Kroemer G, Zamzami N, Susin SA (1997) Mitochondrial control of apoptosis. Immunol Today 18: 44-51. doi: 10.1016/S0167-5699(97)80014-X. PubMed: 9018974. [DOI] [PubMed] [Google Scholar]
- 30. Carvalho PA, Chiu ML, Kronauge JF, Kawamura M, Jones AG et al. (1992) Subcellular distribution and analysis of technetium-99m-MIBI in isolated perfused rat hearts. J Nucl Med 33: 1516-1522. PubMed: 1634944. [PubMed] [Google Scholar]
- 31. Mathur A, Hong Y, Kemp BK, Barrientos AA, Erusalimsky JD (2000) Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc Res 46: 126-138. doi: 10.1016/S0008-6363(00)00002-X. PubMed: 10727661. [DOI] [PubMed] [Google Scholar]
- 32. Zhu J, Liu M, Kennedy RH, Liu SJ (2006) TNF-alpha-induced impairment of mitochondrial integrity and apoptosis mediated by caspase-8 in adult ventricular myocytes. Cytokine 34: 96-105. doi: 10.1016/j.cyto.2006.04.010. PubMed: 16730193. [DOI] [PubMed] [Google Scholar]
- 33. Kumagai T, Müller CI, Desmond JC, Imai Y, Heber D et al. (2007) Scutellaria baicalensis, a herbal medicine: anti-proliferative and apoptotic activity against acute lymphocytic leukemia, lymphoma and myeloma cell lines. Leuk Res 31: 523-530. doi: 10.1016/j.leukres.2006.08.019. PubMed: 17007926. [DOI] [PubMed] [Google Scholar]
- 34. Srinivasan S, Stevens M, Wiley JW (2000) Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes 49: 1932-1938. doi: 10.2337/diabetes.49.11.1932. PubMed: 11078462. [DOI] [PubMed] [Google Scholar]
- 35. Bowser DN, Minamikawa T, Nagley P, Williams DA (1998) Role of mitochondria in calcium regulation of spontaneously contracting cardiac muscle cells. Biophys J 75: 2004-2014. doi: 10.1016/S0006-3495(98)77642-8. PubMed: 9746542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC (2005) Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 111: 1601-1610. doi: 10.1161/01.CIR.0000160359.49478.C2. PubMed: 15811867. [DOI] [PubMed] [Google Scholar]
- 37. Sarafian TA, Montes C, Imura T, Qi J, Coppola G et al. (2010) Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLOS ONE 5: e9532. doi: 10.1371/journal.pone.0009532. PubMed: 20224768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tatsuta T, Langer T (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27: 306-314. doi: 10.1038/sj.emboj.7601972. PubMed: 18216873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodriguez-Enriquez S, He L, Lemasters JJ (2004) Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol 36: 2463-2472. doi: 10.1016/j.biocel.2004.04.009. PubMed: 15325585. [DOI] [PubMed] [Google Scholar]
- 40. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT et al. (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232-235. doi: 10.1038/nature07006. PubMed: 18454133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hickson-Bick DL, Jones C, Buja LM (2008) Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol 44: 411-418. doi: 10.1016/j.yjmcc.2007.10.013. PubMed: 18062988. [DOI] [PubMed] [Google Scholar]
- 42. Wrighton KH (2011) Autophagy: shaping the fate of mitochondria. Nat Rev Mol Cell Biol 12: 344-345. doi: 10.1038/nrm3116. PubMed: 21527952. [DOI] [PubMed] [Google Scholar]
- 43. Mai S, Muster B, Bereiter-Hahn J, Jendrach M (2012) Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 8: 47-62. doi: 10.4161/auto.8.1.18174. PubMed: 22170153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290: 1717-1721. doi: 10.1126/science.290.5497.1717. PubMed: 11099404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mizushima N (2004) Methods for monitoring autophagy. Int J Biochem Cell Biol 36: 2491-2502. doi: 10.1016/j.biocel.2004.02.005. PubMed: 15325587. [DOI] [PubMed] [Google Scholar]
- 46. Barth S, Glick D, Macleod KF (2010) Autophagy: assays and artifacts. J Pathol 221: 117-124. doi: 10.1002/path.2694. PubMed: 20225337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306: 990-995. doi: 10.1126/science.1099993. PubMed: 15528435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B et al. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672-676. doi: 10.1038/45257. PubMed: 10604474. [DOI] [PubMed] [Google Scholar]
- 49. Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M et al. (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9: 1142-1151. doi: 10.1038/ncb1634. PubMed: 17891140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132: 27-42. doi: 10.1016/j.cell.2007.12.018. PubMed: 18191218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bahro M, Pfeifer U (1987) Short-term stimulation by propranolol and verapamil of cardiac cellular autophagy. J Mol Cell Cardiol 19: 1169-1178. doi: 10.1016/S0022-2828(87)80527-8. PubMed: 3443984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The autophagy did not change with β1-AR-ECII in the rat cardiomyocyte-derived cell line H9c2. H9c2 cells were incubated with β1-AABs and β1-AR-ECII for 24 hours and then lysised, and mRNA levels of LC3 (A) and Beclin-1 (B) were detected with Real-time PCR. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05.
(TIF)
β1-AR-ECII did not significantly alter survival of H9c2 cells. After being stimulated for 24 hours by β1-AABs, the level of H9c2 cell survival declined significantly and had no change in the absence of β1-AR-ECII. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05.
(TIF)
RAPA can induce autophagy in β1-AAB-treated rats. (A and B) The differences in LC3 and Beclin-1 protein and mRNA expression after treatment with RAPA. (n=6 per group) (C) Confocal images of Beclin-1 and LC3. The red punctate pots recovered by RAPA. *P < 0.05 vs. Vehicle; #P < 0.05 vs. β1-AAB group. Scale bar: C=10 μm.
(TIF)
Pretreatment with RAPA increased the level of autophagy in β1-AAB-treated H9c2 cardiomyocytes. The differences in LC3 and Beclin-1 protein expression after treatment with RAPA. Data are expressed as Mean ± SD (n=6 per group). *P < 0.05 vs. Vehicle; #P < 0.05 vs. β1-AAB group.
(TIF)
Supplemental materials and methods.
(DOC)