

Abstract
It is believed that the diabetic myocardium is refractory to cardioprotection by ischemic preconditioning (IPC) mainly because of impaired insulin signaling to posphatidylinositol 3-kinase (PI3K) and protein kinase B (PKB or Akt). However, human as well as animal studies have clearly showed that hearts of type 2 diabetic humans and animals may exhibit increased signaling through PI3K-Akt but yet are resistant to cardioprotection by IPC or ischemic post-conditioning. Therefore, this study was designed to determine whether activation of insulin signaling prior to IPC is detrimental for cardioprotection and to assess the role of insulin receptors (IRs) and Akt in mediating this effect. Wild-type (WT) hearts, hearts lacking IRs or hearts expressing an active form of Akt (myrAkt1) were perfused ex vivo using a Langendorff preparation and were subjected to IPC (3 cycles of 5 min ischemia followed by 5 min reflow before 30 min no flow ischemia and then by 45 min reperfusion) in the presence or absence of 1 nmol/L insulin. Interestingly, whereas insulin was protective against I/R (30 min no flow ischemia and 45 min reperfusion), it completely abolished cardioprotection by IPC in WT hearts but not in mice lacking insulin receptors (IRs) in cardiomyocytes (CIRKO) or in all cardiac cells (TIRKO). The suppression of IPC-mediated cardioprotection was mediated through downstream signaling to Akt and Gsk3β. In addition, transgenic induction of Akt in the heart was sufficient to abrogate IPC even when insulin was absent, further confirming the involvement of Akt in insulin’s suppression of cardioprotection by IPC. These data provide evidence that excessive insulin signaling to Akt is detrimental for cardioprotection by IPC and could explain the failure of the diabetic myocardium to precondition.
Keywords: insulin, cardioprotection, insulin signaling, ischemia, reperfusion
1. Introduction
The global incidence of diabetes mellitus is increasing with the estimated number reaching 366 million worldwide by 2030. Cardiovascular disease is the major cause of mortality among patients with diabetes, accounting for nearly 60–80% of the deaths [1–2]. Epidemiological studies and clinical trials have clearly shown that both type 1 and type 2 diabetic individuals are more prone to developing ischemic heart disease, including acute myocardial infarction and post-infarct complications. Moreover, mortality from acute myocardial infarction is almost doubled in diabetic patients compared with non diabetic individuals [3–4]. Despite the burden of ischemic heart disease among diabetic individuals, effective treatment is currently unavailable.
Ischemic preconditioning (IPC) is considered one of the most protective mechanisms known to reduce ischemic damage in humans and animals [5–7]. IPC consists of subjecting the heart to brief cycles of ischemia and reperfusion (I/R) prior to a more prolonged ischemic period [8]. Recently, post-conditioning, which consists of applying very brief cycles of I/R in the early phase of reperfusion, has also been shown to protect the heart, and this protocol can easily be applied in the clinical setting. Similar to IPC, insulin and insulin-like growth factor 1 (IGF-1) protects the heart from I/R injury as evidenced by their ability to reduce infarct size when given prior to ischemia or at the reperfusion [9–11]. However, despite its cardioprotective property in animal studies, several clinical observations showed that insulin had either no effect or worsened cardiovascular events in type 2 diabetic patients [12–14]. For example, the UK Prospective Diabetes Study (UKPDS) showed that the use of intensive glucose lowering therapy with insulin over 10 years, reduced the frequency of micro-vascular endpoints but had minimal effects on diabetes-related mortality or myocardial infarction [15]. Furthermore, in a separate study by Mellbin et al. [16], post hoc proportional hazards regression analysis showed that although cardiovascular mortality was not affected, the risk of nonfatal myocardial infarction and stroke was 73% higher in patients who were on insulin therapy, and these differences persisted in a separate analysis of patients for whom insulin was newly started and in those randomly assigned to insulin in the study. In addition, many investigations have now showed that the failure to precondition the diabetic heart is not always associated with reduced insulin signaling. Indeed, insulin-mediated signal transduction to phosphoinositide 3-kinase (PI3K) and Akt is elevated in the hearts of patients with type 2 diabetes and in obese diabetic ob/ob mice [17–18]. Despite this activated insulin signaling pathway, cardioprotection by ischemic postconditioning was not achieved in ob/ob mice, suggesting that chronic activation of insulin signaling is rather detrimental for cardioprotection. Indeed, excessive insulin signaling exacerbated systolic dysfunction in mice subjected to pressure overload, an effect that was prevented either by lowering hyperinsulinemia or by reducing insulin signaling [19]. Therefore, the main objective of this study is to determine whether activation of insulin signaling prior to IPC is detrimental for cardioroptection and to investigate the mechanisms involved.
2. Methods
2.1. Animals
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85 23, revised 1996) and was approved by the Institutional Animal Care and Use Committee of the University of Utah. Male Ttr-Insr −/− (TIRKO) mice (on a C57B6j background) and cardiomyocyte-specific insulin receptor knockout (CIRKO) mice (mixed background), previously described [20–21], and their respective wild-type (WT) control mice were used between 10–14 weeks of age. Mice harboring α-MHC-tTA and Tet-myrAkt1 genes (Tet-off Akt transgenic: abbreviated as Akt TG), previously described [22], were fed 1g/kg body weight doxycycline chow (DOX) for 8 weeks before the chow was removed for 2 weeks to allow cardiac-specific induction of the myrAkt1 transgene. Wild-type, Tet-myrAkt1 TG and α-MHC-tTA TG mice were used as controls and were all on a mixed background.
2.2. Heart perfusion
Mice were anesthetized by intra-peritoneal injection of 15 mg of chloral hydrate, and the heart was rapidly excised and arrested in ice-cold buffer. The aorta was then cannulated and retrogradely perfused at constant pressure of 60 mmHg with 37°C Krebs buffer containing (in mmol/L) NaCl 118, KCl 4.7, NaHCO3 25, MgSO4 1.2, KH2PO4 1.2, and CaCl2 2, glucose 11, gassed with 95% O2 and 5% CO2. Left ventricular pressure was monitored from a water-filled balloon placed through the left atrial appendage and connected to a Millar transducer (Millar Instruments, Houston, TX). The balloon was inflated to achieve an end-diastolic pressure of 7–10 mmHg.
2.3. Perfusion protocols
Hearts from TIRKO, CIRKO, Akt TG and their corresponding WT mice were allowed to stabilize for 20 min before basal hemodynamic parameters and coronary flow were recorded (baseline). Hearts were then subjected to an ischemic preconditioning (IPC) protocol (Fig. 1) consisting of three cycles of 5 min ischemia followed by 5 min reperfusion before 30 min no flow ischemia followed by 45 min reperfusion during which hemodynamic parameters and coronary flow were again recorded. Perfusions were performed in the presence or absence of 1 nmol/L insulin (Human insulin 100U/ml stock, Novolin, Novo Nordisk Inc. Princeton, NJ), that was added to the perfusate and kept for the entire perfusion protocol. In subsequent experiments, WT hearts were stabilized for 20 min before 30 min of no flow ischemia was applied and then followed by 45 min reperfusion (I/R group). Finally, some hearts were used for glycogen assay and western-blotting were perfused with or without insulin for 50 min or subjected to three cycles of IPC (5 min each) and then snap-frozen for analysis.
Fig. 1.
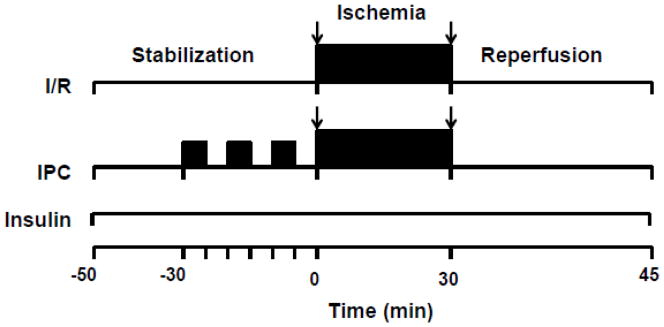
Perfusion protocol. Male mice were perfused ex vivo using Langendorff perfusion system. Hearts were allowed to stabilize for 20 min before contractile parameters were measured (baseline) and then subjected to either an ischemia/reperfusion protocol consisting of 30 min no flow ischemia followed by 45 min reperfusion or preconditioned by three cycles of 5 min no flow ischemia followed by 5 min reperfusion each, before a 30 min no flow ischemia followed by 45 min reperfusion. Insulin (1 nmol/L) was absent or present during the entire protocol Stab: stabilization; I/R: ischemia/reperfusion; IPC: ischemic preconditioning.
2.4. Infarct size measurement
Triphenyltetrazolium chloride (TTC) staining was used to assess myocardial tissue viability and to determine myocardial infarct size. Hearts were subjected to IPC as shown in Fig. 1 without the insertion of the balloon. At the end of the 45 min reperfusion, hearts were perfused with 1% TTC in PBS, pH 7.4, at 37°C for 5 min and then collected and sectioned. The tissue slices were then incubated in 1% TTC in PBS, pH 7.4, at 37°C for 20 min. Tissues were fixed in 10% PBS-buffered formalin overnight at 4°C. Both sides of each TTC-stained tissue slice were photographed and digital photographs were acquired. The infracted area (unstained) was measured using the Image-Pro Plus software package, version 5.0 (Media Cybernetics; Silver Springs, USA) and was expressed as % of risk zone.
2.5. Immuno-blotting analysis
Total proteins were extracted from hearts that were frozen either at the end of the 45 min reperfusion period (full IPC protocol) or at the end of the three cycles of IPC (prior to the 30 min ischemia). Hearts were initially pulverized under liquid nitrogen and then homogenized with a Polytron in sample buffer containing (in mmol/L) HEPES 30, pH7.4, sodium pyrophosphate 50, sodium fluoride 100, EDTA 1, sodium orthovanadate 10 supplemented with 1% Triton X-100 and protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Protein concentration was measured using Micro BCA reagent (Pierce, Rockford, IL). Protein extracts were resolved by SDS-PAGE and electro-transferred onto an Immobilon PVDF membrane (Millipore Corp., Bedford, MA). Membranes were probed with the following primary antibodies: mouse anti-Akt1, rabbit anti-p-Akt (Ser 473) and rabbit anti-p-Akt (Thr 308) (1/1000; 1/750; 1/1000 respectively, Cell Signaling Technology Inc. Danvers, MA), mouse anti-Gsk3α/β and rabbit anti-Phospho-Gsk3β (Ser 9) (1/1000 and 1/2000 respectively, Cell Signaling Technology Inc., Danvers, MA), rabbit-anti LC3 (1/1000, Sigma-Aldrich, St. Louis, MO), rabbit anti-p38MAPK, rabbit anti-Phosphop38MAPK, rabbit anti-MEK and rabbit anti-PhosphoMEK (1/1000, Cell Signaling Technology Inc., Danvers MA) and mouse anti-actin (1/1000, Sigma-Aldrich, St. Louis, MO). Alexafluor anti-Rabbit 680 (Invitrogen, Carlsbad, CA) and anti-Mouse 800 (VWR International, West Chester, PA) were used as secondary antibodies and fluorescence was quantified using the LI-COR Odyssey imager (LI-COR, Lincoln, NE).
2.6. Glycogen assay
For glycogen measurements, three different sets of hearts from both TIRKO and WT mice were used (1) hearts that were perfused for 50 min with or without 1 mmol/L insulin; (2) hearts that were subjected to three cycles of IPC but no prolonged ischemia or reperfusion and (3) hearts that were subjected to full I/R or IPC protocols in the absence or presence of 1 nmol/L insulin as outlined in Fig. 1. At the end of each perfusion protocol, hearts were snap-frozen in liquid nitrogen and stored at − 80°C until further analysis. Total glycogen was measured by acid extraction followed by enzymatic hydrolysis with amylo-α-1,4-α-1,6-glucosidase and measurement of glucose as previously described [23]. Total glycogen was expressed in μmol glucose/g wet tissue weight.
2.7. Metabolomic analysis
Extraction of metabolites
only hearts that were perfused with full I/R or full IPC in the absence or presence of 1 nmol/L insulin were used. At the end of the 45 min reperfusion, hearts were snap-frozen in liquid nitrogen and stored at − 80°C for further analysis. Frozen heart samples (~50 mg) were homogenized in cold methanol (−20 °C) containing D4-succinate as an internal standard at a final concentration of 80% MeOH (aq) and 20% tissue homogenate. Samples were vortexed and incubated for one hour at −20°C to precipitate protein. Following incubation, cell debris was pelleted by centrifugation (14,000 g for 5 min at 4°C) and the supernatant reserved. A second extraction of the pellet was performed by the addition of −20°C MeHO (aq) to a final concentration of 50% MeOH (aq) and 50% tissue homogenate. Each sample was mixed by vortex, incubated for one hour at −20°C, and centrifuged (14,000 g for 5 min at 4°C) to remove cell debris. The two extracts were combined and dried en vacuo.
Gas chromatography-mass spectrometry (GC-MS) analysis
GC-MS analysis was performed with a Waters GCT Premier mass spectrometer fitted with an Agilent 6890 gas chromatograph and a Gerstel MPS2 autosampler. Dried samples were suspended in 40 μL of 40 mg/mL O-methoxylamine hydrochloride (MOX) in pyridine and incubated for one hour at 30°C. 25μL of this solution was added to autosampler vials, then 20μL of N-methyl-N-trimethylsilyltrifluoracetamide (MSTFA) was added using the autosampler and incubated for 60 minutes at 37°C with shaking. 1 μL of the sample was injected to the gas chromatograph inlet in the split mode at a 10:1 split ratio with the inlet temperature held at 250°C. The gas chromatograph had an initial temperature of 95°C for one minute followed by a 40°C/min ramp to 110°C and a hold time of 2 minutes. This was followed by a second 5°C/min ramp to 250°C, a third ramp to 350°C, then a final hold time of 3 minutes. A 30 m Phenomenex-ZB5MSi column with a 5 m long guard column was employed for chromatographic separation. Data was collected using MassLynx 4.1 software (Waters). Known metabolites were identified and their peak area was recorded using QuanLynx. Data was transferred to an Excel file where each sample was normalized to the internal standard D4-succinate.
2.8. Statistical analysis
All values were expressed as means ± standard error of the mean (sem). Comparisons between two groups were evaluated by Student’s t-test. To compare results from more than two groups, a two-way ANOVA followed by Bonferroni test was performed using GraphPad Prizm software, version 5 (GraphPad Software, Inc. La Jolla, CA). A p value of less than 0.05 was considered statistically significant.
3. Results
3.1. Insulin suppressed cardioprotection by IPC in wild-type mice but not in TIRKO mice
To determine the role of insulin receptors in cardioptotection, we subjected TIRKO and wild-type (WT) hearts to I/R or IPC in the absence or presence of 1 nmol/L insulin, present for the entire duration of I/R or IPC protocol (Fig. 1). Percent (%) recovery of left ventricular developed pressure (LVDP) and rate pressure product (RPP) was determined. Consistent with previously published results showing that the presence of insulin at the reperfusion protected rat hearts from I/R injury [10], we demonstrated that the presence of insulin prior to ischemia and during reperfusion protected WT hearts from ischemia. Thus, % recovery of LVDP and RPP was significantly (p < 0.05) elevated in WT hearts subjected to I/R with insulin compared to WT hearts subjected to I/R without insulin (Fig. 2A, B). In contrast, the protective effect of insulin against I/R was lost in TIRKO hearts, consistent with absent insulin signaling. Of note, the absence of insulin receptors in TIRKO hearts did not exacerbate ischemic injury relative to WT hearts perfused without insulin. Three cycles of IPC in the absence of insulin protected WT and TIRKO hearts from ischemia as evidenced by the increase in % recovery of LVDP and RPP compared to the I/R without insulin (Fig. 2A, B). Interestingly, when insulin was present for the entire IPC protocol, cardioprotection was lost in WT hearts as indicated by poor recovery of contractile function. In contrast, IPC protected TIRKO hearts from ischemia despite the presence of insulin, indicating that abolition of IPC by insulin requires the presence of insulin receptors in the heart.
Fig. 2.

Insulin protected against I/R and abolished cardioprotection by IPC in WT but not in TIRKO hearts. Hearts were isolated from mice lacking insulin receptors in the entire heart (TIRKO) and their corresponding WT controls were subjected to I/R or IPC in the absence or presence of 1 nmol/L insulin. Contractile parameters were recorded at the end of the stabilization period (baseline) and 10 min before the end of the reperfusion and are expressed as % from baseline values. (A) % recovery of left ventricular developed pressure (LVDP); (B) % recovery of rate pressure product (RPP) and (C) infarct size expressed as % of risk zone in TIRKO and their corresponding WT controls. Data are mean ± SEM (n = 3 mice per group for I/R and n = 7–9 mice per group for IPC). * p<0.05; **p<0.005 versus WT perfused under the same condition; # p<0.05; ##p<0.005 versus (-insulin) within the same genotype.
To determine the effect of insulin on infarct size, a separate group of WT and TIRKO hearts were subjected to I/R or IPC in the absence or presence of 1 nmol/L insulin as outlined in Fig. 1, without insertion of the balloon. Consistent with the functional data, infarct size, expressed as % of risk zone, was significantly (p < 0.05) bigger in WT hearts subjected to I/R without insulin or to IPC with insulin (Fig. 2C). In contrast, while TIRKO hearts subjected to I/R were susceptible to infarction, IPC was extremely efficient in reducing infarct size in these animals independently of insulin (Fig. 2C).
3.2. Insulin’s suppression of IPC-mediated cardioprotection requires cardiomyocyte insulin receptors
Having demonstrated that insulin abolished cardioprotection by IPC and that this effect was abolished in hearts lacking the insulin receptors (IRs) in the entire heart, we next sought to determine whether this effect was mediated via cardiomyocyte IRs. To achieve this goal, we subjected hearts that lack IRs specifically in cardiomyocytes (isolated from CIRKO mice) to I/R or IPC in the absence or presence of 1 nmol/L insulin and measured functional recovery and infarct size. Similar to TIRKO mice, CIRKO mice exhibited reduced heart size (Table 1), confirming the previously known role of insulin signaling in cardiac growth regulation [21]. Independently of the background strain, insulin protected wild-type hearts from I/R as evidenced by enhanced functional recovery of LVDP and RPP and reduced infarct size (Fig. 3A, B). However, the beneficial effect of insulin on functional recovery and infarct size reduction after I/R was completely abolished in CIRKO mice. Similar to results obtained in TIRKO mice, insulin suppressed IPC in WT mice (different background strain) as evidenced by worse functional recovery and bigger infarct size after ischemia. In contrast, IPC protected the heart from ischemic injury in CIRKO mice despite the presence of insulin during the perfusion protocol (Fig. 3A, B). Thus insulin’s suppression of IPC ia mediated via cardiomyocyte IRs because CIRKO hearts were refractory to insulin’s abolition of IPC as suggested by their enhanced functional recovery when insulin was present. It is important to note that cardiac function at baseline as assessed by echocardiography is indistinguishable between TIRKO, CIRKO and their respective WT controls (data not shown). In addition, the responses of TIRKO and CIRKO mice to IPC in the absence of insulin were similar to their controls, excluding the possibility of additional protective mechanisms related to the absence of IRs in the heart.
Table 1.
Morphometric parameters of TIRKO and CIRKO mice and their corresponding wild-type controls.
| WT | TIRKO | WT | CIRKO | |
|---|---|---|---|---|
| N | 14 | 17 | 9 | 8 |
| BW (g) | 26.9 ± 0.5 | 27.2 ± 1 | 31.7 ± 1.1 | 34.7 ± 2.3 |
| HW (mg) | 173.3 ± 4.6 | 149.6 ± 5** | 171.3 ± 7.4 | 146.1 ± 7.5 |
| HW/BW (mg/g) | 6.5 ± 0.2 | 5.5 ± 0.1** | 5.4 ± 0.1 | 4.2 ± 0.12** |
BW: body weight; HW: heart weight; HW/BW: heart weight/body weight ratio. Data are mean ± STE.
p<0.005 versus WT of the same strain.
Fig. 3.

Insulin’s abolition of contractile recovery is mediated through cardiomyocyte insulin receptors. Hearts isolated from mice lacking insulin receptors specifically in cardiomyocytes (CIRKO) and their corresponding WT controls were subjected to I/R or IPC in the absence or presence of 1 nmol/L insulin during the entire perfusion protocol. (A): % recovery of LVDP; (B): % recovery of RPP and (C) infarct size in CIRKO mice respectively and their corresponding WT controls subjected to I/R or IPC in the absence or presence of 1 nmol/L insulin. Data are mean ± SEM (n = 3–6 mice per group for I/R and n = 7–8 mice per group for IPC). * p<0.05; **p<0.005 versus WT perfused under the same condition; # p<0.05; ##p<0.005 versus (-insulin) within the same genotype.
3.3. Insulin’s abolition of IPC occurs despite elevated Akt phosphorylation
Cardioprotection by insulin was previously shown to involve downstream signaling molecules such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (PKB or Akt) and glycogen synthase 3β (Gsk3β) [10, 24–25]. To explore whether insulin’s effect on IPC required the activation of downstream signaling to Akt and Gsk3β, we first measured Akt(Ser473) and Gsk3β(Ser9) phosphorylation at the end of the 45 min perfusion in TIRKO, CIRKO and their corresponding WT hearts subjected to I/R or IPC in the absence or presence of 1 nmol/L insulin. Second, we determined whether insulin-mediated activation of downstream signaling occurred prior to ischemia by measuring both Akt(Ser473) and Gsk3β(Ser9) phosphorylation at the end of the three cycles of IPC. As expected, Akt(Ser473) and Gsk3β(Ser9) phosphorylation at the end of the reperfusion was significantly (p < 0.005) enhanced in WT hearts subjected to I/R in the presence of insulin compared to WT hearts subjected to I/R without insulin (Fig. 4). The effect of insulin on the phosphorylation of these two downstream effectors of insulin signaling was abolished in TIRKO hearts (Fig. 4). Similarly, while insulin was able to enhance Akt phosphorylation on Ser473 in WT hearts, it failed to do so in CIRKO hearts (Fig. 1S). Interestingly, when compared to I/R, IPC attenuated Akt and Gsk3β phosphorylation independently of insulin. However, WT hearts subjected to IPC with insulin still exhibit higher phospho Akt/total Akt and phospho Gsk3β/total Gsk3β ratios compared to WT hearts subjected to IPC without insulin (Figs. 4, 1S).
Fig. 4.

The effect of insulin on Akt and Gsk3β phosphorylation in TIRKO and their corresponding WT controls. (A) Representative western-blots of Akt phosphorylation on Ser473 and total Akt expression and Gsk3β phosphorylation on Ser9 and total Gsk3β expression in TIRKO and WT hearts subjected to I/R or to a full IPC protocol in the absence or presence of 1 nmol/L insulin. (B) and (C) are the corresponding densitometry of phosphoAkt/total Akt ratios and phosphoGsk3β /total Gsk3β ratios. Data are mean ± SEM (n = 4 mice per group f or I/R and n = 4 mice per group for IPC). * p<0.05; **p<0.005 versus WT perfused under the same condition; # p<0.05; ##p<0.005 versus (-insulin) within the same genotype.
When applied prior to 30 min ischemia, insulin phosphorylated Akt and Gsk3β in WT hearts but not in TIRKO and CIRKO hearts respectively (Fig. 2S). However, although significant (p < 0.05), this effect of insulin on downstream effectors was less pronounced compared to WT hearts examined at the end of the reperfusion. Taken together, these results suggest that insulin’s activation of downstream signaling is involved in its suppression of cardioprotection by IPC.
3.4. Insulin suppression of IPC-mediated cardioprotection is Akt dependent
To further investigate the involvement of Akt in mediating insulin’s effect on IPC, we used animals with 2 weeks induction of an activated form of Akt in the heart. As shown in Figs. 5A–D, withdrawal of the DOX chow for two weeks resulted in small but significant increase in Akt signaling and one of its downstream targets S6 as evidenced by elevated phosphorylated Akt (Ser473 or Thr308)/total Akt and phosphorylated S6 (Ser253/236)/total S6 ratios. It is worth noting that total Akt was increased by 15% (p=0.06) in Akt TG hearts compared to WT hearts, which was associated with a modest but significant 6% increase in heart weight (Fig. 5E) and heart weight/body weight ratio (Fig. 5F), consistent with the previously known role of Akt in promoting cardiac growth [26]. The presence of insulin resulted in an equivalent increase in phospho Akt/ total Akt (~5 fold) and phospho S6/ total S6 (~2 fold) ratios respectively in WT and Akt TG (Figs. 5A). When Akt TG mice and their littermate controls were subjected to IPC in the absence of insulin, Akt TG hearts exhibited significantly (p < 0.05) lower recovery of LVDP compared to their control hearts perfused under the same conditions. Furthermore and similar to control hearts perfused with insulin, % recovery of LVDP and RPP was reduced in Akt TG hearts (Fig. 5G and H). Taken together, these data suggest that similar to the effect of insulin, short-term activation of Akt1 in the heart abolished cardioprotection by IPC.
Fig. 5.

Akt activation in the heart abolished cardioprotection by IPC. Hearts isolated from Akt TG and WT mice were subjected to the full IPC protocol as shown in Fig. 1A in the presence or absence of 1 nmol/L insulin. (A) Representative western-blots of Akt phosphorylation on Ser473 and Thr308, total Akt, S6 phosphorylation on Ser235/236 and total S6 protein; (B), (C) and (D) the corresponding densitometry of phosphoSer473Akt/total Akt, phosphoThr308Akt/total Akt and phosphoSer235/236 S6/total S6 ratios respectively. (E) heart weights; (F) heart weight/body weight ratios ×103; (G) % recovery of left ventricular developed pressure (LVDP); (H) % recovery of rate pressure product (RPP) in Akt TG and WT mice. Data are mean ± SEM (4 mice per group and per genotype for western-blots and n = 5–8 mice per group and per genotype for functional recovery). *p<0.05; **p<0.005 versus WT perfused in the same condition; #p<0.05; ##p<0.005 versus (− insulin) within the same genotype.
3.5. Insulin’s suppression of IPC does not involve MAP kinase pathway
In addition to Akt activation, we investigated the effect of insulin on MAP kinase pathway. As shown in Fig. 6A–C, MAP kinase pathway, as measured by the phosphorylation of p38MAPK or its downstream target MEK, was not modified by insulin in WT hearts. By contrast, we observed a small but significant (p < 0.005) increase in the ratio of phosphorylated p38MAPK/ total p38MAPK in Akt TG hearts subjected to IPC in the presence of insulin (Fig. 6B). However, this elevation did not translate into the activation of its downstream target MEK (Fig. 6C).
Fig. 6.

Insulin does not modulate mitogen-activated protein kinase (MAPK) pathway during IPC. (A) Representative western-blots of phosphop38MAPK, phosphoMEK, total p38MAPK and total MEK expression in Akt TG and WT hearts subjected to the full IPC protocol in the presence or absence of 1 nmol/L insulin. (B) and (C) The corresponding densitometry of phosphop38/total p38 and phosphoMEK/total MEK ratios respectively. Data are mean ± SEM (n = 4 mice per group and per genotype). **p<0.005 versus WT perfused in the same condition; #p<0.05 versus (− insulin) within the same genotype.
3.6. Insulin’s suppression of IPC is associated with less glycogen depletion and more lactate production at the reperfusion
To evaluate the involvement of metabolic regulation by insulin in I/R and IPC, we first measured pre-ischemic cardiac glycogen content in TIRKO hearts and their WT controls before ischemia. As shown in Fig. 7A, insulin increased glycogen content in WT hearts but not in TIRKO hearts. When both TIRKO and WT hearts were subjected to the IPC stimulus, glycogen levels dropped significantly (Fig. 7B). However, insulin was able to significantly enhance glucogen content by 37% in WT hearts. Interestingly, after IPC stimulus, glycogen levels were high in TIRKO hearts independently of insulin (Fig. 7B). Next, we measured post-ischemic glycogen content and lactate levels in TIRKO and WT hearts at the end of the 45 min reperfusion. As shown in Fig. 7C and consistent with higher Gsk3β phosphorylation (Fig. 4C), WT hearts subjected to I/R in the presence of insulin had 58% (p < 0.05) more glycogen compared to WT hearts subjected to I/R without insulin. Similarly, the combination of insulin with IPC in WT hearts increased glycogen content by 3.8 fold (p=0.08) compared to IPC without insulin (Fig. 7C). In the other hand, lactic acid trended higher in WTI/R (+ insulin) compared to WTI/R without insulin (Fig. 7D). By contrast, there was a modest increase in lactic acid in WTIPC (+insulin) compared to WTIPC (-insulin) that did not reach statistical significance (Fig. 7D). It is worth noting that lactic acid levels were overall reduced within the IPC groups compared to the I/R groups independently of the genotype.
Fig. 7.

Glycogen and lactate levels in TIRKO and WT heats. (A) Pre-ischemic glycogen content in TIRKO and WT hearts subjected to 50 min perfusion without IPC in the absence or presence of 1 nmol/L insulin. (B) Same as (A) but hearts received three cycles (5 min each) of IPC. (C) Post-ischemic (at the end of the 45 min reperfusion) glycogen content in TIRKO and WT hearts subjected to I/R or IPC with or without 1 nmol/L insulin. (D) Lactate levels measured by GC-MS at the end the reperfusion in TIRKO and WT hearts subjected to I/R or IPC in the absence or presence of 1 nmol/L insulin. Data are mean ± SEM (n = 3–4 mice per group for A and B and n = 4–7 mice per group for C and D. *p<0.05 versus WT perfused in the same condition; #p<0.05 versus (− insulin) within the same genotype.
4. Discussion
This study was designed to determine the effect of insulin on ischemic preconditioning (IPC)-induced cardioprotection in mice. The major results of this work are: (1) insulin suppressed functional recovery and enhanced infarct size in wild-type mice subjected to IPC; (2) insulin’s abolition of IPC required the presence of cardiomyocyte insulin receptors (IRs); (3) the effect of insulin on cardioprotection by IPC is mediated through downstream signals to Akt but is independent of MAPK signaling. Taken together, these findings demonstrate for the first time that persistent activation of Akt signaling by insulin is detrimental to cardioprotection by IPC, which may explain in part the failure to precondition the diabetic heart.
4.1. Insulin suppressed IPC-mediated cardioprotection
IPC is a potent cardioprotective phenomenon that has been shown to limit infarct size in dogs [8] an d post-conditioning with repetitive I/R cycles at the onset of reperfusion is as effective as IPC in protecting the heart from a prolonged ischemic insult [27], which could be of great relevance in the clinical setting. Similar to IPC and post-conditioning, insulin was shown to protect the heart from ischemic injury when administered at the onset of reperfusion [10]. However, the effect of insulin on IPC has never been explored before and could be of great importance for the clinical utility of IPC or post-conditioning in patients with type 2 diabetes, who most often exhibit hyperinsulinemia. In addition, although it is still debated, several studies found that the diabetic heart is refractory to cardioprotection by IPC or required a greater IPC stimulus to achieve this protection [24, 28–34] and that this resistance to IPC was mainly related to impaired PI3K-Akt signaling [24, 35]. However, recent studies have clearly challenged this idea by showing that instead of reduced insulin signaling, PI3K-Akt signaling is rather enhanced in the hearts of type 2 diabetic humans and animals [17–18]. Thus, we hypothesized that insulin’s activation of PI3K-Akt signaling prior to IPC could be detrimental to cardioprotection. Using a combination of pharmacologic and genetic approaches, we demonstrated that insulin administered prior to IPC abolished its cardioprotection in Langendorff-perfused mouse heart. This was evidenced by worse functional recovery and increased infarct size in WT hearts subjected to IPC in the presence of 1 nmol/L insulin. Although this is the first study to directly assess the acute effect of insulin on IPC, Drenger et al. [36] showed that correction of hyperglycemia in streptozotocin-induced diabetic rats using insulin not only increased infarct size but also abolished ischemic and sevoflurane post-conditioning. Similarly, post-conditioning failed to protect the hearts of obese and diabetic ob/ob mice despite enhanced basal insulin signaling to Akt [17]. By contrast, our data are not supported by a prior study on isolated human right atrial trabeculae obtained from patients taking insulin and in which IPC was shown to be effective in recovering developed force [37]. This could be due in part to differences in the responses of right atrial tissue versus whole heart to the IPC stimulus or to the experimental protocol of IPC (hypoxic, glucose-free buffer with pacing versus Langenforff-perfused heart with 11 mmol/L glucose and no pacing).
4.2. Insulin’s abolition of IPC required the presence of cardiomyocyte IRs
We next sought to examine the involvement of the insulin receptors (IRs) in mediating insulin’s suppression of IPC and asked whether IRs of specific cell types within the heart were responsible for this effect. We used mice with either a complete ablation of IRs in the heart (TIRKO) or with cardiomyocyte-specific deletion of IRs (CIRKO) and showed that the negative effect of insulin on IPC required the presence of IRs in cardiomyocytes as CIRKO mice exhibited the same cardoprotection by IPC in the presence of insulin as TIRKO mice (Figs. 2 and 3). Importantly, our data demonstrated that the efficacy of IPC to reduce infarct size was actually potentiated when IRs are absent in the hearts as evidenced by reduced infarct size in TIRKO and CIRKO hearts subjected to IPC in the absence of insulin (Figs. 2C and 3C). These results on functional recovery and infarct size limitation in CIRKO mice are unique to the IPC stimulus and contrast with previous findings showing that functional recovery after myocardial infarction (induced by coronary artery ligation) in CIRKO mice was impaired [38]. The failure to protect the CIRKO heart from myocardial infarction was attributed to limited substrate utilization and mitochondrial dysfunction. However, in the context of IPC, the lack of cardiomyocyte IRs might be beneficial.
4.3. Insulin’s suppression of IPC requires downstream activation of Akt and GsK3β
After establishing the involvement of IRs in mediating insulin’s abolition of cardioprotection by IPC, we next examined downstream signaling to Akt and Gsk3β in TIRKO and CIRKO hearts subjected to I/R or IPC in the absence or presence of insulin. Whether examined at the end of reperfusion or at the end of the three IPC cycles, acute insulin treatment induced Akt phosphorylation on Ser473 and Gsk3β phosphorylation on Ser9 in hearts of TIRKO and CIRKO controls (Figs. 4, 1S and 2S). However, the fold induction of Akt phosphorylation was much higher when examined at the end of the reperfusion, suggesting that insulin’s abolition of IPC-mediated cardioprotection may have occurred during the reperfusion period. Indeed, insulin mediated cardioprotection again I/R in the absence of IPC is believed to be mainly mediated during early reperfusion [10, 39]. Downstream of Akt, phosphorylation and inactivation of Gsk3β has been implicated as one of the main mechanisms by which IPC confers protection [40–41]. However, the use of animal models that lack the phosphorylation site on Gsk3β that is required for its inactivation did not confirm a role for this protein in cardioprotection [42]. Our results demonstrated that despite increased Gsk3β phosphorylation and inhibition by insulin, the heart could no longer be protected by this protocol, suggesting the existence of Gsk3β-independent mechanisms mediating the insulin’s effect on IPC.
4.4. Moderate induction of Akt in the heart abolished cardioptotection by IPC
To directly assess the role of Akt activation in cardioprotection by IPC, we used hearts with inducible activation of Akt (myrAkt 1) [22]. We hypothesized that if insulin-abolished cardioprotection was associated with downstream activation of Akt signaling, then Akt TG hearts, with moderate expression of myrAkt1, should be refractory to cardioprotection by IPC even in the absence of insulin. As detailed in the methods section, we induced the myrAkt transgene for two weeks, which resulted in a modest but significant increase in Akt expression and one of its downstream targets S6 (Figs. 5B–D) and enhanced cardiac growth. The induction of Akt and its downstream target S6 were not influenced by the IPC stimulus as equivalent changes in their expression were also observed in these animals at baseline when the hearts were not perfused [43]. As predicted, Akt1 induction in the heart was sufficient to abrogate IPC-mediated cardioprotection as evidenced by the poor functional recovery of the Akt TG hearts (Figs. 5G and H) in the absence of insulin. The mechanisms by which activation of Akt abolished IPC-mediated cardioprotection have not been fully investigated in the present study but could involve the inhibition of glucose uptake via feedback impairment of insulin receptor substrate (IRS)1/2. Although, we did not assess IRS 1/2 expression or glucose uptake in WT and Akt TG hearts subjected to IPC, a recent study showed that moderate induction of myrAkt1 in the heart (equivalent to ours) did not impair insulin-mediated IRS 1/2 but reduced glucose uptake [43]. It should be noted however that our data on Akt TG are not in agreement with a recent study by Kunuthur et al. [44] showing that mice deficient in Akt1 were resistant to infarct limitation by IPC. However, it should be noted that in this study, Akt phosphorylation on Thr308 was enhanced when normalized to total Akt, which could explain their resistance to IPC. This increase in Akt phosphorylation on Thr308 is probably caused by Akt2 activation.
4.5. Metabolic changes associated with insulin’s abrogation of IPC
To explore the involvement of metabolic regulation in insulin’s suppression of IPC, we first assessed glycogen content in TIRKO and WT hearts at different time points of perfusion. Next, we performed a metabolomic analysis of key metabolites using GC-MS. When glycogen was measured before ischemia and in the absence of the IPC stimulus, insulin increased glycogen synthesis in WT but not in TIRKO hearts (Fig. 7A). This could be due to enhanced Gsk3β phosphorylation on serine 9, leading to less inactivation of glycogen synthase as previously suggested [45]. Interestingly, TIRKO hearts exhibited less glycogen content compared to WT hearts independently of insulin, which could be due to higher glycogen breakdown. Our results contrast prior report showing that glycogen depletion prior to ischemia is beneficial for functional recovery after no flow ischemia [46], which is mainly due to less lactate accumulation during ischemia and thus less cellular acidosis. In the present study, we did not measure lactate levels at the onset of ischemia but at the end of the 45 min reperfusion and found that insulin did not significantly enhanced lactate levels in WT hearts despite higher glycogen content. This could be related to stimulation of glucose oxidation by insulin and reduced anaerobic glycolysis. One other possibility could be that the beneficial effect of insulin on functional recovery and infarct size is independent of insulin’s effect on glycogen or lactate. Indeed, Jonassen et al. [10] demonstrated that the protective effect of insulin on I/R is independent of glucose presence at the reperfusion because the use of pyruvate in the reperfusion instead of glucose did not negate insulin-mediated cardioprotection. Therefore, the authors concluded that cardioprotection by insulin is mediated via Akt and p70S6 kinase but does not involve insulin-stimulated glucose uptake. Compared to control perfusion, three cycles of IPC significantly reduced glycogen content in the heart independently of the genotype (Fig. 7B). This observation is supported by prior studies highlighting glycogen depletion as one of the mechanisms by which IPC protects the heart [47–48]. Despite glycogen depletion by the IPC stimulus, insulin induced a modest but significant (p < 0.05) increase in glycogen content in WT hearts, but whether this increase is causal for poor functional recovery remains to be determined. When glycogen and lactate were examined at the reperfusion, we also observed a higher glycogen content in WT hearts perfused in the presence of insulin. Interestingly, although glycogen accumulation was similar between WT I/R (+insulin) and WTIPC (+insulin), functional recovery was completely different, suggesting that glycogen content does not correlate with functional recovery. Furthermore, we analyzed several other metabolites in the heart at the end of the 45 min reperfusion and showed that lactate was higher in the I/R groups compared to the IPC groups and that tricarboxylic (TCA) cycle intermediates such as pyruvate, malate and fumarate accumulated more in the I/R groups compared to IPC groups. (Fig. 3S). However, none of these metabolites showed a significant variation in the IPC group with the exception of succinate, which was significantly (p < 0.05) increased in WTIPC (+insulin) compared to WTIPC (−insulin). Succinate can be used as by complex II of the electron transport chain, which can lead to higher production of reactive oxygen species (ROS) [49], possibly exacerbating cellular damage in this group. The exact mechanisms by which these insulin-mediated metabolic changes affect functional recovery after IPC required further investigations. Finally, other mechanisms could explain the detrimental effect of insulin on IPC including inhibition of autophagy, alteration of nitric oxide or calcium signaling, modulation of the mitochondrial KATP channel and perturbation of Na+/K+ ATPase activity.
4.6. Study limitation
There are limitations in the present study. We used an ex-vivo perfusion system to assess cardioprotection by IPC, although this is an established method to conduct such studies, it does not recapitulate the in-vivo situation such as changes in the systemic milieu that could affect cardioprotection. This is particularly true for TIRKO mice which are known to be hyperinsulinemic between 20–25 weeks of age [50]. In addition, only glucose (11 mmol/L) was used as substrate without fatty acids, which are known to be the main substrate for the post-ischemic heart [51]. Furthermore, only one dose of insulin was used in this study (1 nmol/L), which is somewhat supraphysiologic, but was aimed to mimic the hyperinsulinemic state observed in diabetes. In addition, signaling molecules downstream of Akt that mediate insulin’s effect on IPC have not yet been identified and could be the subject of future investigations. Finally, lactate levels were only assessed at the end of the 45 min reperfusion and not during ischemia.
5. Conclusions
The present study suggests that insulin may inhibit cardioprotection by IPC through downstream activation of PI3K-Akt signaling, which could in part explain the resistance of the diabetic heart to cardioprotection by IPC in the context of increased insulin signaling. Additional work is needed to determine the mechanisms by which insulin inhibits IPC-mediated cardioprotection and to examine whether reducing insulin signaling in the hearts of type 2 diabetic animals could sensitize their hearts to IPC.
Supplementary Material
Highlights.
Insulin abrogates ischemic preconditioning (IPC)-mediated cardioptotection
Insulin’s suppression of IPC is mediated via cardiomyocyte insulin receptors
Insulin’s abolition of IPC is Akt-dependent
Acknowledgments
This work was supported by the Utah Biomedical Research Core Center (P30) in Metabolism and Cardiovascular Disease Grant P30 HL101310 from the National Institute of Health to E. Dale Abel and Sihem Boudina and R01DK092065 and U01 HL087947 to EDA. The authors would like to thank Dr. Sandra Sena from GREF INSERM U1053 (Bordeaux, France) for her technical assistance.
Footnotes
Disclosure statement
None of the authors has any conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241:2035–8. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- 2.Turner R, Cull C, Holman R. United Kingdom Prospective Diabetes Study 17: a 9-year update of a randomized, controlled trial on the effect of improved metabolic control on complications in non-insulin-dependent diabetes mellitus. Ann Intern Med. 1996;124:136–45. doi: 10.7326/0003-4819-124-1_part_2-199601011-00011. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar D, Solomon SD, Kober L, Rouleau JL, Skali H, McMurray JJ, Francis GS, Henis M, O’Connor CM, Diaz R, Belenkov YN, Varshavsky S, Leimberger JD, Velazquez EJ, Califf RM, Pfeffer MA. Newly diagnosed and previously known diabetes mellitus and 1-year outcomes of acute myocardial infarction: the VALsartan In Acute myocardial iNfarcTion (VALIANT) trial. Circulation. 2004;110:1572–8. doi: 10.1161/01.CIR.0000142047.28024.F2. [DOI] [PubMed] [Google Scholar]
- 4.Stevens RJ, Coleman RL, Adler AI, Stratton IM, Matthews DR, Holman RR. Risk factors for myocardial infarction case fatality and stroke case fatality in type 2 diabetes: UKPDS 66. Diabetes Care. 2004;27:201–7. doi: 10.2337/diacare.27.1.201. [DOI] [PubMed] [Google Scholar]
- 5.Edwards RJ, Saurin AT, Rakhit RD, Marber MS. Therapeutic potential of ischaemic preconditioning. Br J Clin Pharmacol. 2000;50:87–97. doi: 10.1046/j.1365-2125.2000.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–51. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 7.Kloner RA, Rezkalla SH. Preconditioning, postconditioning and their application to clinical cardiology. Cardiovasc Res. 2006;70:297–307. doi: 10.1016/j.cardiores.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 9.Vogt AM, Htun P, Kluge A, Zimmermann R, Schaper W. Insulin-like growth factor-II delays myocardial infarction in experimental coronary artery occlusion. Cardiovasc Res. 1997;33:469–77. doi: 10.1016/s0008-6363(96)00212-x. [DOI] [PubMed] [Google Scholar]
- 10.Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 2001;89:1191–8. doi: 10.1161/hh2401.101385. [DOI] [PubMed] [Google Scholar]
- 11.Fuglesteg BN, Tiron C, Jonassen AK, Mjos OD, Ytrehus K. Pretreatment with insulin before ischaemia reduces infarct size in Langendorff-perfused rat hearts. Acta Physiol (Oxf) 2009;195:273–82. doi: 10.1111/j.1748-1716.2008.01901.x. [DOI] [PubMed] [Google Scholar]
- 12.Peoples-Sheps MD, Siegel E, Suchindran CM, Origasa H, Ware A, Barakat A. Characteristics of maternal employment during pregnancy: effects on low birthweight. Am J Public Health. 1991;81:1007–12. doi: 10.2105/ajph.81.8.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anselmino M, Ohrvik J, Malmberg K, Standl E, Ryden L. Glucose lowering treatment in patients with coronary artery disease is prognostically important not only in established but also in newly detected diabetes mellitus: a report from the Euro Heart Survey on Diabetes and the Heart. Eur Heart J. 2008;29:177–84. doi: 10.1093/eurheartj/ehm519. [DOI] [PubMed] [Google Scholar]
- 14.Barmpouletos D, Stavens G, Ahlberg AW, Katten DM, O’Sullivan DM, Heller GV. Duration and type of therapy for diabetes: impact on cardiac risk stratification with stress electrocardiographic-gated SPECT myocardial perfusion imaging. J Nucl Cardiol. 2010;17:1041–9. doi: 10.1007/s12350-010-9293-4. [DOI] [PubMed] [Google Scholar]
- 15.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- 16.Mellbin LG, Malmberg K, Norhammar A, Wedel H, Ryden L. The impact of glucose lowering treatment on long-term prognosis in patients with type 2 diabetes and myocardial infarction: a report from the DIGAMI 2 trial. Eur Heart J. 2008;29:166–76. doi: 10.1093/eurheartj/ehm518. [DOI] [PubMed] [Google Scholar]
- 17.Bouhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B. Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol Heart Circ Physiol. 2008;295:H1580–6. doi: 10.1152/ajpheart.00379.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, Strickland N, Matsui T, Das S, Rosenzweig A, Punjabi P, Camici PG. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J. 2010;31:100–11. doi: 10.1093/eurheartj/ehp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, Koh GY, Akazawa H, Shiojima I, Kahn CR, Abel ED, Komuro I. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest. 2010;120:1506–14. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okamoto H, Nakae J, Kitamura T, Park BC, Dragatsis I, Accili D. Transgenic rescue of insulin receptor-deficient mice. J Clin Invest. 2004;114:214–23. doi: 10.1172/JCI21645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–39. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–18. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Passonneau JV, Lauderdale VR. A comparison of three methods of glycogen measurement in tissues. Anal Biochem. 1974;60:405–12. doi: 10.1016/0003-2697(74)90248-6. [DOI] [PubMed] [Google Scholar]
- 24.Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes. 2005;54:2360–4. doi: 10.2337/diabetes.54.8.2360. [DOI] [PubMed] [Google Scholar]
- 25.Kunuthur SP, Mocanu MM, Hemmings BA, Hausenloy DJ, Yellon DM. The Akt1 isoform is an essential mediator of ischemic preconditioning. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2011.01491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 28.Tosaki A, Pali T, Droy-Lefaix MT. Effects of Ginkgo biloba extract and preconditioning on the diabetic rat myocardium. Diabetologia. 1996;39:1255–62. doi: 10.1007/s001250050567. [DOI] [PubMed] [Google Scholar]
- 29.Tosaki A, Engelman DT, Engelman RM, Das DK. The evolution of diabetic response to ischemia/reperfusion and preconditioning in isolated working rat hearts. Cardiovasc Res. 1996;31:526–36. [PubMed] [Google Scholar]
- 30.Kersten JR, Toller WG, Gross ER, Pagel PS, Warltier DC. Diabetes abolishes ischemic preconditioning: role of glucose, insulin, and osmolality. Am J Physiol Heart Circ Physiol. 2000;278:H1218–24. doi: 10.1152/ajpheart.2000.278.4.H1218. [DOI] [PubMed] [Google Scholar]
- 31.Ravingerova T, Stetka R, Pancza D, Ulicna O, Ziegelhoffer A, Styk J. Susceptibility to ischemia-induced arrhythmias and the effect of preconditioning in the diabetic rat heart. Physiol Res. 2000;49:607–16. [PubMed] [Google Scholar]
- 32.Ghosh S, Standen NB, Galinianes M. Failure to precondition pathological human myocardium. J Am Coll Cardiol. 2001;37:711–8. doi: 10.1016/s0735-1097(00)01161-x. [DOI] [PubMed] [Google Scholar]
- 33.Lee TM, Chou TF. Impairment of myocardial protection in type 2 diabetic patients. J Clin Endocrinol Metab. 2003;88:531–7. doi: 10.1210/jc.2002-020904. [DOI] [PubMed] [Google Scholar]
- 34.Kristiansen SB, Lofgren B, Stottrup NB, Khatir D, Nielsen-Kudsk JE, Nielsen TT, Botker HE, Flyvbjerg A. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia. 2004;47:1716–21. doi: 10.1007/s00125-004-1514-4. [DOI] [PubMed] [Google Scholar]
- 35.Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM. Preconditioning the diabetic human myocardium. J Cell Mol Med. 2010;14:1740–6. doi: 10.1111/j.1582-4934.2009.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH. Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology. 2011;114:1364–72. doi: 10.1097/ALN.0b013e31820efafd. [DOI] [PubMed] [Google Scholar]
- 37.Cleveland JC, Jr, Meldrum DR, Cain BS, Banerjee A, Harken AH. Oral sulfonylurea hypoglycemic agents prevent ischemic preconditioning in human myocardium. Two paradoxes revisited Circulation. 1997;96:29–32. doi: 10.1161/01.cir.96.1.29. [DOI] [PubMed] [Google Scholar]
- 38.Sena S, Hu P, Zhang D, Wang X, Wayment B, Olsen C, Avelar E, Abel ED, Litwin SE. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol. 2009;46:910–8. doi: 10.1016/j.yjmcc.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baines CP, Wang L, Cohen MV, Downey JM. Myocardial protection by insulin is dependent on phospatidylinositol 3-kinase but not protein kinase C or KATP channels in the isolated rabbit heart. Basic Res Cardiol. 1999;94:188–98. doi: 10.1007/s003950050142. [DOI] [PubMed] [Google Scholar]
- 40.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ Res. 2002;90:377–9. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 41.Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, Duplaa C, Jaspard-Vinassa B. GSK-3beta at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res. 2011;90:49–56. doi: 10.1093/cvr/cvr002. [DOI] [PubMed] [Google Scholar]
- 42.Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M, Marber MS. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–14. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Y, Pereira RO, O’Neill BT, Riehle C, Ilkun O, Wende AR, Rawlings TA, Zhang YC, Zhang Q, Klip A, Shiojima I, Walsh K, Abel ED. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol Endocrinol. 2013;27:172–84. doi: 10.1210/me.2012-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kunuthur SP, Milliken PH, Gibson CL, Suckling CJ, Wadsworth RM. Tetrahydrobiopterin analogues with NO-dependent pulmonary vasodilator properties. Eur J Pharmacol. 2011;650:371–7. doi: 10.1016/j.ejphar.2010.09.070. [DOI] [PubMed] [Google Scholar]
- 45.Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–8. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neely JR, Grotyohann LW. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischemic hearts. Circ Res. 1984;55:816–24. doi: 10.1161/01.res.55.6.816. [DOI] [PubMed] [Google Scholar]
- 47.Soares PR, de Albuquerque CP, Chacko VP, Gerstenblith G, Weiss RG. Role of preischemic glycogen depletion in the improvement of postischemic metabolic and contractile recovery of ischemia-preconditioned rat hearts. Circulation. 1997;96:975–83. doi: 10.1161/01.cir.96.3.975. [DOI] [PubMed] [Google Scholar]
- 48.Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–88. doi: 10.1161/01.cir.99.4.578. [DOI] [PubMed] [Google Scholar]
- 49.Moreno-Sanchez R, Hernandez-Esquivel L, Rivero-Segura NA, Marin-Hernandez A, Neuzil J, Ralph SJ, Rodriguez-Enriquez S. Reactive oxygen species are generated by the respiratory complex II--evidence for lack of contribution of the reverse electron flow in complex I. FEBS J. 2013;280:927–38. doi: 10.1111/febs.12086. [DOI] [PubMed] [Google Scholar]
- 50.Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang QJ, Gale D, Wilson LJ, Abel ED. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res. 2009;104:1085–94. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, el Alaoui-Talibi Z, Clanachan AS, Schulz R, Lopaschuk GD. Uncoupling of contractile function from mitochondrial TCA cycle activity and MVO2 during reperfusion of ischemic hearts. Am J Physiol. 1996;270:H72–80. doi: 10.1152/ajpheart.1996.270.1.H72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.