

Abstract
Background
Advanced paternal age at birth has been linked to several psychiatric disorders in offspring (e.g., schizophrenia), and genetic mechanisms are thought to underlie these associations. This study is the first to investigate whether advanced paternal age at birth is associated with eating disorder risk using a twin study design capable of examining both phenotypic and genetic associations.
Methods
In a large, population-based sample of female twins ages 8–17 years in mid-puberty or beyond (N = 1,722), we investigated whether advanced paternal age was positively associated with disordered eating symptoms and an eating disorder history (i.e., anorexia nervosa, bulimia nervosa, or binge eating disorder) in offspring. Biometric twin models examined whether genetic and/or environmental factors underlie paternal age effects for disordered eating symptoms.
Results
Advanced paternal age was positively associated with disordered eating symptoms and an eating disorder history, where the highest level of pathology was observed in offspring born to fathers ≥ 40 years old. Results were not accounted for by maternal age at birth, body mass index, socioeconomic status, fertility treatment, or parental psychiatric history. Twin models indicated decreased genetic, and increased environmental, effects on disordered eating with advanced paternal age.
Conclusions
Advanced paternal age increased risk for the full spectrum of eating pathology, independent of several important covariates. However, contrary to leading hypotheses, environmental rather than genetic factors accounted for paternal age-disordered eating associations. These data highlight the need to explore novel (potentially environmental) mechanisms underlying the effects of advanced paternal age on offspring eating disorder risk.
Keywords: advanced paternal age, eating disorders, disordered eating, genetic, environmental, twin study
Advanced paternal age at birth is a risk factor for psychiatric disorders in offspring, including schizophrenia, autism, and bipolar disorder (Malaspina et al., 2001, Reichenberg et al., 2006, Frans et al., 2008). These relationships have been confirmed in meta-analytic studies (Wohl and Gorwood, 2007, Hultman et al., 2011, Miller et al., 2011) and are independent of important confounds (i.e., maternal age at birth, socioeconomic status (SES), parental psychiatric history) that increase psychiatric risk and are associated with later entry into parenthood (Byrne et al., 2003, Croen et al., 2007, Menezes et al., 2010). Recent data also link paternal age to obesity (Eriksen et al., 2012). Despite these robust effects, advanced paternal age has never been examined as a risk factor for eating disorders in offspring. Examining this possibility could provide new insights into the complex etiology of eating disorders.
Leading theories propose that de novo genetic mutations are primary mechanisms underlying paternal age-psychiatric risk associations (Malaspina et al., 2002). The increased probability of DNA copy error problems with each sperm replication (~610 replications by age 40), and the accumulation of mutations in the germline of older fathers, are thought to lead to disease phenotypes in offspring (Crow, 2000). Genetic studies indicate that offspring de novo mutation rates are influenced by paternal age at conception (Kong et al., 2012), and de novo mutations are implicated in schizophrenia and autism (Awadalla et al., 2010, Xu et al., 2011, Iossifov et al., 2012). However, to our knowledge, no study has directly confirmed that de novo mutations underlie paternal age-offspring psychiatric risk associations.
Twin studies can be used to indirectly examine the role of de novo mutations in paternal age-psychiatric disorder relationships. Because monozygotic (MZ) twins share 100% of their genes, and dizygotic (DZ) twins share, on average, half of their segregating genetic material, MZ twins would be expected to share all de novo mutations, whereas DZ twins would almost never share genetic mutations given their rarity (Zhao et al., 2007, Liu et al., 2010, Ronald and Hoekstra, 2011). If de novo mutations underlie paternal age–psychiatric risk associations, larger differences between MZ and DZ twin correlations should be observed with advancing paternal age since DZ co-twins should become less similar as mutation rates increase.
One previous study examined these processes by investigating twin similarity for autism across paternal age (Lundström et al., 2010). Importantly, differences in MZ/DZ concordance for autism decreased with advanced paternal age, implicating environmental rather than genetic processes. Although results were limited by a small number of cases in each age category (Ns = 3–44), they highlight the need to test hypotheses regarding mechanisms for paternal age effects. Twin registries are excellent, low-cost resources for such investigations, as they can indirectly examine whether de novo mutations or other competing processes most likely underlie paternal age-psychiatric risk associations.
The current study investigated phenotypic and etiologic associations between advanced paternal age and disordered eating symptoms (e.g., weight/shape concerns) in a large, population-based sample of female twins. These symptoms are core features of eating disorders and prospectively predict the development of diagnoses (Jacobi et al., 2004); thus, results should inform risk models of eating disorders. Exploratory analyses examining associations between advanced paternal age and an eating disorder history (anorexia nervosa (AN), bulimia nervosa (BN), binge eating disorder (BED)) were also conducted to investigate whether results generalize across categorical and continuous eating disorder dimensions (American Psychiatric Association, 2000; Insel et al., 2010). Several covariates were included (e.g., maternal age, body mass index (BMI) percentile, SES, fertility treatment, parental psychiatric history) to ensure that associations were not due to confounding factors. Finally, we compared twin similarity for disordered eating across paternal age to explore mechanisms (i.e., genetic/environmental) underlying paternal age-disordered eating associations.
Methods
Participants
Participants were 1,722 female twins (488 (28%) MZ, 611 (35%) same-sex DZ, and 623 (36%) opposite-sex) between the ages of 8–17 years (M (S.D) = 13.86 (2.39)) from the on-going Michigan Twins Project (MTP), a study conducted within the Michigan State University Twin Registry (MSUTR; Klump and Burt, 2006, Burt and Klump, 2013). Twins were recruited using birth records and driver’s license databases (see Klump & Burt (2006)). MTP recruitment began in 2008, and response rates (~56%) are on par with those for other population-based twin registries (Kendler et al., 1992, Lichtenstein, 2002).
Because increases in genetic influences on disordered eating are observed during puberty in females (Klump et al., 2007, Culbert et al., 2009), we only included twins who were in mid-puberty or beyond (i.e., score ≥ 2.5 on the Pubertal Development Scale; Petersen et al., 1988). Twins spanned a range of racial/ethnic backgrounds (e.g., Caucasian, Black/African American, Asian/Pacific Rim), but consistent with the region (see http://www.michigan.gov), most (87%) were Caucasian.
Measures
All data came from the MTP questionnaire completed by the twins’ mother or father (93% biological mothers, 6% biological fathers, 1% step/adoptive parents).
Zygosity
Similar to other twin registries (Kendler et al., 1992, Lichtenstein, 2002), zygosity was determined using five physical similarity items. These items have demonstrated over 95% accuracy when compared to genotyping (Lykken et al., 1990).
Parental Age at Birth
Paternal and maternal ages at offspring birth were calculated using birth dates. Average ages at birth (Paternal M (S.D) = 32.70 (5.92), range = 15–55; Maternal M (S.D) = 30.49 (4.77), range = 17–48) were similar to those from other studies examining parental age effects (e.g., Paternal M (S.D) = 31.5 (6.8), range = 13–70; Maternal M (S.D) = 28.8 (5.9), range = 12–53) (Croen et al., 2007).
Disordered Eating Symptoms
Disordered eating was assessed using 9 items from the Eating Disorder Examination-Questionnaire (EDE-Q; Fairburn and Beglin, 1994) (see Table 1). The EDE-Q assesses core eating disorder symptoms including weight/shape concerns, dietary restraint, binge eating, and compensatory behaviors. EDE-Q items were included if they: 1) represented key attitudinal and behavioral symptoms; 2) exhibited significant correlations with the EDE-Q Global Score (mean r = .76); and 3) were developmentally appropriate for pre- and early-adolescent participants. Importantly, these symptoms are well-established risk factors for eating disorders (Jacobi et al., 2004).
Table 1.
Disordered Eating Items and Frequency of Item Endorsement
| Disordered Eating Items | Item Endorsement Frequency (% of twins)
|
||
|---|---|---|---|
| 0 (Not True) | 1 (Somewhat True) | 2 (Certainly True) | |
| 1. Feels fat | 70.7 | 21.4 | 7.9 |
| 2. Definite fear of gaining weight or of becoming fat | 66.3 | 25.3 | 8.5 |
| 3. Has a strong desire to lose weight | 77.1 | 17.1 | 5.9 |
| 4. Diets to control weight | 86.2 | 11.2 | 2.6 |
| 5. Very dissatisfied with his/her body weight or shape | 65.1 | 24.9 | 10.0 |
| 6. Body weight and/or shape influences how thinks about (judges) him/herself as a person | 69.7 | 25.1 | 5.2 |
| 7. Feels guilty about what he/she eats because of the effect on body weight and/or shape | 80.1 | 16.6 | 3.3 |
| 8. Eats lots of food and can’t stop | 89.5 | 8.7 | 1.9 |
| 9. Has vomited to control his/her weight | 99.4 | .20 | .30 |
EDE-Q items were modified for use in this large-scale, mail-in twin registry. Parent, rather than child, reports were collected in order to keep the MTP questionnaire brief and easy to complete. This was necessary for recruiting as many families as possible into the registry (which serves as a participant bank – see Burt and Klump (2013)). Although parent reports of disordered eating are less commonly used than parent reports of other psychiatric symptoms, data from another on-going MSUTR study (Klump et al., 2010) show that the parent-child correlation for the MTP disordered eating items (r = .52) is similar or better than parent-child correlations for externalizing/internalizing symptoms (r’s = .14–.48) (Kolko and Kazdin, 1993, Youngstrom et al., 2000). EDE-Q parent reports also demonstrated expected correlations with external correlates in the Klump et al. (2010) sample (i.e., BMI, r = .58; depressive symptoms, r = .35).
Parents rated their children’s disordered eating based on what they “generally are like” using a three-point scale (“not true”, “sometimes true”, “certainly true”) rather than the original EDE-Q format (i.e., number of days a symptom was present in past month). This was necessary to match other MTP items and to index trait- rather than state-levels of disordered eating. Our data suggest that this rating format does not substantially affect the validity of the EDE-Q. In a sub-sample of twins whose mothers rated disordered eating using the MTP EDE-Q and the original EDE-Q, ratings were highly correlated (r = .59) despite occurring an average of 1.5 years apart (SD = 0.77; range = 0.23–2.90 years).
Internal consistency for the MTP items was excellent (α = .86). There was significant variability in disordered eating, as the rate at which most symptoms were “sometimes” or “certainly” true ranged from 10–35% (see Table 1). Levels of disordered eating were on par with those from other MSUTR studies examining a similar age range, but using the original EDE-Q (item endorsement = 7–42%) (Klump et al., 2010, Klump et al., 2013). As would be expected in a population-based, adolescent sample (Wade et al., 2008), attitudinal symptoms (e.g., fear of fatness) were the most highly endorsed, although behaviors (e.g., dieting, binge eating) also showed sufficient variation. Correlations with BMI were in the low-to-moderate range (range = .009–.42; mean r = .25; average percent variance shared = 6%), suggesting that our measure taps disordered eating symptoms that are not merely a reflection of weight status/obesity.
Eating Disorder Diagnosis
Lifetime histories of eating disorders (AN, BN, or BED) were assessed via parent report. Of the 1,668 twins with available data, 11 (1%) had a history of an eating disorder (6/11 (55%) AN, 4/11 (36%) BED, 1/11 (9%) AN and BN), of which 7 (64%) received previous treatment and 4 (36%) did not. These percentages are on par with the prevalence of eating disorders in female children/adolescents (Merikangas et al., 2010, Swanson et al., 2011). As expected, twins with an eating disorder history had substantially higher MTP disordered eating scores (M (SD) = 7.81 (5.98)) than those with no history (M (SD) = 2.31 (3.16); p = .01; Cohen’s d = 1.15). Disordered eating scores did not meaningfully differ between patients with a history of AN (or AN/BN) (M (S.D) = 8.14 (7.40)) vs. BED (M (S.D) = 7.25 (2.98)).
Covariates
Several covariates were examined to ensure that associations between paternal age and eating pathology were not accounted for by confounding factors. Covariates included: twin age, ethnicity, and BMI percentile (i.e., BMI adjusted for age); SES; parental fertility treatment and psychiatric history. Selected covariates have been associated with disordered eating and/or later paternal age at birth (Hare and Moran, 1979, Lilenfeld et al., 1998, O’Dea and Caputi, 2001, Croll et al., 2002, Doornbos et al., 2007, Sobotka, 2010, Eriksen et al., 2012). Parent reports of twin height and weight, which correlate highly with laboratory measurements (r’s = .79–.81; Huybrechts et al., 2011), were used to calculate BMI. BMI values were transformed to percentiles (see www.cdc.gov) in order to capture age-related variation in our child/adolescent sample. Family yearly income and parental education (i.e., highest level of education achieved by mother or father) were used to assess SES. A history of depression, anxiety disorder (i.e., panic, obsessive-compulsive, post-traumatic stress, separation anxiety), or eating disorder (i.e., AN, BN, BED) in the biological mother and/or father was assessed using a family history checklist coded “yes” if at least one parent suffered from one or more disorder or “no” if neither parent reported a history of the disorders.
Statistical Analyses
Phenotypic Associations
Paternal age at birth was examined as a predictor of disordered eating symptoms and history of an eating disorder. Paternal age was modeled continuously as well as categorically (i.e., < 25 years, 25–29 years, 30–34 years, 35–39 years, ≥ 40 years) to increase statistical power and identify particularly high-risk age thresholds (e.g., paternal age > 35 years - see Wohl and Gorwood (2007)).
Generalized linear mixed models (GLMMs) were used as they could account for the non-independence of twin data (i.e., by nesting a level 1 variable (individual twin) within a level 2 unit (family)) and could examine both continuous and categorical outcomes. GLMM linear models (normal distribution with identity link) and binary logistic regressions (binomial distribution with logit link) were used to examine disordered eating and eating disorder history, respectively. We standardized all variables prior to analysis in order to interpret unstandardized coefficients as standardized coefficients.
GLMMs were run twice: 1) controlling only for maternal age at birth, and 2) controlling for all covariates. This two-step process allowed us to identify the effects of covariates on paternal age-eating pathology associations, while always controlling for maternal age, since maternal age correlated highly with paternal age (r = .72)1 and is associated with other psychiatric disorders (Croen et al., 2007, Menezes et al., 2010). Controlling for maternal age also ensured that our independent variable (i.e., paternal age at birth) and dependent variables (i.e., parent-reported eating pathology) were not confounded, as 93% of MTP questionnaires were completed by biological mothers.2
Genetic Associations
We investigated genetic influences on paternal age effects by comparing MZ and DZ twin similarity for disordered eating as a function of paternal age at birth. Given that very few participants had an eating disorder history, analyses focused on disordered eating symptoms. Further, although same-sex and opposite-sex twins were included in phenotypic analyses, twin analyses included same-sex twins only, as male co-twins of opposite-sex females were not examined due to low eating disorder prevalence in males (Swanson et al., 2011).
Twin intraclass correlations were calculated in the full sample and each paternal age category to provide an initial indication of genetic/environmental effects on disordered eating across paternal age. Additive genetic effects (A; genetic influences that add across genes) are indicated if the MZ twin correlation is approximately twice the DZ twin correlation, while non-additive genetic effects (D; interaction of genetic effects at same locus) are suggested if MZ correlations are more than double DZ correlations. De novo mutations are considered a non-additive genetic process in twin studies, as DZ twins would share far less than half of their mutations because of their rarity (Liu et al., 2010). Therefore, if de novo mutations underlie paternal age-disordered eating relationships, we would expect larger MZ/DZ differences with advanced paternal age.
Twin studies also decompose variance into two forms of environmental effects. Shared environmental influences (C; factors that make co-twins similar to one another) are inferred if MZ and DZ twin correlations are approximately equal, while non-shared environmental influences (E; factors that make co-twins different from one another, including measurement error) are implied if the MZ correlation is less than 1.0. If differences between MZ/DZ correlations decrease with paternal age, shared environmental processes would be implicated, as co-twins would increase in similarity regardless of genetic sharing. Finally, decreases in the MZ correlation would indicate non-shared environmental influences on paternal age-disordered eating effects.
We then used twin moderation models (see Purcell, 2002) to statistically quantify the degree to which genetic/environmental influences on disordered eating vary by paternal age (the moderator). ACE and ADE models were examined in order to test for differences in both additive and non-additive genetic effects. Model fitting was conducted using full information maximum-likelihood raw data techniques with Mx statistical software (Neale, 1997).
Following previous recommendations (Purcell, 2002), we fit “full” ACE and ADE moderator models that included linear and non-linear moderators to directly test whether genetic/environmental estimates vary linearly or non-linearly with paternal age. We compared full models to two more restrictive models: 1) linear moderation models that dropped non-linear moderation coefficients, and 2) no moderation models that estimated only genetic/environmental paths. To minimize the number of models fit, we did not drop individual moderation coefficients one by one. Instead, we determined whether each etiologic influence varied by paternal age by examining whether confidence intervals for moderator estimates overlapped with zero.
The best fitting model was determined using both the difference in minus twice the log likelihood (Δ-2lnL) and Akaike’s Information Criteria (AIC). Δ-2lnL was used to compare full and nested moderation models, and AIC was used to compare the unnested ACE and ADE models. Statistically significant differences in-2lnL suggest that dropping moderator coefficients results in significantly worse model fit, whereas lower AIC values indicate better model fit.
Although definition variables can be used to account for covariate effects in twin models (Neale et al., 2006), it was not possible to simultaneously include all eight covariates as definition variables in one model, as this would reduce statistical power to detect differences in etiologic effects across paternal age (Agrawal et al., 2010). Consequently, we ran individual models that included each covariate as a definition variable one by one to examine individual covariate effects. Because findings were unchanged in each run of the model, we focused our results on models that included maternal age at birth as the definition variable in order to mirror our first set of phenotypic analyses described above.
Results
Phenotypic Associations
Consistent with hypotheses, paternal age was positively associated with disordered eating symptoms in female offspring, even after controlling for several important covariates (e.g., maternal age, BMI percentile; see Table 2). Similar to other disorders (Byrne et al., 2003), a threshold effect was detected, such that mean levels of disordered eating were significantly higher in offspring of fathers ≥ 40 years compared to offspring of younger fathers (see Table 2). Effect sizes indicate that the phenotypic effect of paternal age on disordered eating is small-to-medium in magnitude (beta’s =.08–.09; d’s =.18–.41).3
Table 2.
Paternal Age at Birth as a Predictor of Disordered Eating Symptoms and Lifetime Eating Disorder History in Offspring
| Paternal Age | Maternal Age Only | Full Covariate Model | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| n (%) | Coefficient (S.E.) | t (df) | p | d | Coefficient (S.E.) | t (df) | p | d | ||
| Disordered Eating Symptoms | ||||||||||
| Continuous Categorical | 1722 | .09 (.04) | 2.37 (1719) | .02 | -- | .08 (.03) | 2.44 (1709) | .01 | -- | |
| < 25 | 128 (7.4) | −.02 (.10) | −2.08 (1716) | .04 | .28 | −.06 (.10) | −2.53 (1706) | .01 | .30 | |
| 25–29 | 439 (25.5) | −.15 (.05) | −4.14 (1716) | <.001 | .41 | −.10 (.06) | −3.98 (1706) | <.001 | .31 | |
| 30–34 | 589 (34.2) | .01 (.04) | −3.10 (1716) | .002 | .27 | .04 (.06) | −2.89 (1706) | .004 | .18 | |
| 35–39 | 383 (22.2) | .04 (.06) | −2.80 (1716) | .005 | .32 | .05 (.07) | −2.85 (1706) | .004 | .18 | |
| ≥ 40 | 183 (10.6) | .29 (.08) | -- | -- | -- | .28 (.09) | -- | -- | -- | |
| Lifetime Eating Disorder (ED) History | ||||||||||
| ED n (%) | No ED n (%) | |||||||||
| Continuous Categorical | 11 | 1, 657 | .64 (.32) | 2.01 (1666) | .04 | -- | .67 (.31) | 2.14 (1655) | .03 | -- |
| < 25 | 1 (9.1) | 122 (7.4) | -- | -- | -- | -- | -- | -- | -- | -- |
| 25–29 | 0 (0) | 422 (25.5) | -- | -- | -- | -- | -- | -- | -- | -- |
| 30–34 | 5 (45.5) | 566 (34.2) | -- | -- | -- | -- | -- | -- | -- | -- |
| 35–39 | 2 (18.2) | 369 (22.3) | -- | -- | -- | -- | -- | -- | -- | -- |
| ≥ 40 | 3 (27.3) | 178 (10.7) | -- | -- | -- | -- | -- | -- | -- | -- |
The “Maternal Age Only” model controlled for maternal age at birth only. The “Full Covariate” model controlled for maternal age at birth, child age, child ethnicity, child body mass index percentile, family education and income, fertility treatment, and parental psychiatric history. Coefficient = standardized beta in continuous paternal age models; standardized mean in categorical paternal age models. The ≥ 40 years group was coded as the reference group for t-test comparisons in categorical paternal age models.
Exploratory analyses revealed that paternal age at birth, measured continuously, also predicts a lifetime eating disorder history. Small sample sizes prevented examining paternal age as a categorical predictor of eating disorder diagnosis; however, there were a disproportionate number of cases in older age categories (see Table 2), and this effect was not driven by a specific diagnosis (i.e., AN and BED were both present in the older age groups).
Genetic Associations
Similar to previous research in pubertal females (Klump et al., 2007, Culbert et al., 2009), higher MZ than DZ correlations and an MZ correlation less than 1.0 indicated the presence of genetic and non-shared environmental influences on disordered eating in the full sample. However, differences in MZ/DZ correlations across paternal age suggested significant heterogeneity in etiologic effects (see Table 3). Before age 35, the MZ correlation was twice the DZ correlation, suggesting additive genetic effects but little-to-no shared environmental or non-additive genetic influences. By contrast, after age 35, the DZ correlation increased and was comparable to most of the MZ correlations, indicating a complete lack of additive or non-additive genetic effects but significant shared environmental influences. The lower MZ correlation in the oldest paternal age group could reflect greater non-shared environmental effects, although conclusions are hard to draw given the group’s small sample size (n = 16 pairs) and the fact that this correlation does not significantly differ from MZ correlations in most younger paternal age groups (as evidenced by overlapping confidence intervals; see Table 3). Overall, twin correlations suggest increasing shared environmental influences on disordered eating after age 35, arguing against increases in genetic or de novo mutation effects with advanced paternal age.
Table 3.
Twin Correlations for Disordered Eating by Paternal Age Category
| MZ (95% CI) [N] | DZ (95% CI) [N] | Z | p | |
|---|---|---|---|---|
| Full sample | .69 (.63, .75) [227] | .43 (.35, .50) [269] | 4.28 | <.001 |
| Paternal age | ||||
| < 25 | .73 (.50, .87) [26] | .32 (−.03, .66) [17] | 1.76 | .04 |
| 25–29 | .67 (.53, .79) [67] | .36 (.21, .50) [77] | 2.54 | .005 |
| 30–34 | .77 (.67, .84) [72] | .33 (.18, .46) [88] | 4.18 | <.001 |
| 35–39 | .65 (.50, .79) [46] | .57 (.41, .70) [54] | 0.62 | .24 |
| ≥ 40 | .30 (−.01, .56) [16] | .64 (.46, .78) [33] | −1.35 | .99 |
MZ = monozygotic twins; DZ = dizygotic same-sex twins; CI = confidence interval; N = number of twin pairs; Z = test of equality examining whether MZ correlation is higher than DZ twin correlations. p = one-tailed significance value for Z test of equality. Maternal age at birth was regressed from each twin’s disordered eating score, and standardized residual scores were used to calculate twin correlations.
Because twin correlations suggested no differences in etiologic effects until age 35, we created two paternal age groups for analyses (< 35 years versus ≥ 35 years) to maximize power and capture key etiologic differences across paternal age.4 Non-linear moderation cannot be examined with only two moderator levels, so we focused on linear and no moderation models. ACE models fit better than ADE models (i.e., lower AICs), which is not surprising given that non-additive genetic effects were non-significant (i.e., 95% confidence intervals included 0).
Within ACE models, a statistically significant Δ-2lnL indicated that the linear moderation model fit better than the no moderation model and that genetic/environmental influences on disordered eating varied by paternal age. Following previous recommendations (Purcell, 2002), we report unstandardized path and moderator estimates in Table 4 and Figure 1 in order to depict absolute changes in genetic/environmental influences rather than changes in proportions of total variance. Nonetheless, in order to compare genetic/environmental estimates to previous twin studies of disordered eating, we also report standardized estimates (calculated by squaring the path coefficients in the < 35 group, and squaring the sum of the moderator estimates plus the path coefficients in the ≥ 35 group; see percentages below).
Table 4.
Unstandardized Parameter Estimates and Test Statistics for the Comparison of Linear Moderation and No Moderation Models
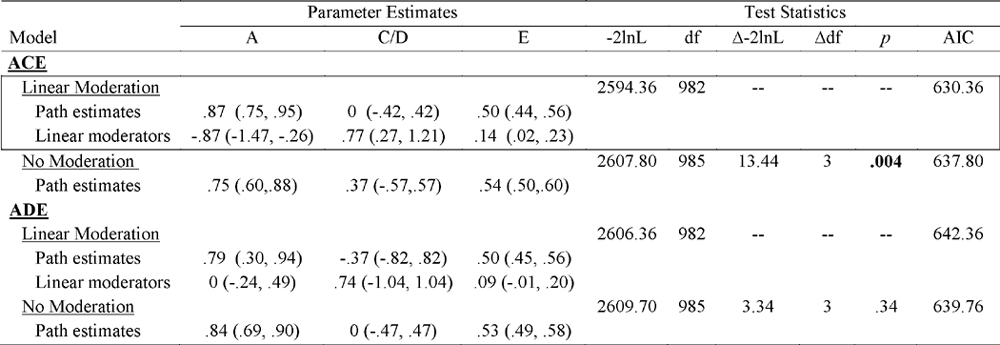
|
A = additive genetic; C = shared environmental; D = non-additive genetic; E = non-shared environmental; −2lnL = minus twice the log likelihood; df = degrees of freedom; Δ-2lnL = change in minus twice the log likelihood; Δdf = change in degree of freedom; AIC = Akaike Information Criteria; Linear Moderation = genetic, shared environmental, and non-shared environmental parameter estimates vary linearly by paternal age at birth; No Moderation = genetic, shared environmental, and non-shared environmental parameter estimates do not vary by paternal age at birth; Best-fitting model is denoted by boxed-in text. Significant paths and moderator estimates are indicated by confidence intervals that do not overlap with 0.
Figure 1. Unstandardized Additive Genetic, Shared Environmental, and Non-shared Environmental Variance Contributions to Disordered Eating by Paternal Age at Birth.
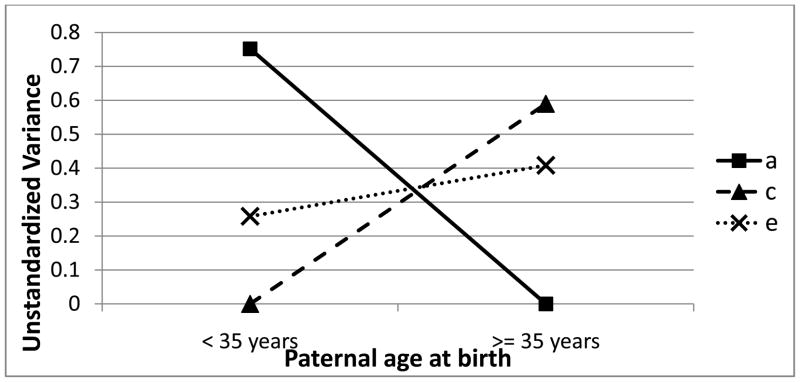
a = additive genetic; c = shared environment; e =non-shared environment
As shown in Table 4, a, c, and e moderator estimates were statistically significant, suggesting that all three differ across paternal age. The most substantial change occurs for additive genetic and shared environmental influences (see Figure 1). Before age 35, additive genetic effects primarily contributed to disordered eating (A = 75%), with the remaining variance due to the non-shared environment (E = 25%). In stark contrast, after age 35, the variance in disordered eating was entirely due to shared and non-shared environmental factors (C = 59%; E = 41%).5
Discussion
Using a large, population-based sample of female twins, we demonstrated for the first time that advanced paternal age at birth is a significant risk factor for offspring eating pathology. This phenotypic effect was quite robust, in that it was present for eating disorder symptoms and diagnoses, and effects were independent of several important covariates (e.g., maternal age, BMI percentile, SES, parental psychiatric history). We also examined differences in etiologic influences across paternal age to investigate whether genetic factors (more specifically, de novo mutations) might underlie paternal age-disordered eating relationships. However, results strongly suggested that genetic contributions to disordered eating decreased, rather than increased, with paternal age.
Findings contribute to a growing body of literature implicating advanced paternal age in offspring psychiatric risk, as indicated for schizophrenia, autism, and bipolar disorder (Malaspina et al., 2001, Reichenberg et al., 2006, Frans et al., 2008). A relationship with disordered eating is novel and suggests that eating disorders may share some, perhaps more general, etiologic risk factors with these disorders. This is important given that socio-cultural influences (e.g., pressures for thinness) have led to views that eating disorders are etiologically distinct from “neuropsychiatric” illnesses (Klump et al., 2009). Instead, eating disorders may be uniquely associated with both the internalizing and neuropsychiatric spectrums of psychopathology. This possibility is being explored by researchers investigating social/cognitive similarities between AN and autism (Zucker et al., 2007) as well as other neuropsychiatric disorders (Steinglass et al., 2007).
De novo mutations have been proposed as a potential biological mechanism underlying paternal age-psychiatric risk associations, although our results argue against de novo mutation effects for paternal age-disordered eating relationships. We observed a sharp decrease in the magnitude of genetic influences on disordered eating in the paternal age ≥ 35 group, as DZ twins (who would not share de novo mutations) became more rather than less similar with paternal age. Our results converge with those of an autism twin study (Lundström et al., 2010), and together, suggest that de novo mutations may not account for paternal age effects. However, if only the frequency (rather than the location) of mutations confers risk, paternal-age related mutations could theoretically increase the similarity of DZ twins born to older fathers, as DZ twins may have a similar mutation burden but, unlike MZ twins, would not share specific mutations. We presumed that sharing specific mutations, rather than frequency alone, would influence twin similarity for eating pathology, consistent with the prediction that de novo mutations increase heritability estimates in twin studies (Liu et al., 2010, Ronald and Hoekstra, 2011). Nonetheless, we cannot definitively rule out the importance of de novo mutations. Future twin and molecular genetic studies are needed to further examine the mutation hypothesis for paternal age-psychiatric disorder relationships.
Another leading mechanism proposed to underlie paternal age-psychiatric disorder associations is disruptions in epigenetic processes (i.e., activation-inactivation of genes and/or changes to chromatin structure) that occur with advanced paternal age (Perrin et al., 2007). While epigenetic mechanisms have been implicated in eating disorders and other psychiatric illnesses (e.g., Schanen, 2006, Frieling et al., 2010), epigenetic disruptions that accumulate in the male germline and are passed to twin offspring should increase MZ/DZ twin differences. Indeed, MZ twins, formed from one sperm, should share these epigenetic errors at a higher rate than DZ twins who develop from two different sperm. Thus, similar to studies showing greater epigenetic similarity for MZ vs. DZ twins (Kaminsky et al., 2009), paternal-age related epigenetic errors would be expected to increase genetic influences on disordered eating. We instead observed an increase in DZ twin similarity, and thus shared environmental effects with paternal age, arguing against epigenetic mechanisms. Of course, replication, particularly with molecular genetic designs, is needed.
Several covariates examined (e.g., maternal age, SES) could contribute to paternal age effects by decreasing genetic and increasing shared environmental influences on disordered eating. However, these factors did not account for paternal age-disordered eating associations and are unlikely to be strong mechanistic candidates. Additional unexamined factors that could account for paternal age effects and increased shared environmental influences include birth/obstetric complications and paternal weight concerns/behaviors. First, older paternal age has been associated with several birth/obstetric complications in offspring (e.g., pre-term birth, pre-eclampsia, cesarean section) (Harlap et al., 2002, Tang et al., 2006, Sartorius and Nieschlag, 2010, Shah, 2010), and birth/obstetric complications are linked to risk for eating disorders and other psychiatric illnesses (Cannon et al., 2002, Favaro et al., 2006). These paternal age and birth/obstetric complication associations are present after controlling for maternal age (Harlap et al., 2002; Tang et al., 2006), suggesting that they might account for the unique effects of paternal age on offspring psychiatric risk. However, maternal age has been shown to be a stronger predictor of birth/obstetric complications than paternal age (Harlap et al., 2002; Shah, 2010), and the lack of association between maternal age and disordered eating in our data makes it less likely that birth/obstetric complications could entirely account for our findings.
Second, longitudinal research suggests that weight concerns and dieting increase with age in men, such that these attitudes/behaviors are higher in older adulthood than earlier adulthood (Keel et al., 2007). In older fathers, weight concerns and dieting may be highest when children are being raised and could be transmitted environmentally to offspring in the form of paternal expectations and/or criticism. Importantly, although older women continue to report significant weight/shape concerns and eating disorder behaviors in mid-life (Gagne et al., 2012), longitudinal data suggest that women experience a relative decrease in weight concerns/behaviors with advancing age (Keel et al., 2007). Sex differences in these longitudinal trajectories could potentially explain differential relationships between paternal and maternal ages at birth and offspring eating pathology. Clearly, these possibilities are speculative, particularly given that paternal weight concerns/behaviors may have genetic (and/or gene-environment interaction) effects on offspring disordered eating as well. Future research is necessary to examine whether these types of mechanisms environmentally contribute to paternal age-eating pathology associations.
Despite notable strengths of our study (e.g., large, population-based twin sample, examination of confounding factors), our study was not without limitations. Given that the maximum age of our participants was 17 years, we were unable to capture the entire period of risk for eating disorders which can extend up until at least age 25 (Lewinsohn et al., 2000). However, disordered eating symptoms are present as early as childhood and increase across the pubertal period in females (Maloney et al., 1989, Killen et al., 1992). Therefore, meaningful variance in disordered eating was likely captured in our sample of twins who are in mid-puberty and beyond. Nonetheless, future research should examine young adult samples to ensure results generalize to other periods of risk.
Eating disorder symptoms and diagnoses, as well as covariates, were assessed via parent report. Parent reports allowed us to collect data on a large sample of twins, which was necessary for twin models investigating mechanisms underlying paternal age effects. However, parent reports may miss significant eating disorder symptoms/diagnoses that would be captured by self-reports, and limiting assessment of all covariates to a single parent report could introduce error due to shared method variance. Fortunately, our identified eating disorder cases performed as expected on measures of disordered eating and external correlates, and initial MSUTR data suggest that parent-child concordance for disordered eating is better than that observed for other phenotypes routinely assessed via parent report (see Methods). Future studies should nevertheless confirm that advanced paternal age predicts eating pathology when examining self-reported symptoms/diagnoses using questionnaire and interview data. Using multiple methods to assess covariates (e.g., laboratory height/weight measures) could also help minimize potential influences of shared method variance.
Finally, given the small number of twins with eating disorder histories, phenotypic links between paternal age and eating disorder diagnoses could only be cursorily examined, and paternal age-diagnosis relationships were not investigated using twin models. Although the symptoms we examined are risk factors for eating disorders (Jacobi et al., 2004), additional studies with larger samples are needed to investigate phenotypic and etiologic relationships between paternal age and the broad eating disorder category as well as the specific diagnoses of AN, BN, and BED. Such analyses could increase understanding of the role of paternal age in eating disorder risk and identify specific mechanisms contributing to differential symptom presentations.
Acknowledgments
Data collection was supported by grants from Michigan State University (Drs. Klump and Burt). Data analysis was supported by grants from the Canadian Institutes of Health Research (MDR-96630; Ms. Racine) and the National Institute of Mental Health (5 T32 MH082761 and 1 F31 MH0844701; Dr. Culbert) (1 R01 MH0820-54 and 1 R01 MH092377-01; Drs. Klump and Burt). The content is solely the responsibility of the authors and does not necessarily represent the official views of Michigan State University, the Canadian Institutes of Health Research, or the National Institute of Mental Health. Parts of this manuscript were presented at the Eating Disorders Research Society meeting, Edinburgh, Scotland, September 22-24, 2011.
Footnotes
We tested for multi-collinearity due to the high correlation between paternal and maternal age at birth, but tolerance and the variance inflation factor (VIF) were well within the acceptable range (tolerance = .50, threshold = < .10; VIF = 2.01, threshold = >10).
Results remained unchanged when excluding participants whose biological father completed the questionnaire (6% of the sample; data not shown).
Advanced maternal age at birth was not associated with offspring eating pathology. Maternal age exhibited a modest, negative association with disordered eating (b = −.06, p = .03), but this association became non-significant after controlling for covariates (b = −.03, p =.15). Further, maternal age at birth did not significantly predict history of an eating disorder diagnosis (b = .21; p = .48).
We also tested the five paternal age categories in order to ensure that dichotomizing paternal age did not unduly influence results. Results were identical to those presented herein (data not shown), where genetic effects decreased, and shared environmental effects increased, with older paternal age.
We examined whether the same pattern of results (i.e., decrease in additive genetic effects, increase in shared environmental effects, with paternal age) was observed when twins in the paternal age >40 year group were excluded from analyses given the low MZ correlation in this age group. Results were identical, in that the ACE linear moderation model was best fitting, and both additive genetic and shared environmental moderation coefficients were significant and in the same direction as in the original models.
None of the authors have biomedical financial conflicts of interest or other potential conflicts of interest to disclose.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. American Psychiatric Association; Washington: 2000. [Google Scholar]
- Agrawal A, Balasubramanian S, Smith EK, Madden PA, Bucholz KK, Heath AC, Lynskey MT. Peer substance involvement modifies genetic influences on regular substance involvement in young women. Addiction. 2010;105:1844–1853. doi: 10.1111/j.1360-0443.2010.02993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, Côté M, Henrion E, Spiegelman D, Tarabeux J, Piton A, Yang Y, Boyko A, Bustamante C, Xiong L, Rapoport JL, Addington AM, DeLisi JLE, Krebs M, Joober R, Millet B, Fombonne E, Mottron L, Zilversmit M, Keebler J, Daoud H, Marineau C, Roy-Gagnon M, Dube M, Eyre-Walker A, Drapeau P, Stone EA, Lafreniere RG, Rouleau GA. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. The American Journal of Human Genetics. 2010;87:316–324. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA, Klump KL. The Michigan State University Twin Registry: An update. Twin Research and Human Genetics. 2013;16:344–350. doi: 10.1017/thg.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M, Agerbo E, Ewald H, Eaton WW, Mortensen PB. Parental age and risk of schizophrenia: A case-control study. Archives of General Psychiatry. 2003;60:673–678. doi: 10.1001/archpsyc.60.7.673. [DOI] [PubMed] [Google Scholar]
- Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: Historical and meta-analytic review. American Journal of Psychiatry. 2002;159:1080–1092. doi: 10.1176/appi.ajp.159.7.1080. [DOI] [PubMed] [Google Scholar]
- Croen LA, Najjar DV, Fireman B, Grether JK. Maternal and paternal age and risk of autism spectrum disorders. Archives of Pediatrics and Adolescent Medicine. 2007;161:334–340. doi: 10.1001/archpedi.161.4.334. [DOI] [PubMed] [Google Scholar]
- Croll J, Neumark-Sztainer D, Story M, Ireland M. Prevalence and risk and protective factors related to disordered eating behaviors among adolescents: Relationship to gender and ethnicity. Journal of Adolescent Health. 2002;31:166–175. doi: 10.1016/s1054-139x(02)00368-3. [DOI] [PubMed] [Google Scholar]
- Crow JF. The origins, patterns and implications of human spontaneous mutation. Nature Reviews Genetics. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- Culbert KM, Burt SA, McGue M, Iacono WG, Klump KL. Puberty and the genetic diathesis of disordered eating attitudes and behaviors. Journal of Abnormal Psychology. 2009;118:788–796. doi: 10.1037/a0017207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doornbos ME, Maas SM, McDonnell J, Vermeiden JPW, Hennekam RCM. Infertility, assisted reproduction technologies and imprinting disturbances: A Dutch study. Human Reproduction. 2007;22:2476–2480. doi: 10.1093/humrep/dem172. [DOI] [PubMed] [Google Scholar]
- Eriksen W, Sundet JM, Tambs K. Paternal age at birth and the risk of obesity in young adulthood: A register-based birth cohort study of norwegian males. American Journal of Human Biology. 2012;25:29–34. doi: 10.1002/ajhb.22333. [DOI] [PubMed] [Google Scholar]
- Fairburn CG, Beglin SJ. Assessment of eating disorders: Interview or self-report questionnaire? International Journal of Eating Disorders. 1994;16:363–370. [PubMed] [Google Scholar]
- Favaro A, Tenconi E, Santonastaso P. Perinatal factors and the risk of developing anorexia nervosa and bulimia nervosa. Archives of General Psychiatry. 2006;63:82–88. doi: 10.1001/archpsyc.63.1.82. [DOI] [PubMed] [Google Scholar]
- Frans EM, Sandin S, Reichenberg A, Lichtenstein P, Langstrom N, Hultman CM. Advancing paternal age and bipolar disorder. Archives of General Psychiatry. 2008;65:1034–1040. doi: 10.1001/archpsyc.65.9.1034. [DOI] [PubMed] [Google Scholar]
- Frieling H, Römer KD, Scholz S, Mittelbach F, Wilhelm J, De Zwaan M, Jacoby GE, Kornhuber J, Hillemacher T, Bleich S. Epigenetic dysregulation of dopaminergic genes in eating disorders. International Journal of Eating Disorders. 2010;43:577–583. doi: 10.1002/eat.20745. [DOI] [PubMed] [Google Scholar]
- Gagne DA, Von Holle A, Brownley KA, Runfola CD, Hofmeier S, Branch KE, Bulik CM. Eating disorder symptoms and weight and shape concerns in a large web-based convenience sample of women ages 50 and above: Results of the gender and body image (GABI) study. International Journal of Eating Disorders. 2012;45:832–844. doi: 10.1002/eat.22030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare E, Moran P. Raised parental age in psychiatric patients: Evidence for the constitutional hypothesis. The British Journal of Psychiatry. 1979;134:169–177. doi: 10.1192/bjp.134.2.169. [DOI] [PubMed] [Google Scholar]
- Harlap S, Paltiel O, Deutsch L, Knaanie A, Masalha S, Tiram E, Caplan LS, Malaspina D, Friedlander Y. Paternal age and preeclampsia. Epidemiology. 2002;13:660–667. doi: 10.1097/00001648-200211000-00010. [DOI] [PubMed] [Google Scholar]
- Hultman C, Sandin S, Levine S, Lichtenstein P, Reichenberg A. Advancing paternal age and risk of autism: New evidence from a population-based study and a meta-analysis of epidemiological studies. Molecular Psychiatry. 2011;16:1203–1212. doi: 10.1038/mp.2010.121. [DOI] [PubMed] [Google Scholar]
- Huybrechts I, Himes JH, Ottevaere C, De Vriendt T, De Keyzer W, Cox B, Van Trimpont I, De Bacquer D, De Henauw S. Validity of parent-reported weight and height of preschool children measured at home or estimated without home measurement: A validation study. BMC Pediatrics. 2011;11:1–8. doi: 10.1186/1471-2431-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR, Cuthbert BN, Garvey MA, Heinssen RK, Pine DS, Quinn KJ, Sanislow CA, Wang PS. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. American Journal of Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi C, Hayward C, de Zwaan M, Kraemer HC, Agras WS. Coming to terms with risk factors for eating disorders: Application of risk terminology and suggestions for a general taxonomy. Psychological Bulletin. 2004;130:19–65. doi: 10.1037/0033-2909.130.1.19. [DOI] [PubMed] [Google Scholar]
- Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GHT, Wong AHC, Feldcamp LA, Virtanen C, Halfvarson J, Tysk C, McRae AF, Visscher PM, Montgomery GW, Gottesman II, Martin NG, Petronis A. DNA methylation profiles in monozygotic and dizygotic twins. Nature Genetics. 2009;41:240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- Keel PK, Baxter MG, Heatherton TF, Joiner TE. A 20-year longitudinal study of body weight, dieting, and eating disorder symptoms. Journal of Abnormal Psychology. 2007;116:422–432. doi: 10.1037/0021-843X.116.2.422. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Heath AC, Neale MC, Kessler RC, Eaves LJ. A population-based twin study of alcoholism in women. JAMA: The Journal of the American Medical Association. 1992;268:1877–1882. [PubMed] [Google Scholar]
- Killen J, Hayward C, Litt I, Hammer L, Wilson D, Miner B, Taylor C, Varady A, Shisslak C. Is puberty a risk factor for eating disorders? Archives of Pediatrics and Adolescent Medicine. 1992;146:323–325. doi: 10.1001/archpedi.1992.02160150063023. [DOI] [PubMed] [Google Scholar]
- Klump KL, Bulik CM, Kaye WH, Treasure J, Tyson E. Academy for eating disorders position paper: Eating disorders are serious mental illnesses. International Journal of Eating Disorders. 2009;42:97–103. doi: 10.1002/eat.20589. [DOI] [PubMed] [Google Scholar]
- Klump KL, Burt SA. The Michigan State University Twin Registry (MSUTR): Genetic, environmental and neurobiological influences on behavior across development. Twin Research and Human Genetics. 2006;9:971–977. doi: 10.1375/183242706779462868. [DOI] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Racine SE, Burt SA, Neale M, Sisk C, Boker S, Hu JY. The interactive effects of estrogen and progesterone on changes in emotional eating across the menstrual cycle. Journal of Abnormal Psychology. 2013;122:131–137. doi: 10.1037/a0029524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Sisk C, Burt SA. Preliminary evidence that estradiol moderates genetic influences on disordered eating attitudes and behaviors during puberty. Psychological Medicine. 2010;40:1745–1753. doi: 10.1017/S0033291709992236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Perkins PS, Alexandra Burt S, McGue M, Iacono WG. Puberty moderates genetic influences on disordered eating. Psychological Medicine. 2007;37:627–34. doi: 10.1017/S0033291707000189. [DOI] [PubMed] [Google Scholar]
- Kolko DJ, Kazdin AE. Emotional/behavioral problems in clinic and nonclinic children: Correspondence among child, parent and teacher reports. Journal of Child Psychology and Psychiatry. 1993;34:991–1006. doi: 10.1111/j.1469-7610.1993.tb01103.x. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WSW, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinsohn PM, Striegel-Moore RH, Seeley JR. Epidemiology and natural course of eating disorders in young women from adolescence to young adulthood. Journal of the American Academy of Child and Adolescent Psychiatry. 2000;39:1284–1292. doi: 10.1097/00004583-200010000-00016. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P. The Swedish Twin Registry: A unique resource for clinical, epidemiological and genetic studies. Journal of Internal Medicine. 2002;252:184–205. doi: 10.1046/j.1365-2796.2002.01032.x. [DOI] [PubMed] [Google Scholar]
- Lilenfeld LR, Kaye WH, Greeno CG, Merikangas KR, Plotnicov K, Pollice C, Rao R, Strober M, Bulik CM, Nagy L. A controlled family study of anorexia nervosa and bulimia nervosa: Psychiatric disorders in first-degree relatives and effects of proband comorbidity. Archives of General Psychiatry. 1998;55:603–10. doi: 10.1001/archpsyc.55.7.603. [DOI] [PubMed] [Google Scholar]
- Liu K, Zerubavel N, Bearman P. Social demographic change and autism. Demography. 2010;47:327–343. doi: 10.1353/dem.0.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström S, Haworth C, Carlström E, Gillberg C, Mill J, Råstam M, Hultman CM, Ronald A, Anckarsäter H, Plomin R, Lichtenstein P, Reichenberg A. Trajectories leading to autism spectrum disorders are affected by paternal age: Findings from two nationally representative twin studies. Journal of Child Psychology and Psychiatry. 2010;51:850–856. doi: 10.1111/j.1469-7610.2010.02223.x. [DOI] [PubMed] [Google Scholar]
- Lykken D, Bouchard T, McGue M, Tellegen A. The Minnesota Twin Family Registry. Acta Geneticae Medicae et Gemellologiae. 1990;39:35–70. doi: 10.1017/s0001566000005572. [DOI] [PubMed] [Google Scholar]
- Malaspina D, Corcoran C, Fahim C, Berman A, Harkavy-Friedman J, Yale S, Goetz D, Goetz R, Harlap S, Gorman J. Paternal age and sporadic schizophrenia: Evidence for de novo mutations. American Journal of Medical Genetics. 2002;114:299–303. doi: 10.1002/ajmg.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina D, Harlap S, Fennig S, Heiman D, Nahon D, Feldman D, Susser ES. Advancing paternal age and the risk of schizophrenia. Archives of General Psychiatry. 2001;58:361–367. doi: 10.1001/archpsyc.58.4.361. [DOI] [PubMed] [Google Scholar]
- Maloney MJ, McGuire J, Daniels SR, Specker B. Dieting behavior and eating attitudes in children. Pediatrics. 1989;84:482–489. [PubMed] [Google Scholar]
- Menezes P, Lewis G, Rasmussen F, Zammit S, Sipos A, Harrison G, Tynelius P, Gunnell D. Paternal and maternal ages at conception and risk of bipolar affective disorder in their offspring. Psychological Medicine. 2010;40:477–485. doi: 10.1017/S003329170999064X. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, He JP, Brody D, Fisher PW, Bourdon K, Koretz DS. Prevalence and treatment of mental disorders among US children in the 2001–2004 NHANES. Pediatrics. 2010;125:75–81. doi: 10.1542/peds.2008-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B, Messias E, Miettunen J, Alaräisänen A, Järvelin MR, Koponen H, Räsänen P, Isohanni M, Kirkpatrick B. Meta-analysis of paternal age and schizophrenia risk in male versus female offspring. Schizophrenia Bulletin. 2011;37:1039–1047. doi: 10.1093/schbul/sbq011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale M. MX: Statistical modeling. Author; Richmond, VA: 1997. [Google Scholar]
- Neale MC, Aggen SH, Maes HH, Kubarych TS, Schmitt JE. Methodological issues in the assessment of substance use phenotypes. Addictive Behaviors. 2006;31:1010–1034. doi: 10.1016/j.addbeh.2006.03.047. [DOI] [PubMed] [Google Scholar]
- O’Dea JA, Caputi P. Association between socioeconomic status, weight, age and gender, and the body image and weight control practices of 6-to 19-year-old children and adolescents. Health Education Research. 2001;16:521–532. doi: 10.1093/her/16.5.521. [DOI] [PubMed] [Google Scholar]
- Perrin MC, Brown AS, Malaspina D. Aberrant epigenetic regulation could explain the relationship of paternal age to schizophrenia. Schizophrenia Bulletin. 2007;33:1270–1273. doi: 10.1093/schbul/sbm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen AC, Crockett L, Richards M, Boxer A. A self-report measure of pubertal status: Reliability, validity, and initial norms. Journal of Youth and Adolescence. 1988;17:117–133. doi: 10.1007/BF01537962. [DOI] [PubMed] [Google Scholar]
- Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Research. 2002;5:554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Reichenberg A, Gross R, Weiser M, Bresnahan M, Silverman J, Harlap S, Rabinowitz J, Shulman C, Malaspina D, Lubin G, Knobler HY, Davidson M, Susser E. Advancing paternal age and autism. Archives of General Psychiatry. 2006;63:1026–1032. doi: 10.1001/archpsyc.63.9.1026. [DOI] [PubMed] [Google Scholar]
- Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2011;156:255–274. doi: 10.1002/ajmg.b.31159. [DOI] [PubMed] [Google Scholar]
- Sartorius GA, Nieschlag E. Paternal age and reproduction. Human Reproduction Update. 2010;16:65–79. doi: 10.1093/humupd/dmp027. [DOI] [PubMed] [Google Scholar]
- Schanen NC. Epigenetics of autism spectrum disorders. Human Molecular Genetics. 2006;15:R138–R150. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- Shah PS. Paternal factors and low birthweight, preterm, and small for gestational age births: A systematic review. American Journal of Obstetrics and Gynecology. 2010;202:103–123. doi: 10.1016/j.ajog.2009.08.026. [DOI] [PubMed] [Google Scholar]
- Sobotka T. Shifting parenthood to advanced reproductive ages: trends, causes and consequences. In: Tremmel JC, editor. A Young Generation Under Pressure? Springer; New York: 2010. pp. 129–154. [Google Scholar]
- Steinglass JE, Eisen JL, Attia E, Mayer L, Walsh BT. Is anorexia nervosa a delusional disorder? An assessment of eating beliefs in anorexia nervosa. Journal of Psychiatric Practice. 2007;13:65–71. doi: 10.1097/01.pra.0000265762.79753.88. [DOI] [PubMed] [Google Scholar]
- Swanson SA, Crow SJ, Le Grange D, Swendsen J, Merikangas KR. Prevalence and correlates of eating disorders in adolescents: Results from the national comorbidity survey replication adolescent supplement. Archives of General Psychiatry. 2011;68:714–723. doi: 10.1001/archgenpsychiatry.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CH, Wu MP, Liu JT, Lin HC, Hsu CC. Delayed parenthood and the risk of cesarean delivery—Is paternal age an independent risk factor? Birth. 2006;33:18–26. doi: 10.1111/j.0730-7659.2006.00070.x. [DOI] [PubMed] [Google Scholar]
- Wade TD, Byrne S, Bryant-Waugh R. The Eating Disorder Examination: Norms and construct validity with young and middle adolescent girls. International Journal of Eating Disorders. 2008;41:551–558. doi: 10.1002/eat.20526. [DOI] [PubMed] [Google Scholar]
- Wohl M, Gorwood P. Paternal ages below or above 35 years old are associated with a different risk of schizophrenia in the offspring. European Psychiatry. 2007;22:22–26. doi: 10.1016/j.eurpsy.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S, Gogos JA, Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nature Genetics. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngstrom E, Loeber R, Stouthamer-Loeber M. Patterns and correlates of agreement between parent, teacher, and male adolescent ratings of externalizing and internalizing problems. Journal of Consulting and Clinical Psychology. 2000;68:1038–1050. doi: 10.1037//0022-006x.68.6.1038. [DOI] [PubMed] [Google Scholar]
- Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, Law P, Qiu S, Lord C, Sebat J, Ye K, Wigler M. A unified genetic theory for sporadic and inherited autism. Proceedings of the National Academy of Sciences. 2007;104:12831–12836. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker NL, Losh M, Bulik CM, LaBar KS, Piven J, Pelphrey KA. Anorexia nervosa and autism spectrum disorders: Guided investigation of social cognitive endophenotypes. Psychological Bullentin. 2007;133:976–1006. doi: 10.1037/0033-2909.133.6.976. [DOI] [PubMed] [Google Scholar]