

Abstract
Fluorescence microscopy allows direct visualization of fluorescently tagged proteins within cells. However, the spatial resolution of conventional fluorescence microscopes is limited by diffraction to ~250 nm, prompting the development of super-resolution microscopy which offers resolution approaching the scale of single proteins, i.e., ~20 nm. Here, we describe protocols for single molecule localization-based super-resolution imaging, using focal adhesion proteins as an example and employing either photoswitchable fluorophores or photoactivatable fluorescent proteins. These protocols should also be easily adaptable to imaging a broad array of macromolecular assemblies in cells whose components can be fluorescently tagged and assemble into high density structures.
Keywords: Super-resolution microscopy, Focal adhesions, Localization microscopy, TIRF, PALM, Single molecules, Photoswitchable fluorophores, Photoactivatable fluorescent proteins
1 Introduction
Fluorescence microscopy has proven an indispensable tool in modern cell biology, offering unparalleled abilities to interrogate the presence and spatial distribution of specific biomolecules within cells or tissues, as well as determine how they change during the course of biological functions or in response to perturbations. These days, researchers can visualize most classes of biomolecules by tapping into a wide array of fluorescent labelling strategies, ranging from antibodies conjugated to fluorescent dyes, to proteins fused to green fluorescent protein (GFP) or spectral variants, to oligonucleotides tagged with fluorophores (fluorescent in situ hybridization: FISH), to genetically encoded protein linkers that allow covalent linkage of synthetic fluorophores (SNAP(C) and HALO(C) tags). These methods are capable of exquisite sensitivity, in many cases to single or few copies of molecules. Furthermore, they are generally widely available (commercially in many cases), well-developed, and well-documented. Nevertheless, despite these strengths, conventional fluorescence microscopy is still limited in one important aspect, that is, in its diffraction-limited spatial resolution. Even though a fluorescent source can be reliably detected at the single molecule level by modern detectors such as the EMCCD (Electron Multiplying Charge Coupled Device) cameras, due to the fundamental properties of light, their images appear as diffraction-limited spots, i.e., the so-called point spread function (PSF). For light microscopes operating in the visible spectral range, the PSF dimension is more than an order of magnitude larger than the size of typical protein molecules, at ~250 nm laterally (x, y) and >500 nm axially (z). The PSF dimension determines the microscope’s resolving power; spatial features within the cells finer than ~250 nm are essentially unresolvable.
In the past decade or so, physicists have exploited several optical and photophysical phenomena to circumvent the diffraction limit, giving rise to a range of new Super-Resolution Microscopy (SRM) techniques. These techniques and their applications in cell biology have gained rapidly in recognition in recent years [1–5]. The three most well-known SRM approaches include STED (STimulated Emission Depletion) microscopy [6, 7], SIM (Structured Illumination Microscopy) [8], and single molecule-based Localization Microscopy [9–11]. In this chapter, we focus on Localization Microscopy, which encompasses techniques known by acronyms such as PALM (PhotoActivated Localization Microscopy) [9] and STORM (STochastic Optical Reconstruction Microscopy) [11], as well as several others [10, 12]. These are particularly attractive and practical for many cell biology labs, in no small part due to their comparatively simple hardware requirements similar to that of Total Internal Reflection Fluorescence (TIRF) microscopy. Indeed such accessibility is likely one major factor in the wide adoption of Localization Microscopy compared to other SRM modalities.
The protocols described in this chapter are geared toward setting up and performing Localization Microscopy for visualizing proteins in Focal Adhesions (FAs). These are dense plaques of proteins associated with plasma membrane sites where cells adhere to the extracellular matrix (ECM) and form linkages between the ECM and the actin cytoskeleton. While the molecular complexity and high protein density in FAs have long impeded protein-specific ultrastructural analysis by immuno-EM approaches, FAs are quite suitable for Localization Microscopy [13–16]. This is due to the high molecular specificity of fluorescent labelling as well as the proximity of the FAs to an ECM-coated cover glass surface, which allows high signal-to-noise, low background single molecule detection. Note that although the FA is the example used in this chapter, the protocols described herein should also be easily applied to imaging any cellular macromolecular assembly whose components can be fluorescently tagged and assemble into high density structures.
In the following sections of the introduction, we further discuss the fundamentals and experimental considerations important for designing a Localization Microscopy experiment. This background is particularly pertinent for new practitioners since Localization Microscopy is a recent and highly interdisciplinary technique, and some of the key concepts are borrowed from physical chemistry and thus may be unfamiliar to cell biology-oriented researchers. In Subheading 2, we describe a procedure for setting up a customized microscope system for Localization Microscopy imaging, as well as protocols for preparing ultraclean fiducialed substrates for culturing cells for Localization Microscopy. Finally, in the Subheading 3 we present a protocol for the acquisition and processing of Localization Microscopy data. Where necessary, expanded discussions are included as Notes at the end of the chapter.
1.1 Basic Principles and Considerations for Localization Microscopy
The diffraction limit of the conventional light microscope can be illustrated by the well-known Rayleigh criterion (Fig. 1) which posits that two fluorescent molecules situated closer than the PSF dimension cannot be resolved (Fig. 1b). However, this holds true only if both fluorescent molecules are observed simultaneously. While this applies for most situations in conventional fluorescence microscopy, single molecule studies in the 1990s revealed that many fluorophores ranging from GFP to synthetic dyes exhibit more complicated photophysics such as blinking or photoswitching [17, 18]. Although this phenomenon was initially considered a nuisance in single molecule enzymology experiments, it was soon recognized that this effect can be exploited for super-resolution imaging [19]—closely spaced molecules that would otherwise overlap can be detected separately in time, due to their stochastic blinking/switching. Three key requirements necessary for Localization Microscopy are: (1) single molecule contrast: the brightness of the fluorophore (characterized as photon numbers) relative to background noise is critical for accurate determination of the position of each fluorophore; (2) sparsity: to detect molecules individually, they must be sufficiently isolated spatially from one another; (3) high total spatial density: high density of labelling is needed to sample the spatial profile of the underlying structure (known as Nyquist–Shannon sampling criterion [20]).
Fig. 1.
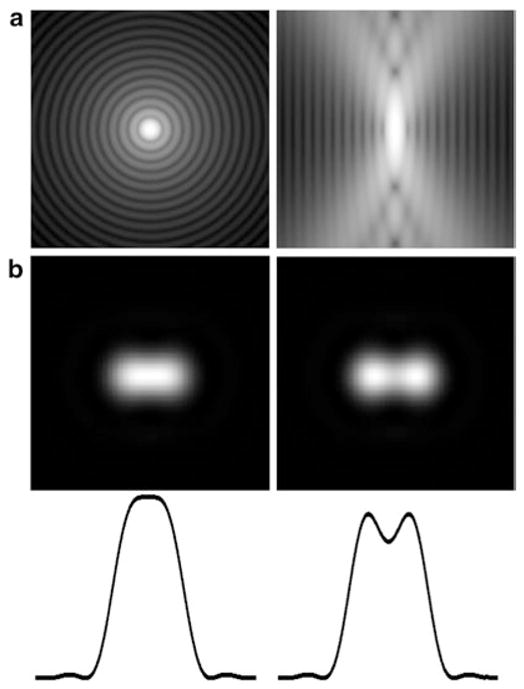
Microscope point spread function and Rayleigh resolution criterion. The dimension of the microscope point spread function (PSF) (a, left: lateral or x, y; vertical x, z view) limits the resolution of the conventional light microscope. Two fluorophores in close proximity below the size scale of the central PSF intensity peak will appear to merge (b left) and thus cannot be resolved as distinct fluorophores. The limit at which the molecules appear separate as judged by the intensity cross-section (bottom) is called the Rayleigh criterion. (a) is in log scale to show the structure of the PSF, whereas in (b), linear intensity scale is used
To meet these requirements, photoswitchable (or photoactivatable) fluorophores are needed for Localization Microscopy. A well-known example in cell biology of such photoswitchable fluorophores is PA-GFP (photoactivatable-GFP), which was originally developed as an optical highlighter for tracking subcellular organelles [21], but has since been used successfully for Localization Microscopy [9, 22, 23]. PA-GFP is a photoswitchable variant of GFP, whereby, prior to photoactivation, it exists in the non-activated state, with negligible emission in the GFP channel (520 nm). Upon illumination by photoactivating blue light at 405 nm, PA-GFP turns into a photoactivated form which now absorbs and fluoresces in the GFP channel. In order to fulfil the sparsity requirement above, researchers can control the amount of photoactivating blue light (405 nm), i.e., by controlling the duration or the intensity, so that only a sparse subset of PA-GFP is photoactivated at a given instance. With proper excitation intensity and a sensitive EMCCD camera, a random subset of isolated PA-GFP molecules can be readily observed as spatially segregated diffraction-limited spots. These activated PA-GFPs typically appear for a brief period of a few seconds while being imaged before disappearing. Such so-called “photobleaching” serves to clear out the molecules that have been imaged, maintaining sparsity, while newly activated molecules become visible. Note that the appearance and disappearance of the fluorophores is a stochastic process, such that the molecules appear to blink randomly and there is a spread in how long they remain fluorescent during their activated state, and likewise in their brightness. By collecting a series of images of such blinking over a sufficiently long time, the cellular structures labelled by PA-GFP are spatially and cumulatively sampled, fulfilling the high spatial density requirement.
1.2 Localization Analysis of Single Molecule Fluorescence
The imaging protocols for Localization Microscopy consist of the acquisition of raw data and subsequent image processing and reconstruction steps. Upon the acquisition of high quality single molecule datasets, peak detection and localization analysis algorithms are employed to extract the molecular coordinate of each observed fluorophore. Although an individual fluorescent molecule is detected as a diffraction-limited spot spread over several pixels on the detector (Fig. 2a), the centroid position of such a spot can be “localized” by fitting to a model of point-spread function, and assumed to be the position of the emitting molecules. A 2-dimensional Gaussian function is commonly used as it provides a close approximation while being fast to compute [24]. The Gaussian approximation usually suffices for most applications of Localization Microscopy, providing that the fluorophores are freely rotating during the imaging time scale. However, it should be noted that there are special cases, for example where the fluorophores are directionally constrained and the emission dipole is not well averaged, such as when the dye is embedded in the membrane. In these cases, a more advanced (and computationally demanding) model is required [25]. The precision at which the lateral molecular coordinate (i.e., the xy-centroid) can be localized is dependent upon the brightness of the molecule relative to the background signals. Localization uncertainty (σxy) is commonly computed [24] based on the following equation (further derivation and refinement in ref. 34): σxy= ((s2+ a2/12)/N +(8πs4b2/(a2N2)))1/2 (N: detected photons; s: width of the PSF; a: pixel size; b: background noise). Thus, the brighter the fluorophore, the more precise the localization. Likewise, lower background is also desirable for high localization precision. On average, PA-FP (photoactivatable-fluorescent proteins) typically emit 500 or more detected photons before undergoing photobleaching, allowing localization precision of ~20 nm. Some organic fluorophores such as Cy5 or AlexaFluor 647 are brighter, thus giving rise to higher localization precision [26]. Note that since the photoswitching of fluorophores is a stochastic process influenced by both the intrinsic fluorophore properties and other environmental factors (such as redox potential [27] and oxygen level [28]), the brightness and hence localization precision could vary between preparations. Also, since the molecules in the sample exhibit varying brightness, this results in a distribution of their localization uncertainties (σxy). Thus, rather than a uniform single resolution number for the entire image, the spatial resolution of Localization Microscopy varies depending on local density, fluorophore brightness, and background noise.
Fig. 2.

Schematic diagram of single molecule localization analysis and localization microscopy. Isolated single fluorophores such as EosFP can be detected as diffraction limited spots by an EMCCD camera (a, top: 3-D plot of pixel intensity, bottom: 2-D view). These can be fitted by a model of the point-spread function (PSF) such as a 2-dimensional Gaussian (b, top). Localization analysis computationally minimizes the residual (b, bottom), by determining the centroid position as well as the width of the observed PSF. The resulting centroid coordinate represents the position of the molecule with much better precision than the PSF. The position of the molecule can be rendered as a 2-D Gaussian with the width corresponding to the localization uncertainty (c). Flowchart for Localization Microscopy is shown in (d), whereas in (e) the super-resolved image (right) of Paxillin-tdEos is shown next to diffraction-limited TIRF image (left), demonstrating the significant gain in resolution due to Localization Microscopy. Bottom panels show zoomed-in views. Bright spots in the top left panel are due to the gold nanoparticle fiducials
Due to the large volume of raw data sets (gigabytes), and the numbers of iterations needed for localizing each molecule, it is highly recommended that a sufficiently powerful computer is used for image processing. Thanks to the increasingly wide adoption of Localization Microscopy, several software packages for performing single molecule detection and localization analyses have been published and are freely distributed [29–31].
1.3 Labelling Proteins for Localization Microscopy
The requirement for high spatial sampling density and localization also has implications for the nature of the fluorescent probes used for labelling the molecules of interest. While in conventional fluorescence microscopy, the size of the probe molecule itself is rarely an issue since the spatial resolution is an order of magnitude coarser than the molecular length scale, in Localization Microscopy, the resolution of the imaging method approaches the molecular length scale, and thus judicious selection of the labelling strategy is important.
For imaging of proteins, two common labelling approaches are protein fusion with PA-FP and antibodies conjugated with synthetic fluorophores. These two approaches offer different advantages and disadvantages. PA-FP fusion is genetically encoded and is inherently compatible with live cells. Furthermore, many PA-FP are also useful for other cell biology experiments. Similar to GFP, the PA-FP tag is a highly compacted globular domain of <300 amino acids, with a diameter of <5 nm across. Thus, in principle a large number of fusion proteins can be packed together into a small volume, providing higher sampling density. However, in practice typical cells also express unlabelled endogenous protein which effectively dilutes the labelling density of the expressed fusion protein. Thus, where necessary, this should be addressed by using genetic knock-out or RNAi-mediated knockdown in cells to gain a more complete replacement of the endogenous protein by the fusion construct. Of note, recent gene editing technologies such as TALEN (Transcription-Activator-Like Effector Nuclease) [32] may also offer a promising solution for a complete replacement of endogenous protein with labelled proteins. Finally, the main disadvantage of PA-FP is that their photophysical properties are not as good as the best of the synthetic fluorophores.
The major strength of the antibody-based synthetic fluorophore approach is the high brightness of such fluorophores. Additionally, where available, antibodies allow detection of diverse post-translationally modified forms of proteins, such as phosphorylation. However, in practice, where the antibodies are not commercially available for the target proteins, a greater amount of resources and effort is required to produce such antibodies. Furthermore, the antibodies are multi-domain proteins much larger than FPs (>10 nm for an IgG). This size is compounded by the fact that common immunofluorescence (IF) protocols use unlabelled primary antibodies to detect the proteins and fluorescent-tagged secondary antibodies as probes for the primary antibodies. The large combined size of the probes (on the order of ~20 nm) therefore limits how densely these probes can be packed into a given volume, as well as how well the probes can access tightly packed structures. Also, the labelling of cells with antibodies generally requires fixation and permeabilization and thus is not compatible with live cells.
1.4 Visual Representation of Localization Microscopy Datasets
Unlike most other imaging modalities, the so-called “finished product” for Localization Microscopy is not the super resolved image per se. Rather, the result is essentially a long list of single molecule attributes, containing entries such as the spatial coordinates: x, y, and/or z; uncertainty of the spatial coordinates: σx, σy, σz; amplitudes; brightness; fit quality and so on. A standard convention for Localization Microscopy is to represent each molecule with a normalized 2-dimension Gaussian function, with the width corresponding to the coordinate uncertainty (σ). In other words, the intensity shown in the super-resolution reconstructed image is proportional to the probability of finding a molecule at a particular coordinate. Thus, a bright and well localized molecule will be portrayed as a sharp and narrow point with a relatively high peak intensity, whereas a dimmer and less well localized molecule will be shown as being less distinct with a smaller peak intensity.
2 Materials
Localization Microscopy requires a research-grade TIRF microscope capable of detecting single molecule fluorescence. Instructions below describe a custom-built open-beam microscope system equipped with moderately high-powered lasers (~100 mW) and coupled with an EMCCD camera and appropriate filter sets (Fig. 3). Custom-built systems offer more flexibility and savings on equipment cost, although familiarity with optical instrumentation will be required. As a guideline, examples of manufacturers and parts numbers are given below for each component. We found that commercial TIRF microscopes are also usually adequate (available from most major manufacturers: Nikon, Olympus, Zeiss, Leica). These typically offer fiber-coupled lasers, which simplify much of the alignment work, albeit with reduced power output and higher cost. In addition to the microscope, appropriate sample preparation is essential for successful imaging. Protocols later on in this section describe the preparation of fiducialed cover glasses, pre-treatment of these cover glasses for cell culture, as well as the preparation of fixed cell samples for Localization Microscopy.
Fig. 3.

Schematic diagram of a microscope configured for Localization Microscopy. Laser beams (wavelength of 488, 561, 640 nm) are directed by mirror (M) and combined by laser merging dichroic mirror (LM) and coupled into an AOTF (Acousto-Optical Tunable Filter). The 405 nm laser is combined separately due to AOTF bandwidth limitation. The combined laser beam is expanded by a beam expander (BE) and focused onto the back focal plane of the high NA objective via a lens (f ~ 300 mm), mounted on a translatable platform. The illumination laser is reflected toward the sample by a dichroic mirror (DM). The fluorescence emission passes through the dichroic mirror and is filtered by the emission filter (EF), before being focused by the tube lens and/ or magnification changer lens (L2). For 3-D Localization Microscopy, a weak cylindrical lens (L3) could be mounted on a translatable platform in front of the EMCCD camera. The microscope body is denoted by the dashed line. The sample is mounted on a microscope stage (optional: Z-piezo for 3-D Localization Microscopy). For examples of manufacturers and part numbers, see Subheading 2
2.1 Instrumentation for Localization Microscopy
-
Suggested components:
Research grade inverted microscope (e.g., Nikon Eclipse Ti).
100× High numerical aperture (NA) objective lens (Nikon, 100× Apo TIRF NA 1.49).
Piezoelectric Z-translation stage (optional) (Mad City Labs, #Nano-Bio100).
Vibration-damped optical table (at least 3 ft × 6 ft, e.g., Newport Corp. #RS-2000-36-8).
Lasers with the following wavelengths and power: 405 nm (100 mW, Coherent, #1142279), 488 nm (200 mW, Coherent, #1137964), 561 nm (200 mW, Coherent, #1137977), 640 nm (100 mW, Coherent, #1185055).
Acousto-Optic Tunable Filter (A-A Optoelectronic, #AOTFnCVis).
Heatsink mounts for lasers and AOTF, custom-machined from Aluminum.
Dichroic mirror (for filter cube, DM in Fig. 3: Semrock, #Di01-R405/488/561/635-25x36; for combining laser beams: #LM01-613-25, #LM01-503-25, and #LM0-427-25; LM1, LM2, LM3 in Fig. 3, respectively).
Emission filter (Semrock, green, #FF01-525/50-25; red, #FF01-609/57-25; far-red, #FF01-676/37-25; EF in Fig. 3).
Steerable mirror mounts (ThorLabs, #KS1).
Neutral density filter wheel (ThorLabs, #NDC-25-C).
First surface mirrors (New Focus, #5101; M in Fig. 3).
10× beam expander (ThorLabs, # BE10M; BE in Fig. 3).
Achromatic doublet lens (f = 300 mm, CVI MellesGriot, #LAO-300.0-25.0; L1 in Fig. 3).
Cylindrical lens (optional, f = 1,000 mm, CVI MellesGriot, #SCC-25.4-508.6-C; L3 in Fig. 3).
Translational stage (ThorLabs, #PT1).
EMCCD camera (Andor, #DU-897U-CS0#BV).
Desktop Workstation PC.
Secure an inverted microscope on a research-grade optical table with good vibration damping. Ensure that the room housing the microscope has good temperature stability. (Note the position of the air vent, and if necessary install a deflector or put the microscope in an enclosure to minimize mechanical instability.)
On the optical table, set up continuous wave (CW) solid state lasers with the following wavelengths and power (see above for specific model suggestions): 405 nm (50–100 mW, for photoactivation), 488 nm (~100 mW or greater, for imaging of PA-GFP [21] or Dronpa [33]), 561 nm (~100 mW or greater, for imaging EosFP [34], Dendra2 [35], or PAmCherry [22]), and 640 nm (~100 mW or greater, for imaging of Cy5 or AlexaFluor 647 [11]). To simplify alignment, obtain custom-machined heatsink (these can be machined from standard aluminum block by any local machine shop) with the appropriate height so that all the lasers beams are at the same heights above the optical table (see Notes 1 and 2).
Combine laser beams using appropriate dichroic mirrors in suitable steerable mounts. The longest wavelength line in the system, such as 640 nm, should be used as the primary beam against which all other lasers are aligned. It is generally useful to install a filter wheel with neutral density (ND) filters in front of the laser heads, for an added degree of intensity control. Align the mirrors and dichroic so that the laser beams are colinear. Depending on laser regulations at different institutions, it may be advisable to build an opaque enclosure to contain stray light.
Position an Acousto-Optic Tunable Filter (AOTF) on the optical table. Make sure that the AOTF optical opening is well matched to the laser beam height. If needed, obtain a custom-machined aluminum pedestal of appropriate height. Position a mirror to direct the filtered beam into a 5–10× beam expander (see Note 3).
Install a 100× high NA oil-immersion objective lens (NA > 1.45) on the microscope objective turret (see Note 4).
To illuminate the sample in epifluorescence geometry, focus the expanded laser beam onto the back focal plane of the microscope objective. This can be achieved by mounting a 300 mm achromatic doublet lens on a 2-axis translational stage near the illumination port of the microscope (see Note 5).
Select the appropriate dichroic mirror and emission filters and install them in the appropriate filter cubes of the microscope (see Note 6).
Install an EMCCD camera (see Note 7) at the microscope emission port. Securely fasten the camera to the optical table to minimize vibration.
(Optional) For 3-D Localization Microscopy using astigmatism [36], install a piezoelectric Z-translation stage (e.g., Nano-Bio100 from Mad City Labs, Madison, WI). Place a defocusing lens (a weak cylindrical lens with long focal length of 1,000 mm or more) before the camera. Mount the lens on a translation stage with good repeatability. Make sure to build a light-tight enclosure for this assembly to protect the EMCCD from stray light. The distance between the cylindrical and the EMCCD chip can be calculated from the formula for calculating back focal length of a compound lens system: S =(f2(d − f1))/ (d −f1− f2), where S is the back focal length, f1 and f2 the focal length of the first (in this case, the microscope tube lens) and the second lens (cylindrical lens), respectively, and d the distance between the two lenses. As an example, for a relative defocused difference of 400 nm, the cylindrical lens (f = 1,000 mm), should be placed at d ~179.8 mm behind the tube lens (f = 200 mm for Nikon microscope).
Set up a desktop computer for controlling the microscope. Configure the microscope and peripheral control software. Commercial software such as Metamorph (Molecular Devices, Sunnyvale, CA) can be used. Alternatively, if cost is an important consideration, MicroManager, a freely available microscope control software should be considered [37] (see Note 8).
Configure the image processing computer. A desktop computer with multiple-core CPU and a 64-bit operating system is recommended in order to take advantage of large memory—16 GB or more may be required for large datasets. Alternatively, the data can be processed in smaller chunks on a more modest machine and pieced together afterward. Typically, several hours of processing on a workstation level machine are needed for a single data set. Alternatively, many academic institutions have a centralized high performance computing cluster which can speed up the processing steps significantly. While initial setup work may be needed to configure the software for distributed computing, this may be a more efficient solution in the long run.
2.2 Preparation of Cover glasses for Cell Culture
Stringent cleaning of the cover glasses is mandatory for Localization Microscopy since any background fluorescent signal will interfere with the fluorescence from labelled molecules. Of equal importance, since acquisition time is on the order of several minutes or more, mechanical drift is inevitable and must be corrected for. We recommend using fluorescent beads or fluorescent nanoparticles affixed to the coverslip as fiducial marks. Since the dynamic range of the EMCCD camera is limited in the EM-gain mode, an important consideration is to ensure that the fiducials are not so excessively bright as to become saturated. Our choices for the fiducials are gold or bimetallic gold–silver nanoparticles whose plasmonic emissions are within similar spectral ranges to PA-FPs. These also exhibit appropriate brightness and are resistant to photobleaching. In the simplest preparation, these nanoparticles can be added into the sample prior to imaging and allowed to adsorb to the cover glass via non-specific adhesion. However, fiducials affixed this way typically still exhibit thermal fluctuations and can be dislodged over time. In the protocol below, we describe a method for immobilizing pre-attached fiducials with a thin layer of SiO2 which allows for much more stable fiducials and reliable drift correction. Note that fiducialed cover glasses have recently become commercially available (www.hestzig.com) which provide an alternative for cases where access to a sputter coating instrument is difficult.
-
Suggested equipment and materials:
Plasma cleaner (e.g., Harricks, #PDC/32-G).
Laminar flow hood with UV lamp.
Hotplate/Stirrer.
Shaker/Rotator.
Ultrasonic Bath.
Sputter coater (Denton Vacuum Technologies).
Cover glass (#1.5, 22 mm square, Fisher Scientific, #12-541B).
Porcelain staining rack (Thomas Scientific, #8542E40).
Adhesive spacer (BioscienceTools, #UTIC-21).
Dry N2 gas.
Ethanol.
Acetone.
Dulbecco’s Phosphate Buffered Saline (DPBS): 2.7 mM KCl, 1.5 mM KH2PO4, 136.9 mM NaCl, 8.9 mM Na2HPO4·7H2O.
Poly-L-lysine solution: 0.1 % w/v solution of poly-L-lysine (m.w. 150,000–300,000) in double deionized water (ddH2O). Alternatively, ready-made solution is also commercially available (e.g., Sigma, #P8920).
100 nm gold nanoparticle solution: Resuspend gold nanoparticles solution (Corpuscular Inc., #790122), then dilute 1:1,000 in DPBS.
Fibronectin solution: Dilute 1 mg/mL fibronectin stock solution (Millipore, #FC010) in sterile DPBS to 2–10 μg/mL. Perform the dilution in sterile laminar flow hood.
1 M HNO3: Add 63.3 mL of concentrated nitric acid (15.8 M) into approximately 500 mL of ddH2O. Adjust volume to 1 L. Make sure to perform dilution in a chemical fume hood, with appropriate protection equipment.
Select #1.5 cover glasses individually and arrange them on an acid-resistant porcelain rack. Sonicate for 15–30 min in the following solutions in the following order: ddH2O, acetone, 100 % ethanol. Rinse in ddH2O and immerse in 1 M HNO3 (in chemical fume hood). Heat the nitric acid to a boil and let cool overnight. Rinse the cover glasses in ddH2O multiple times and then dry by compressed air or N2. Clean the cover glasses for 5 min in a plasma cleaner or a UV–ozone cleaner. If either of these pieces of equipment are not available, RCA etch can be used (see Note 9).
Incubate cleaned cover glass with the poly-L-lysine solution for 10–20 min to aid in the adsorption of fiducials. Rinse thoroughly with ddH2O and let dry. Dilute nanoparticle fiducials to approximately 1:1,000 (the concentration should be empirically adjusted to obtain appropriate density of several fiducials per a field of view of ~5,000 μm2). Incubate individual poly-L-lysine-coated cover glass with 2 mL of the fiducial solution for ~30 min. Rinse away excess fiducials by ddH2O and dry by compressed air or nitrogen gas. Using a sputter coater, deposit 30–50 nm of SiO2 onto the fiducial-affixed side of the cover glass. Plasma clean the cover glass upon finishing.
Place each cleaned cover glass into a 6-well tissue culture plate. Sterilize by UV radiation in a laminar flow hood for 15 min.
Rinse three times with DPBS. Incubate each well with 2 mL of fibronectin solution at desired concentration (typically 2–10 μg/mL). Let the cover glasses incubate overnight at 4 °C, or with gentle rocking at 37 °C for a few hours. Aspirate out the fibronectin solution. Replace with DPBS. The cover glasses can be stored at 4 °C for up to 7–10 days until needed for cell plating.
2.3 Cell Culture and Preparation of Fixed Cells for Imaging
High quality single molecule raw image data is the most important ingredient for successful Localization Microscopy. This is strongly influenced by the properties of the fluorophores. In particular, EosFP (tdEos [34] and mEos2 [38]) and AlexaFluor647 are among the best performing PA-FPs and synthetic fluorophores, respectively. Thus, we recommend that novice users start imaging with these fluorophores. The preparation of cell cultures for imaging, for the most part, is similar to that of standard imaging protocols. Since Localization Microscopy depends on single molecule imaging, it is important to make sure that the expression level is appropriate—due to background auto-activation of PA-FPs in highly expressing cells, the single molecule signals can be too dense even before any photoactivation light is applied. It is highly recommended to compare expression and localization of multiple FP fusions of the same protein, to ascertain that the constructs are minimally perturbative (see Note 10). Potential artifacts include protein aggregation, incorrect localization, and altered cellular morphology. Where possible, test the biological function of the fusion construct and compare the localization with known immunofluorescence staining.
-
Suggested equipment and materials:
Tissue culture incubator (37 °C, 5 % CO2).
Sterile laminar flow biosafety hood.
Transfection reagents and apparatus (e.g., Life Technologies, Neon).
U2OS cell line (ATCC).
McCoy5A media (Life Technologies).
Fetal Bovine Serum (Life Technologies).
Penicillin/Streptomycin (Life Technologies).
Trypsin/EDTA (Life Technologies).
Endofree Plasmid Maxi kit (Qiagen).
DPBS.
Phalloidin-AlexaFluor647 (Life Technologies).
KOH solution (10 M): 74.55 g of KOH added to 100 mL ddH2O (care should be taken as KOH is highly corrosive).
PHEM buffer (2× stock solution): 120 mM PIPES, 50 mM HEPES, 20 mM EGTA, 4 mM MgCl2, pH 7.0 with KOH. Dissolve 6.5 g of HEPES, 3.8 g of EGTA, and 190 mg of MgCl2 in ~300 mL of ddH2O. With vigorous stirring, monitor the pH (should be acidic), and start adding concentrated KOH solution dropwise to bring the pH to approximately 6. Start adding 18.14 g of PIPES in small batches. PIPES will start to dissolve at pH > 6. Adjust the pH to 7.0 with KOH solution, and bring the volume to 500 mL by adding ddH2O. Filter-sterilize and store at 4 °C. Dilute PHEM 2× stock 1:1 with ddH2O before use.
Permeabilization buffer (0.1 % Triton): Dilute 1 mL of Triton X-100 in 1 L of PHEM 1× buffer. Fresh preparation of Triton solution on the day of the experiment is recommended for effective permeabilization and antibody penetration into the cells.
Quenching buffer (50 mM NH4Cl): 2.68 g of NH4Cl in 1 L of PHEM 1× buffer.
Oxygen-depleted Imaging buffers: (a) 1 M of glucose solution: Add 18 g of glucose to 100 mL of PHEM 1× buffer. (b) 1 M (10×) cysteamine solution: Add 770 mg cysteamine to 10 mL of PHEM 1× buffer. (c) 100× stock enzyme mixture: Add 4 mg of catalase and 10 mg of glucose oxidase to 100 μL of PHEM 1× buffer. Mix well by vortexing. Centrifuge for 2–3 min and aliquot supernatant for storage at 4 °C. (d) Right before imaging, mix 75 μL of 1 M glucose solution, 30 μL of 1 M cysteamine solution, 3 μL of 100× stock enzyme mixture, and adjust the volume to 300 μL with PHEM 1× buffer and use immediately after mixing to mount the sample.
VALAP: Melt 100 g each of vaseline, lanolin, and paraffin in a beaker on a hotplate. Use a low heat level for melting (below 100 °C) to prevent ignition of the mixture. Stir until well mixed.
Culture the desired cell type in the appropriate medium and environmental conditions. For example, we culture the U2OS (human osteosarcoma) cell line in McCoy5A media supplemented with 10 % fetal bovine serum and penicillin/streptomycin under 5 % CO2 at 37 °C.
For fluorescent protein labelling, a fusion construct of the protein of interest can be constructed from cDNA of the target protein and a photoactivatable fluorescent protein vector (several cloning vectors can be obtained from the AddGene repository (www.addgene.org)). We highly recommend that standard FP fusions such as GFP or mCherry be made in parallel to the PA-FP constructs (see Note 10).
Verify the sequence of the fusion construct and prepare high-purity endotoxin-free vectors for transfection.
Transfect cells with the fusion protein cDNA. We obtain good results using electroporators such as Amaxa nucleofector (Lonza, Basel, Switzerland) or Neon (Life Technologies, Carlsbad, CA). Alternatively, lipid-mediated approaches (Lipofectamine from Life Technologies or Fugene from Roche) can also be used.
Incubate transfected cells for 6 or more hours or until the desired expression level is attained (determined empirically). Rinse the cells with warm DPBS. Dislodge the cells by incubating with 1–2 mL warm trypsin for 1–2 min. Stop the trypsinization by adding complete (serum containing) media. Count the cells and replate the desired number of cells onto the prepared cover glass (Subheading 2.2). Incubate replated cells in a 37 °C, 5 % CO2 incubator. For typical FA imaging, cells can be fixed 12–24 h after replating.
Fixative: Prepare the fixatives fresh before use. We use pre-packaged methanol-free paraformaldehyde in ampoule format (16 % solution) diluted to 4 % in a cytoskeleton-stabilizing buffer (such as PHEM buffer, warmed to 37 °C).
Start the fixation by aspirating out the media and replace with warm fixative. Incubate in the dark for 12–15 min at 37 °C. Aspirate out the fixative. Rinse with PHEM buffer. If permeabilization is needed, incubate with permeabilization buffer for 15 min after fixation. Quench for 10 min with the quenching buffer.
(Optional) To visualize actin filaments, incubate the samples with phalloidin labelled with AlexaFluor647 (20–30 nM in PHEM buffer) for 30 min to 1 h.
Wash by incubating with PHEM buffer for 5 min and then exchange with fresh buffer. Repeat three times.
Following fixation, fiducial beads or gold nanoparticles can be added if fiducialed cover glasses are not being used. We obtain good results with a starting concentration of 1:1,000 dilution and 20–30 min incubation.
Wash with PHEM buffer to remove excess unbound fiducials.
Throughout the fixation steps, samples should be assessed by light microscopy for the integrity of cell morphology, and/or fluorescence of the target proteins. Make sure to dispense the solutions gently and to the edge of the well and never on top of the cells directly. Perform the change of solutions quickly and never let the cells dry out.
The sample should be imaged preferably as soon as the fixation is concluded. Alternatively, samples can be stored for a day or so at 4 °C. Be aware that the fluorescence of some PA-FPs degrades over time.
For PA-FPs, imaging can be done in PBS or PHEM buffer without additional components. However, in our experience a careful preparation of imaging buffer appears to improve the quality of the image. We typically use PHEM buffer, filtered by 0.22 μm filter. For vacuum-driven filtration, the filtering process will also help to partially degas the buffer. This can be done by tapping the evacuated tube repeatedly to dislodge the bubbles. Additives such catalase (0.04 μg/mL) helps to remove peroxide and radicals that could be generated by the reaction of dissolved oxygen and excited fluorophores.
For imaging synthetic fluorophores such as AlexaFluor647, a reducing and oxygen-depleted imaging buffer is recommended. Prepare fresh imaging buffer just before mounting as above, and ensure that the sample is sealed completely as described below.
Mount the cover glass on a glass slide (3″ × 1″) or another piece of large cover glass. We use double-sided adhesive spacer (Bioscience Tools) to aid in the assembly. The sample is rinsed once with the imaging buffer and then assembled into an enclosed cell. Make sure to perform this step gently to avoid any air bubbles. Afterward, gently blot away excess buffers. Seal the sample by melted VALAP. Clean the surface of the sealed imaging chamber by rinsing with deionized H2O and blow dry with compressed air. The sample can then be mounted onto the microscope for imaging.
3 Methods
Since multiple parameters must be optimized for proper imaging, researchers are encouraged to become familiarized with the acquisition and analysis workflow using easy-to-prepare test samples (such as bare fiducialed cover glass) before embarking on imaging their research samples. Procedures detailed below include the initial optimization needed to ensure proper and stable microscope operation, followed by guidelines for the acquisition of raw datasets from cellular samples and, finally, the image processing required to obtain super-resolved images.
3.1 Initial Characterization of Microscope Stability
Since mechanical stability is important for Localization Microscopy, prior to imaging fixed cell samples, users should take great care in characterizing and optimizing the stability of the microscopy system. This can be carried out by imaging a simple test sample as described below.
A fiducialed cover glass prepared earlier can be used directly as a test sample. Alternatively, the test samples can be prepared by incubating fluorescent beads on poly-lysine coated cover glasses for 30 min, followed by sample assembly as described earlier.
Mount the sample onto the microscope stage, and bring into focus. Make sure the proper filters are in place, and then switch the emission path to the camera, and the excitation path for the lasers.
Configure the camera with moderate EM-gain settings according to the manufacturer’s recommendation. Set the readout mode to the highest bit depth available. Set the exposure time to 50 ms for the frame rate of 20 frames per second in frame transfer mode.
Adjust the illumination lens until total internal reflection (TIR) condition is achieved. Adjust the laser intensity so that the fluorescent bead is not saturating the camera (relatively low power is needed for very bright fiducials). Locate an area with sufficient number of fiducials for imaging.
Collect a stream of images of between 1,000 and 10,000 frames, or longer if necessary. In cases where the microscope is not equipped with focus correction, it is recommended to monitor the focal drift and apply gentle and gradual manual adjustment as needed.
Perform localization analysis on the test data sets (see below). Evaluate and apply drift correction. The fiducials should be brought into registration with an uncertainty of <20 nm or so, such that the uncertainty in the drift correction is smaller than the uncertainty of fluorophore localization.
If the magnitude of the drift is larger than a few microns over the acquisition time scale, monitor temperature fluctuation as well as other mechanical disturbances, particularly the air circulation (many electronic equipment and computers contain fans which may inadvertently blow directly on to the microscope), and take remedial action as needed. Also, mechanical drifts tend to be greatest soon after sample mounting. Thus, a period of waiting may lessen the magnitude of the drift.
3.2 Imaging Cells Expressing PA-FP by Localization Microscopy
Upon ensuring the microscope is capable of stable operation, fixed cell samples can be imaged. Typically, higher excitation intensity is needed to visualize single molecule signals. However, too high intensity excitation could lead to saturation in the signal of the fluorescent fiducials, especially due to the limited dynamic range of the EMCCD camera. Thus, laser intensity must be carefully optimized to capture signals from both the labelled molecules and the fiducials without saturation.
Mount the sample, focus, and locate candidate cells for imaging by transmitted light or epifluorescence mode. In the case of photoconvertible proteins such as tdEos or mEos2, the cells can be located in the green fluorescence channel. Record images in the standard epifluorescence and/or transmitted light modes for reference.
Many PA-FPs are highly sensitive to photoactivation. Thus, it is recommended to prevent illumination by 405 nm prior to imaging.
Switch to TIRF mode and make sure that the desired imaging area contains multiple fiducials for the assessment and correction of drift.
For EosFP imaging, excite with the 561 nm laser and observe in the red emission channel. Adjust the laser intensity so that bright single molecules are observable. There should be sparse blinking of molecules that last for a few image frames.
Make sure that the 405 nm photoactivation line is strongly attenuated. Illuminate the sample with 405 nm, starting at the lowest intensity. A rapid increase of single molecule fluorescence in the red emission channel should be observed.
Adjust 561 and 405 nm intensity so that on balance, there are sparse but sufficiently large numbers of molecules appearing in each frame. Make sure that the fiducials are not saturated.
The intensity control of the 405 nm laser should be set up so that it can be controlled independently of the acquisition process. This is recommended since molecules are continually depleted during long acquisitions, and the 405 nm intensity needs to be increased progressively. Note that some models of 405 nm lasers (such as Coherent Cube) are supplied with a USB-based control software that can be used to adjust the laser intensity on-the-fly during the acquisition.
Once proper acquisition conditions are set, start the acquisition stream. An initial number of frames of 20,000 is suggested. For high quality images, obtain as many frames as practical (50,000 frames or more is not uncommon).
Transfer data to the image processing computer.
3.3 Processing and Analysis of Localization Microscopy Data Sets
Localization Microscopy requires intensive computation on a large volume of image data sets, with the results being a list of the coordinates for each single molecule. A typical raw image data set can range from 20,000 to 100,000 or more frames, depending on the labelling density and fluorophore blinking characteristics. Thus, the localization coordinates usually contain a few to several millions of molecular entries. These can be rendered to reconstruct a super-resolved image to be used in subsequent analyses in standard image processing software. Advanced applications and statistical analyses are also possible with the use of a programming language such as MATLAB but is beyond the scope of this protocol (for an example of advanced analysis, see ref. 39). The instructions below describe the processing and analysis of Localization Microscopy data using QuickPALM software, freely available as a plug-in in ImageJ.
Install QuickPALM plug-in in ImageJ (already included on FIJI distribution of ImageJ, www.fiji.sc).
Open single molecule time-series data in ImageJ (or FIJI). Make sure that the data have been saved with the full bit-depth, for example using 16-bit TIFF format.
Open the QuickPALM plug-ins (under Analyze/QuickPALM/Analyze Particles in FIJI).
Enter the three key parameters necessary for particle detection: minimum SNR (signal-to-noise ratio), maximum FWHM (full-width-half-maximum), and Image plane pixel size (nm).
Of these, SNR corresponds to the brightness of the fluorophores relative to the background (which is in turn proportional to the localization precision [24]). Only single molecules with SNR above the minimum threshold are accepted. Depending on the type of fluorophores, and experimental conditions, the minimum SNR can be adjusted for proper balance between sufficient density and rejection of noise (typical starting values of 4–10 can be used). Higher minimum SNR results in sparse detection, while lower SNR gives more localization points but also with increased spurious noise.
The image plane pixel size parameters can be calculated from the camera pixel size and the magnification of the microscope. As an example, for Andor Ixon-Ultra EMCCD camera, the camera pixel size is 16 μm. If a magnification of 150× is used (100× and 1.5× Optovar), the image plane pixel size is then: 16mu;m/150 or 106.7 nm.
Since single molecules appear as diffraction limited spots, their ideal sizes can be estimated from the emission wavelength (λ) and the objective NA according to the formula: FWHM = 0.52λ/ NA. For 600 nm emission and with a NA 1.49 objective, the ideal FWHM is ~205 nm or approximately 2 pixels (assuming an image plane pixel size of 106.7 nm). Thus, a maximum FWHM parameter of 4 pixels can be used as the initial value for rejecting peaks that appear too broad (this could arise when adjacent molecules are activated at the same time).
We recommend an iterative parameter refinement step using a subset of data, for example, just the first 100 frames, in order to arrive at the appropriate parameters in a reasonable amount of time. Subsequently, these parameters can be used to analyze the full datasets. Upon completion, the results of the analysis are shown in a Particle Table window.
To render a super-resolved image, make sure that the Particle Table window is open. Select “Reconstruct Dataset” option (under Analyze/QuickPALM in FIJI), select the appropriate pixel size for the rendered image (5–10 nm is useful as the starting point).
To assess and correct for sample drift, make ROI (Region-of-Interest) selections around selected fiducial regions in the rendered image, and add each ROI to the ROI manager window in ImageJ. Fiducials close to the image center are preferred as optical aberrations can be more pronounced near the edge. Subsequently, perform drift correction (Analyze/QuickPALM/ Correct Particle Drift in FIJI). As shown in Fig. 4, typical drift can be more than several tens of nanometers.
The particle table will be updated with the drift-corrected coordinates and can be saved or exported for further analysis. Reconstruct a drift-corrected super-resolved image using the “Reconstruct Dataset” option as described earlier (see Note 11). The resulting super-resolved image reveals significant details within the FAs relative to the diffraction-limited image (Fig. 2e).
Fig. 4.

Drift Correction by registration of fluorescent fiducial markers. The localized coordinate of fiducial (e.g., Fig. 2e) allows the measurement of mechanical drift of the sample during the acquisition period. An example of a rather high drift is shown for an acquisition of 20,000 frames (a, position of the fiducial; c, fluctuation of the X coordinate of the fiducial; d, fluctuation of the Y coordinate of the fiducial). The sample exhibits large drift along the Y-direction of more than 5 pixels or almost a micron. Such drift will smear the super-resolution image if not corrected for. Drift correction can be calculated by subtracting the moving averaged X and Y trajectories, resulting in a well-registered fiducial (b, using 200 frame moving average correction), and small residuals (e)
Acknowledgments
PK is supported by the Singapore National Research Foundation under the NRF Fellowship (NRFF-2011-04). CMW is supported by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health. We thank Harald Hess and Gleb Shtengel (Howard Hughes Medical Institute, Janelia Farm Research Campus), and Michael Davidson (The Florida State University) for advice, equipment, reagents, and collaboration related to this work.
Footnotes
Localization Microscopy requires sufficiently high excitation intensity for single molecule imaging, and thus lasers are generally used as light sources. The wavelengths 405, 488, 561, and 640 nm are typically adequate for photoactivation and imaging of most fluorophores. Common manufacturers include Coherent, Spectra-Physics, MellesGriot, and Crystalaser. For convenience and versatility, a pre-aligned multi-laser fiber-coupled source can be used. These are typically available from the microscope manufacturer as part of the total internal reflection fluorescence (TIRF) or confocal system. The power output is typically attenuated by the fiber, however, which may result in longer exposure and acquisition times.
Since the acquisition of raw data sets takes at least several minutes, focal drift can be substantial and must be corrected for. Most manufacturers offer automatic focus tracking and correction options. These are based on the detection and tracking of infrared beams reflected off the cover glass surface. Among commercial options, hardware-based models such as the Perfect Focus System from Nikon (Melville, NY) or the CRISP add-on module from Applied Scientific Instrumentation (Eugene, OR) are the most suitable for Localization Microscopy, as it is fully independent of the acquisition software, allowing continuous maintenance of focus during PALM acquisition. Other designs that depend on image processing of the acquisition computer may be usable but will need further modification in the data acquisition control software.
An Acousto-Optic Tunable Filter (AOTF) allows the control of the intensity and the selection of the laser wavelengths. Note that since a typical AOTF has a limited spectral range (e.g., 450–700 nm for model AOTFnCVis from A-A Optoelectronic, Orsay, France), some laser lines outside of this range, such as 405 nm, may need to be modulated, expanded, and combined with the rest of the beams separately after the AOTF. Lasers from some manufacturers (e.g., from Crystalaser, or Cube and Obis models from Coherent) include the option of modulation by TTL pulse (0–5 V) or USB/Serial input, thus an AOTF is not required.
Since the localization precision depends on the brightness of the detected single molecule images, a high NA oil-immersion objective lens is advantageous due to its large light gathering cone. In particular for TIRF, an objective lens with NA > 1.45 is needed. The magnification of the objective lens must also be chosen to match the pixel size of the camera. For a 16 μm pixel, a 100× objective lens used in combination with either a 1× or 1.5× magnification changer lens is commonly used.
Since focal adhesions form via attachment to the extracellular matrix adhered to the cover glass, TIRF excitation is most suitable. This mode of illumination provides high signal to noise ratio, since the remainder of the cell beyond the evanescent field is not excited. Note, however, that certain structures linking up with the focal adhesions, such as actin stress fibers, could extend beyond the TIRF field. Localization Microscopy can also be applied in the wide-field or oblique illumination geometry, although the increased background will degrade the localization precision to some extent.
Altogether, dichroic mirror and emission filters are particularly important, as they serve to reject excitation light and permit the fluorescence emission to reach the camera. High quality multiband dichroic mirrors are available from several manufacturers such as Semrock and Chroma. This allows a single filter cube to be used for multichannel imaging. The dichroic spectral profile should be compared against the laser lines used, as well as the emission spectra of the fluorophores. Another important consideration is the flatness of the dichroic. Depending on how it is mounted, the dichroic could be slightly warped, resulting in unintended astigmatism in the excitation light. For emission filters, if a peripheral emission filter wheel is available, it may be more advantageous to mount only the dichroic in the cube, and place single band emission filters in the filter wheel. A single band filter is generally more economical and better matched to the spectral profile of the fluorophore emission. However, if the emission filter wheel is not available, a multiple band-pass emission filter can be mounted in the filter cube. In this case, users should be aware of the possibility for fluorophore cross-talk. Examples of the filters we use are (all from Semrock, Rochester, NY): green, FF01-525/50-25; red, FF01-609/57-25; far-red, FF01-676/37-25; dichroic mirror, Di01-R405/488/561/635-25×36; Laser Combiners, LM01-503-25, LM0-427-25, and LM01-613-25.
A common hardware for detecting single molecule fluorescence is the electron-multiplying charge coupled device (EMCCD) camera. Models with a back-illuminated sensor (such as the Ixon-Ultra model from Andor or the Evolve model from Photometrics) are capable of peak quantum efficiency of >90 %. Before further processing, the signal readouts from the camera should be converted into the unit of detected photons, using the appropriate calibration factors. The possible value of a 16-bit pixel is between 0 and 65,535. However, the actual signal is offset from zero by a certain amount (such as 200–1,000), depending on the manufacturer. Likewise, the digital value of the pixel is related to the actual photoelectron by a multiplication factor. A number of methods have been published for the calibration of the offset and conversion factor [40].
Because Localization Microscopy outputs large data sets, a computer with a sufficiently large memory bank and storage is recommended. As an example, at a typical frame rate of 20 frames per second, 15 min of acquisition yields 12,000 frames, or more than 6 GB of file size. For ease of configuration and future expansion, 64-bit Windows OS is recommended. The computer used for microscope control should also have sufficient peripheral ports to control camera, AOTF, and laser firing, during the acquisition. This computer should be dedicated for acquisition with a minimum of other software installed, and should have a sufficient amount of memory (4 GB or more) and a sufficiently large hard disk (>1 TB). For rapid boot-up and read/write rate, a solid state hard drive is recommended. The computer should also be connected to the local area network for ease of data transfer.
The cover glasses are first cleaned in ddH2O, followed by 10 min heating in 1:1:5 mixture of NH4OH, H2O2, and ddH2O at 80 °C. Subsequently, the cover glasses are immersed briefly in 2 % hydrofluoric acid solution (1:50 HF in ddH2O) at room temperature, followed by heating in a 1:1:6 mixture of HCl, H2O2, and ddH2O at 80 °C for 15 min. Finally, the cover glasses are rinsed in ddH2O and blown dry with nitrogen gas. Due to the highly corrosive nature of the chemicals used, these steps must be performed in a chemical fume hood. Appropriate safety precaution must also be strictly followed.
We recommend that researchers pay particular attention to the design of PA-FP fusion constructs. Many FA proteins are multi-domain and the function of different parts of the proteins should be taken into consideration when designing the probe, since the location (i.e., amino (N) versus carboxy (C) terminus) of the PA-FP probe can have a strong effect on whether the fusion will localize properly and be functional, or not. Also, in addition to physiological expression levels, in certain cases co-expression of binding partners may be important for proper localization. As an example, integrin is a heterodimeric transmembrane protein. In our experience, we obtain good results by conjugating the PA-FP probe to the C-terminal cytoplasmic tail of the α subunit. We found that N-terminal PA-FP position is problematic since this region contains signal sequence for membrane insertion and transport. Also, we chose the α subunit for tagging since the cytoplasmic tail of the β subunit is well documented to participate in many essential binding interactions, whereas much fewer binding partners have been identified for the α tail. Finally, since αβ heterodimerization is required for export to the plasma membrane, we found that co-transfection with untagged β integrin is often required for proper localization in many cell types—without which a significant fraction of integrin appears to remain in the endoplasmic reticulum.
Because Localization Microscopy data sets essentially are comprised of a list of molecular positions, instead of conventional images, region analysis is not straightforward. To apply conventional image analysis techniques, the super-resolved image should be reconstructed first. Alternatively, advanced users may choose to develop custom programs for analyzing molecular positions data. For instance, to selectively analyze a region of interest such as the FAs, an image map of the dataset can be created from the rendered image, and thresholded to create a binary mask corresponding to FA regions. From the binary mask, morphological properties of the region such as area, aspect ratio, and ellipticity can be calculated. Molecular coordinates that belong to FAs (i.e., fall within the mask region) can then be selected, for example by using the command “ismember” in MATLAB to compare the molecule list against the coordinates of the mask regions.
References
- 1.Patterson G, Davidson M, Manley S, Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galbraith CG, Galbraith JA. Super-resolution microscopy at a glance. J Cell Sci. 2011;124:1607–1611. doi: 10.1242/jcs.080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernández-Suárez M, Ting AY. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 4.Heintzmann R, Gustafsson MGL. Subdiffraction resolution in continuous samples. Nat Photonics. 2009;3:362–364. [Google Scholar]
- 5.Huang B, Bates M, Zhuang Super-resolution fluorescence microscopy. Annu Rev Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440:935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- 7.Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci USA. 2000;97:8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- 9.Betzig E, Patterson GH, Sougrat R, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 10.Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fölling J, Bossi M, Bock H, et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- 13.Shtengel G, Galbraith JA, Galbraith CG, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shroff H, Galbraith CG, Galbraith JA, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci USA. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanchanawong P, Shtengel G, Pasapera AM, et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 18.Levitus M, Ranjit S. Cyanine dyes in biophysical research: the photophysics of polymethine fluorescent dyes in biomolecular environments. Q Rev Biophys. 2011;44:123–151. doi: 10.1017/S0033583510000247. [DOI] [PubMed] [Google Scholar]
- 19.Moerner WE. New directions in single-molecule imaging and analysis. Proc Natl Acad Sci USA. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon C. Communication in the presence of noise. Proc IRE. 1949;37:10–21. [Google Scholar]
- 21.Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 22.Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott-Schwartz J, Verkhusha VV. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lippincott-Schwartz J, Patterson GH. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 2010;7:377–381. doi: 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogelsang J, Kasper R, Steinhauer C, et al. A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew Chem Int Ed Engl. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 28.Aitken CE, Marshall RA, Puglisi JD. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henriques R, Lelek M, Fornasiero EF, Valtorta F, Zimmer C, Mhlanga MM. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7:339–340. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- 30.Wolter S, Endesfelder U, van de Linde S, Heilemann M, Sauer M. Measuring localization performance of super-resolution algorithms on very active samples. Opt Express. 2011;19:7020–7033. doi: 10.1364/OE.19.007020. [DOI] [PubMed] [Google Scholar]
- 31.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller JC, Tan S, Qiao G, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 33.Ando R, Mizuno H, Miyawaki A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- 34.Wiedenmann J, Ivanchenko S, Oswald F, et al. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurskaya NG, Verkhusha VV, Shcheglov AS, et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- 36.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol. 2010;14:14.20.1–14.20.17. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray JM, Appleton PL, Swedlow JR, Waters JC. Evaluating performance in three-dimensional fluorescence microscopy. J Microsc. 2007;228:390–405. doi: 10.1111/j.1365-2818.2007.01861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]