

Abstract
Synaptic plasticity is critical for elaboration and adaptation in the developing and developed brain. It is well established that astrocytes play an important role in the maintenance of what has been dubbed “the tripartite synapse”. Increasing evidence shows that a fourth cell type, microglia, is critical to this maintenance as well. Microglia are the resident macrophages of the central nervous system (CNS). Because of their well-characterized inflammatory functions, research has primarily focused on their innate immune properties. The role of microglia in the maintenance of synapses in development and in homeostasis is not as well defined. A number of significant findings have shed light on the critical role of microglia at the synapse. It is becoming increasingly clear that microglia play a seminal role in proper synaptic development and elimination.
1. Microglia
Microglia constitute approximately 10% of the cells in the CNS. They have been traditionally thought to function as the immunocompetent cells of the brain and spinal cord [1] and to be the sensors of injury and infection in the tissue [2, 3]. They derive from primitive c-kit(+) erythromyeloid precursors from the yolk sac [4–6], migrate into the brain during the period of early embryonic development prior to the formation of the blood-brain barrier (BBB), and remain there once the BBB is formed [7]. It is notable that this population is self-sustaining, and peripheral macrophages only contribute to this population in disease states, in which the BBB becomes compromised [8].
Within the brain, microglia have been described to have the ability to detect and mount an inflammatory response to various insults. Sensing neuronal injury has been ascribed primarily to purinergic and chemokine receptors on the surface of microglia, as they monitor the levels of extracellular ATP and secreted chemokines, respectively [9]. Their reaction to neuronal injury is accomplished as they undergo a process collectively called “activation.” Activation consists of several biological events that include migration to the site of injury, local proliferation, a change in morphology and gene expression, antigen presentation, and phagocytosis of dead cells and cell debris [10, 11]. During activation, some of the changes in gene expression involve the secretion of cytokines and chemokines, which modulate the CNS environment and regulate the state of inflammation. Inflammation in turn affects the progression of neuronal death after CNS insult. Microglia can secrete both proinflammatory mediators, such as tumor-necrosis-factor- (TNF-) α [12, 13] or interleukin- (IL-) 1β, nitric oxide (NO) [14, 15], and glutamate [16], and anti-inflammatory effectors, such as IL-4 and IL-13, which can enhance neuronal survival [17, 18].
Depending on the predominance of factors secreted, microglia have classically been characterized, similarly to macrophages, as M1 (proinflammatory) or M2 (anti-inflammatory) cells [19]. The M1 and M2 distinctions serve to separate activated microglia into the two broad categories. It is argued, however, that no specific marker designates a microglial response as definitively M1 or M2. Moreover, microglia differentially express pro- and anti-inflammatory markers making the M1 and M2 phenotypes the extremes of the categories, respectively, with variable phenotypes seen in between the two [8]. With that in mind, M1 microglia have been associated with neurotoxic and neurodegenerative outcomes, as they are observed in a variety of chronic neurodegenerative diseases, such as Alzheimer's [20], end-stage amyotrophic lateral sclerosis (ALS) [21], and multiple sclerosis (MS) [22]. Stroke [23] and traumatic brain injury [24] show a characteristic accumulation of M1 microglia as well. A shift to an M2 phenotype of activated microglia has been correlated with neuroprotection, recovery, and repair in various disease settings [25–27].
2. Microglia as Regulators of Neuronal Function and Plasticity
Until recently, initial studies to understand neuronal-microglial interactions had described that a variety of neuroactive substances, such as NO [29] and TNF-α [30], have potent effects on neuronal function, in particular, synaptic plasticity. However, the cellular origin of these molecules had not been attributed to microglia but, rather, to astrocytes. The focus was maintained on the effect that inflammatory processes have on synaptic plasticity. In neuroinflammatory diseases, such as experimental autoimmune encephalomyelitis (EAE), a rodent model of MS, alterations in synaptic plasticity have been noted. Specifically, in the hippocampus of diseased animals, there is greater induction of long-term potentiation (LTP), an electrophysiological measurement that relates to the connectivity and strength of synapses. This change in LTP was attributed to the secretion of IL1β from the accumulated microglial cells [31]. Bacterial lipopolysaccharide (LPS) strongly upregulates IL1β secretion by macrophages.
It is notable, however, that prolonged exposure to inflammatory cytokines can result in priming or sensitization of microglia so they more readily adopt an M2, rather than M1, phenotype in response to inflammation. This is quite the opposite response to that in acute exposure [32]. As such, chronic inflammation can be induced by LPS infusion and has been reported to attenuate LTP in the dentate gyrus (DG) of the hippocampus. This is accompanied by the loss of pyramidal neurons [33]. Similarly, using LPS infusion, Min et al. found that LTP, dependent on either NMDA receptors (NMDAR) or on voltage-dependent calcium channels, was impaired [34]. Further work is necessary to elucidate the specific mechanism causing these phenomena.
The cytokine, TNF-α, increases the surface expression of AMPA receptors in neuronal cultures, which is accompanied by the enhancement of synaptic strength [30]. In a model of neuropathic pain in the C fibers of the dorsal horn of the spinal cord, inhibition of microglial activation by minocycline resulted in the induction of long-term depression (LTD) rather than LTP. This change in plasticity was found to involve Src family kinases and to be mediated partially by TNF-α [35]. Some studies have found that microglial activation, when both genetically and pharmacologically induced, results in an increase of AMPAR/NMDAR ratio and an enhanced ratio of AMPAR- over NMDAR-mediated currents [36]. These studies demonstrated that when microglia were activated under pathological inflammatory conditions, they caused synaptic alterations via secretory mediators. The precise role of microglia on synaptic activity in the normal brain remained unclear.
Imaging studies have shown that microglia extend and retract their processes continuously to survey their local environment in the healthy brain [37, 38]. Moreover, interactions between microglia and neuronal synapses in the visual cortex have been directly visualized by electron microscopy (EM) and by in vivo two-photon microscopy. The availability of visual stimuli resulted in enhancement of the duration of these contacts and the preservation of the synapse [39]. These intriguing imaging observations first indicated the possibility that microglia could modulate neuronal functions by direct physical contacts. On the other hand, Wake et al. demonstrated that, under conditions of prolonged ischemia, contact time between microglial processes and synapses increased, associated with a greater chance for elimination of presynaptic boutons [40]. It is likely that the mechanism of synaptic pruning is modulated by a distinct molecular mechanism in each of these states and not simply by the longevity of contact.
3. Mechanisms Governing the Interactions of Neurons and Microglia
Paolicelli et al. [41] explored whether there is a functional role for microglial interactions with synaptic structures during the development of the postnatal brain, using imaging and electrophysiological approaches. They used a transgenic mouse line expressing GFP in microglia, under the control of the chemokine receptor CX3CR1 promoter. Thus, they were able to label and visualize microglia as well as manipulate them. The authors found that the number of synaptic elements and dendritic spines expressing the postsynaptic marker PSD95 in Cx3cr1GFP/+ mice was about 3-fold higher than that in mice deficient in CX3CR1 (Cx3cr1KO/KO). Their result provided some insight into potential roles of microglia in synapse maturation, along with the possibility that this may be a direct CX3CR1-mediated event [42]. These microglial properties, thus, extend beyond immune surveillance and indicate modulatory roles during normal brain development.
Schafer et al. [43] provided direct evidence, via confocal microscopy and electron microscopy, that microglia phagocytose synapses in the dorsal lateral geniculate nucleus (dLGN) as well. The authors proposed that the classical complement cascade, which includes members C1q and C3, was a potential molecular pathway of microglia-synapse interactions in postnatal brain development. According to the experimental data, the synapses that were tagged with C1q and C3 were phagocytosed by microglia that expressed complement receptor CR3. In mice deficient in the receptor or the ligand, higher numbers of synaptic inputs were observed. Moreover, these animals showed deficits in their ability to segregate the territories of each eye. Therefore, the microglia-mediated engulfment was important to drive synaptic stripping during normal development. Together, these observations reveal that complement-mediated phagocytic activity of microglia is crucial in microglia-synapse interactions during normal brain development.
To further address whether microglia contribute to synaptic activity in the normal young adult brain, Ji et al. [28] used an electrophysiological approach in organotypic hippocampal brain slices and primary neuronal cultures. In this system, they manipulated the presence of microglia by either depleting them using clodronate, replenishing them in previously depleted cultures, or by using cocultures of neurons and microglia. The absence of microglia resulted in a robust increase of synaptic frequencies known as spontaneous and miniature excitatory postsynaptic currents (sEPSC and mEPSC) from the CA1 region of the hippocampus. This increase was subsequently reversed when microglia were replenished in the organotypic slices. In the complementary approach, the addition of microglia to neuronal cultures decreased the synaptic activity measured compared to cultures of neurons alone. The change in synaptic activity coincided with changes in synaptic numbers, which suggested that microglia could participate in the control of synaptic activity by regulating synaptic numbers (Figure 1). As shown in previous reports [43], they also observed that the phagocytic activity of microglia drove synapse elimination when microglia were coincubated with neurons. This could be one mechanism by which synapse numbers are regulated in the normal brain; however, it is still undefined whether synaptic pruning and phagocytic engulfment by microglia occur via a universal mechanism under normal and pathological conditions.
Figure 1.
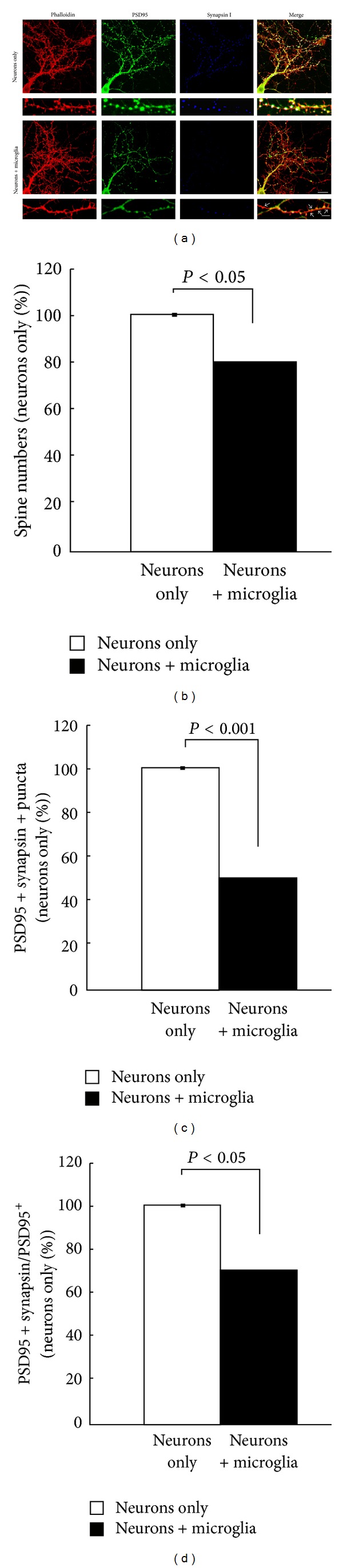
Microglia alter the synaptic density of hippocampal neurons. Hippocampal neurons with or without microglia were stained with PSD95 (green), synapsin I (blue), and phalloidin (red) (a). The smaller boxes show magnified images. Arrows depict PSD95+ synapsin 1− puncta. Scale bars: 20 μm (upper panel); 5 μm (lower panel). Quantification of spine numbers (b), PSD95+synapsin 1+ puncta (c), and PSD95+ synapsin 1+ puncta in total PSD95+ puncta (d) in neurons cultured with or without microglia. Values are presented as mean ± SEM and expressed as a percent of the neurons-only control sample (adapted from [28]).
A proposed mechanism by which microglia could regulate synaptic activity was suggested by the same study [28]. Overall expression of synaptic adhesion molecules, such as protocadherin and SynCAM1, which determine synapse remodeling, stability, and synaptic activity, was decreased in neurons incubated with microglia compared to neurons alone (Figure 1). The decreased levels of the synaptic adhesion molecules were recovered to wild-type levels when neurons were incubated with microglia deficient in the serine protease tissue plasminogen activator (tPA) (Figure 2), potentially implicating serine proteolytic functions in the stability of these proteins.
Figure 2.
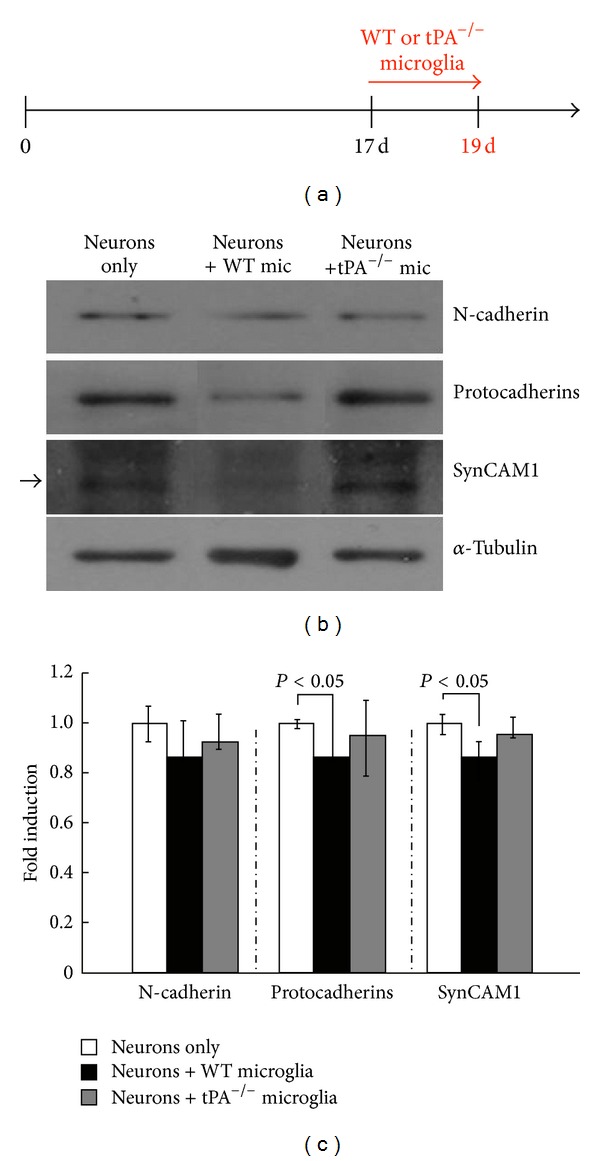
Microglial tPA deficiency preserves the levels of synaptic adhesion molecules. (a) Hippocampal neurons at 19 DIV were cocultured with microglia for 2 days. (b) The western blot analysis of the levels of N-cadherin, pan-γ-protocadherin, and SynCAM-1 in neurons in the absence or presence of microglia from wild-type (WT) or tPAKO(tPA−/−) mice. α-Tubulin was used as a loading control. (c) Quantification was performed using the ImageJ software and normalized against α-tubulin (n = 4). *P < 0.05 compared to neurons alone.
Structural changes at the synapse are closely associated with synaptic stability. In particular, numerous synaptic adhesion molecules, such as classic cadherins (E-cadherin and N-cadherin), protocadherins, and NCAM, have been studied in modulating structural and functional synaptic plasticity. Hippocampal slices pretreated with antibodies against the extracellular domain of N- and E-cadherins or with antagonistic peptides that inhibit cadherin dimerization exhibit a significantly reduced LTP [44]. Moreover, expression of mutant N-cadherin or short hairpin RNA-mediated knockdown of N-cadherin prevents LTP-induced long-term stabilization of synapses [45]. Additionally, Yamagata et al. showed that blocking antibodies to protocadherins or NCAM in hippocampal slices diminished synaptic transmission and LTP induction [46].
Proteases in a synaptic microenvironment are important in the regulation of dynamic changes in the adhesion molecules associated with synaptic plasticity [47]. Proteases, such as matrix metalloproteinases (MMPs) and tPA, secreted from neurons, astrocytes, or microglia under basal or pathological conditions of the CNS have been associated with the targeted degradation or proteolytic processing of extracellular matrix (ECM) and cell adhesion molecules on the cell surface and at the synapse [48–55]. In particular, application of tPA or MMP-9 was shown to be involved specifically in the production of LTP and synaptic growth. Emerging evidence has shown that application of MMP-9 or tPA decreases the levels of N-cadherin and diminishes synaptic transmission [56]. Moreover, tPA regulates MMP activity [57], which leads to the regulation of synaptic plasticity. It is possible that proteases secreted from microglia could regulate synaptic activity by remodeling the ECM which is known to affect synaptic connectivity [58].
4. Connexins and Large Pore Channels
One way of communication among microglia is through connexins and large pore channels. Connexins (Cx) are proteins found in gap junctions, connecting adjacent cells. Each of the connected cells provides an array of Cx isoforms, which form oligomers containing 6 of these Cx proteins. This complex is called a connexon and constitutes a hemichannel [59]. The most common isoforms in mammals are Cx36, Cx43, and Cx45. Connexins are traditionally described as being expressed in astrocytes and in neurons. Cx36 and Cx43 have been reported to be expressed in microglia [60, 61], where they are thought to be involved in the local release of proinflammatory cytokines (TNF-α and IL1β) [62] and metabolites. During inflammatory events, the expression of Cx43 was shown to increase. This increase results in the formation of a functional syncytium among microglial cells, confirmed by the diffusion of the fluorescent dye, Lucifer yellow. However, the syncytium neither forms in nonactivated microglia [63], nor happens if the gap junction formation is inhibited by inhibitors of Cx43, indicating the involvement of Cx43 in the process. Cx36 remains active in resting microglia and does not become upregulated during microglial activation.
In disease settings, it has been reported that blocking Cx hemichannels resulted in the blockade of the microglial release of glutamate [64], which led to the subsequent exaggerated activation of neurons (excitotoxicity). In a model of spinal cord injury (partial cord transection), inhibition of Cx resulted in improved functional recovery [65].
Similar to Cx proteins, large pore channels are formed in microglia and consist primarily of pannexins and P2X channels. They are purinergic and activated by extracellular ATP. Among them, P2X4 is the channel that becomes primarily upregulated in activated microglia [63]. In a recent report, Li et al. state that in the optic tectum of larval zebrafish, neuronal activity drives the activation of pannexin-1 hemichannels. These can then “steer” the processes of resting microglia and facilitate their contact with highly active neurons [66]. In turn, when resting microglia are in contact with neurons or neurites, a decrease in both spontaneous and visually evoked neuronal activities is observed, specifically for the neurons contacted.
These results indicate that connexins and large pore channels could constitute one way by which microglia interact directly with neurons, especially during neuronal insult and inflammation, and could directly affect neuronal activity and survival.
5. Direct Modulation of Neurotransmitter Release and Homeostasis
As mentioned above, microglia can generate neurotransmitters, primarily glutamate. They also respond to changes in neurotransmitters by changing morphology and the motility of their processes. Such responses have been documented both for glutamatergic but also for GABAergic transmission [67]. Application of the glutamate receptor inhibitors NBQX and GYKI, as well as the GABAergic signaling inhibitor bicuculline, has been shown to decrease microglial process motility [68]. Although there is debate on whether microglia express glutamate receptors [68], the presence of GABAA receptors on the surface of human microglia has been documented [69]. However, there is no concrete evidence that microglia respond in an obvious way to direct application of agonists of glutamate or GABA receptors, that is, in a pure microglial cell culture. Rather, they seem to respond indirectly to such application, on a slice or tissue, suggesting that these agonists potentially have indirect effects on the cells. These indirect effects have been postulated to be mediated through ATP's effect on purinergic receptors since they are expressed on the surface of the cells [37]. Although the source of the ATP release is not entirely determined, the most likely mechanism involves release through neuronal pannexin channels [70].
In models of disease, specifically in the EAE model of MS, the presence and accumulation of activated microglia have been correlated with decreases in the Purkinje cell survival, connectivity in the cerebella of the EAE animals, and attenuation of GABAergic transmission [71]. This has also been observed in the EAE striatum [72] and hippocampus, where a decrease in GABAergic interneurons was also noted, accompanied by induction of LTP [31]. These results suggest that, in this context, microglia may be direct regulators of the numbers of GABAergic neurons and the subsequent attenuation of GABAergic inhibitory transmission.
6. Ectosome and Lipid Signaling
A new way of communication in the CNS has been described involving the release of microvesicles, also referred to as shed vesicles or ectosomes, from the plasma membrane [73]. These materials were originally thought to be inert but have been recently recognized as critical in mediating cell-to-cell communication. The vesicles contain lipids, cell surface proteins, and material from the cytoplasm or nucleus of the cell [74]. The vesicles are recognized by the recipient cell through the presence of phosphatidylserine on their surface [75] and interact with the relevant receptors. They can also directly fuse with the recipient cell.
On the surface of microglia, P2X7 receptors, which respond to the release of ATP, mediate the shedding of ectosomes [76]. This process is triggered by the activity of acid sphingomyelinase and involves the activation of the effector protein p38. Although this is not a mechanism exclusive to microglia (as astrocytes also have been shown to express the P2X7 receptors), microglia constitute a significant source of these shed vesicles.
Signaling through these microvesicles has been reported in different systems. One of the factors thought to contribute to such signaling is Annexin A2 [77], a protein expressed by microglia that affects their activation [78, 79]. Annexin A2 has been shown in different systems to affect neuronal ion channels and neuronal functioning [80, 81], either directly or through its interaction with p11 [82].
In a recent report, it was noted that microvesicles derived from microglia were able to increase the frequency and amplitude of EPSCs [74]. This effect required interaction between microglia and neuronal cells and did not involve secretion of cytokines. It did involve, however, an increase in the metabolism of sphingolipids in neuronal cells. This resulted in an acute increase in excitatory neurotransmitter release. Although in a more chronic exposure to these shed particles the release of cytokines as regulators of neuronal activity cannot be excluded, these data provide another possible pathway by which microglia affect neuronal activity.
7. Conclusions
Given the evidence from imaging, cellular, and electrophysiological approaches, the physical proximity between neurons and microglia seems to result in synaptic maturation and synaptic activity (Figure 3). Several different mechanisms, either involving direct contact and interaction between the two cell types, or mediated through chemical ligands and effectors [9], are described as potential regulators of these microglial functions. The findings indicate that microglia affect both the maturation of the CNS during development and the acute and dynamic regulation of neuronal activity in the mature, healthy, or unhealthy CNS and suggest that they are active contributors to a potential quad-partite synapse [83].
Figure 3.
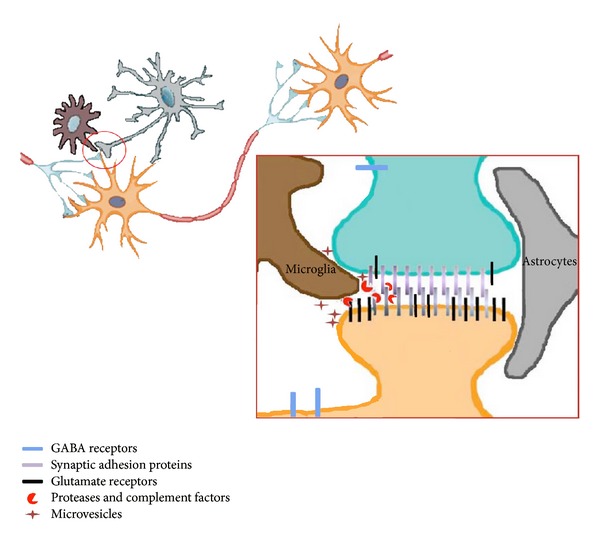
Schematic depicting some of the potential mechanisms through which microglia can affect neuronal activity. Potential interactions between neurons, astrocytes, and microglia at the synapse. The area depicted by the red circle is magnified in inset. Inset: microglial processes in proximity to neuronal synapses can modify neuronal activity via multiple potential pathways. They can secrete proteases to modulate the stability of synaptic adhesion molecules (which in turn influences synaptic transmission) or remove complement-tagged structures. They can release ectosomes that directly interact with the neuronal membranes and initiate signaling cascades. They can also affect directly glutamatergic or GABAergic transmission.
Acknowledgments
The authors thank members of the Tsirka and Wollmuth labs for helpful discussion and comments. This work was partially supported by T32GM008444 (JM) and R0142168 (SET).
References
- 1.Skaper SD. Ion channels on microglia: therapeutic targets for neuroprotection. CNS & Neurological Disorders Drug Targets. 2011;10(1):44–56. doi: 10.2174/187152711794488638. [DOI] [PubMed] [Google Scholar]
- 2.Kreutzberg GW. Microglia, the first line of defence in brain pathologies. Arzneimittel-Forschung. 1995;45(3):357–360. [PubMed] [Google Scholar]
- 3.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 4.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Frontiers in Cellular Neuroscience. 2013;7, article 45 doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu. 1- and Irf8-dependent pathways. Nature Neuroscience. 2013;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 7.Neumann H, Wekerle H. Brain microglia: watchdogs with pedigree. Nature Neuroscience. 2013;16:253–255. doi: 10.1038/nn.3338. [DOI] [PubMed] [Google Scholar]
- 8.Eggen BJ, Raj D, Hanisch UK, Boddeke HW. Microglial phenotype and adaptation. Journal of Neuroimmune Pharmacology. 2013;8:807–823. doi: 10.1007/s11481-013-9490-4. [DOI] [PubMed] [Google Scholar]
- 9.Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 10.Altman J. Microglia emerge from the fog. Trends in Neurosciences. 1994;17(2):47–49. doi: 10.1016/0166-2236(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 11.Aloisi F. Immune function of microglia. Glia. 2001;36(2):165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 12.Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. European Journal of Immunology. 1992;22(9):2429–2436. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- 13.Emmetsberger J, Tsirka SE. Microglial inhibitory factor (MIF/TKP) mitigates secondary damage following spinal cord injury. Neurobiology of Disease. 2012;47:295–309. doi: 10.1016/j.nbd.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. Journal of Immunology. 1992;149(8):2736–2741. [PubMed] [Google Scholar]
- 15.Wu M, Tsirka SE. Endothelial NOS-deficient mice reveal dual roles for nitric oxide during experimental autoimmune encephalomyelitis. Glia. 2009;57(11):1204–1215. doi: 10.1002/glia.20842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. Journal of Neurochemistry. 2001;76(3):846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 17.Shin WH, Lee D-Y, Park KW, et al. Microglia expressing interleukin-13 undergo cell death and contribute to neuronal survival in vivo. Glia. 2004;46(2):142–152. doi: 10.1002/glia.10357. [DOI] [PubMed] [Google Scholar]
- 18.Park KW, Lee DY, Joe EH, Kim SU, Jin BK. Neuroprotective role of microglia expressing interleukin-4. Journal of Neuroscience Research. 2005;81(3):397–402. doi: 10.1002/jnr.20483. [DOI] [PubMed] [Google Scholar]
- 19.Colton CA, Wilcock DM. Assessing activation states in microglia. CNS and Neurological Disorders. 2010;9(2):174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 20.Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. The Journal of Neuroscience. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Experimental Neurology. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao Z, Tsirka SE. Animal models of MS reveal multiple roles of microglia in disease pathogenesis. Neurology Research International. 2011;2011:9 pages. doi: 10.1155/2011/383087.383087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiology of Aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikita J, Dubourdieu-Cassagno N, Deloire MS, et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis: amelioration of clinical status by M2 activated monocyte administration. Multiple Sclerosis. 2011;17(1):2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- 26.Wu M, Nissen JC, Chen EI, Tsirka SE. Tuftsin promotes an anti-inflammatory switch and attenuates symptoms in experimental autoimmune encephalomyelitis. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034933.e34933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shechter R, Schwartz M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer “if” but ‘how’. The Journal of Pathology. 2013;229:332–346. doi: 10.1002/path.4106. [DOI] [PubMed] [Google Scholar]
- 28.Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0056293.e56293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260(5116):1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
- 30.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFα . Science. 2002;295(5563):2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 31.Nistico R, Mango D, Mandolesi G, et al. Inflammation subverts hippocampal synaptic plasticity in experimental multiple sclerosis. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0054666.e54666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ajmone-Cat A, Mancini M, De Simone R, Cilli P, Minghetti L. Microl polarization and plasticity: evidence from organotypic hippocampal slice cultures. Glia. 2013;61:1698–1711. doi: 10.1002/glia.22550. [DOI] [PubMed] [Google Scholar]
- 33.Hauss-Wegrzyniak B, Lynch MA, Vraniak PD, Wenk GL. Chronic brain inflammation results in cell loss in the entorhinal cortex and impaired LTP in perforant path-granule cell synapses. Experimental Neurology. 2002;176(2):336–341. doi: 10.1006/exnr.2002.7966. [DOI] [PubMed] [Google Scholar]
- 34.Min SS, Quan HY, Ma J, Han J-S, Jeon BH, Seol GH. Chronic brain inflammation impairs two forms of long-term potentiation in the rat hippocampal CA1 area. Neuroscience Letters. 2009;456(1):20–24. doi: 10.1016/j.neulet.2009.03.079. [DOI] [PubMed] [Google Scholar]
- 35.Zhong Y, Zhou L-J, Ren W-J, et al. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-α . Brain, Behavior, and Immunity. 2010;24(6):874–880. doi: 10.1016/j.bbi.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Roumier A, Pascual O, Béchade C, et al. Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS ONE. 2008;3(7) doi: 10.1371/journal.pone.0002595.e2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 38.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 39.Tremblay M-Ě, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biology. 2010;8(11) doi: 10.1371/journal.pbio.1000527.e1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. Journal of Neuroscience. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 42.Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Frontiers in Cellular Neuroscience. 2013;7, article 6 doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20(6):1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- 45.Mendez P, De Roo M, Poglia L, Klauser P, Muller D. N-cadherin mediates plasticity-induced long-term spine stabilization. Journal of Cell Biology. 2010;189(3):589–600. doi: 10.1083/jcb.201003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamagata K, Andreasson KI, Sugiura H, et al. Arcadlin is a neural activity-regulated cadherin involved in long term potentiation. Journal of Biological Chemistry. 1999;274(27):19473–19479. doi: 10.1074/jbc.274.27.19473. [DOI] [PubMed] [Google Scholar]
- 47.Shiosaka S, Yoshida S. Synaptic microenvironments: structural plasticity, adhesion molecules, proteases and their inhibitors. Neuroscience Research. 2000;37(2):85–89. doi: 10.1016/s0168-0102(00)00115-2. [DOI] [PubMed] [Google Scholar]
- 48.Kaczmarek L, Lapinska-Dzwonek J, Szymczak S. Matrix metalloproteinases in the adult brain physiology: a link between c-Fos, AP-1 and remodeling of neuronal connections? EMBO Journal. 2002;21:6643–6648. doi: 10.1093/emboj/cdf676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dzwonek J, Rylski M, Kaczmarek L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Letters. 2004;567(1):129–135. doi: 10.1016/j.febslet.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 50.Qian Z, Gilbert M, Colicos M, Kandel E, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 51.Gualandris A, Jones T, Strickland S, Tsirka S. Membrane depolarization induces the Ca2+-dependent release of tissue plasminogen activator. Journal of Neuroscience. 1996;16(7):2220–2225. doi: 10.1523/JNEUROSCI.16-07-02220.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang Y, Bach M, Lipp H, et al. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proceedings of the National Academy of Sciences of the USA. 1996;93:8699–8704. doi: 10.1073/pnas.93.16.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baranes D, Lederfein D, Huang YY, Chen M, Bailey C, Kandel E. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 54.Madani R, Hulo S, Toni N, et al. Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO Journal. 1999;18:3007–3012. doi: 10.1093/emboj/18.11.3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neuhoff H, Roeper J, Schweizer M. Activity-dependent formation of perforated synapses in cultured hippocampal neurons. European Journal of Neuroscience. 1999;11:4241–4250. doi: 10.1046/j.1460-9568.1999.00856.x. [DOI] [PubMed] [Google Scholar]
- 56.Restituito S, Khatri L, Ninan I, et al. Synaptic autoregulation by metalloproteases and γ-secretase. Journal of Neuroscience. 2011;31(34):12083–12093. doi: 10.1523/JNEUROSCI.2513-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Lee S-R, Arai K, et al. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nature Medicine. 2003;9(10):1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 58.Dityatev A, Schachner M. Extracellular matrix molecules and synaptic plasticity. Nature Reviews Neuroscience. 2003;4(6):456–468. doi: 10.1038/nrn1115. [DOI] [PubMed] [Google Scholar]
- 59.Willecke K, Eiberger J, Degen J, et al. Structural and functional diversity of connexin genes in the mouse and human genome. Biological Chemistry. 2002;383(5):725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- 60.Dobrenis K, Chang H-Y, Pina-Benabou MH, et al. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. Journal of Neuroscience Research. 2005;82(3):306–315. doi: 10.1002/jnr.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garg S, Syed MM, Kielian T. Staphylococcus aureus-derived peptidoglycan induces Cx43 expression and functional gap junction intercellular communication in microglia. Journal of Neurochemistry. 2005;95(2):475–483. doi: 10.1111/j.1471-4159.2005.03384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oviedo-Orta E, Evans WH. Gap junctions and connexin-mediated communication in the immune system. Biochimica et Biophysica Acta. 2004;1662(1-2):102–112. doi: 10.1016/j.bbamem.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 63.Mika T, Prochnow N. Functions of connexins and large pore channels on microglial cells: the gates to environment. Brain Research. 2012;1487:16–24. doi: 10.1016/j.brainres.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 64.Maezawa I, Jin L-W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. Journal of Neuroscience. 2010;30(15):5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cronin M, Anderson PN, Cook JE, Green CR, Becker DL. Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Molecular and Cellular Neuroscience. 2008;39(2):152–160. doi: 10.1016/j.mcn.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 66.Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell. 2012;23:1189–1202. doi: 10.1016/j.devcel.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 67.Domercq M, Vazquez-Villoldo N, Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Frontiers in Cellular Neuroscience. 2013;7, article 49 doi: 10.3389/fncel.2013.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong WT, Wang M, Li W. Regulation of microglia by ionotropic glutamatergic and GABAergic neurotransmission. Neuron Glia Biology. 2011;7:41–46. doi: 10.1017/S1740925X11000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee M, Schwab C, Mcgeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59(1):152–165. doi: 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- 70.Fontainhas AM, Wang M, Liang KJ, et al. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015973.e15973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mandolesi G, Grasselli G, Musella A, et al. GABAergic signaling and connectivity on Purkinje cells are impaired in experimental autoimmune encephalomyelitis. Neurobiology of Disease. 2012;46(2):414–424. doi: 10.1016/j.nbd.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 72.Rossi S, Muzio L, De Chiara V, et al. Impaired striatal GABA transmission in experimental autoimmune encephalomyelitis. Brain, Behavior, and Immunity. 2011;25(5):947–956. doi: 10.1016/j.bbi.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 73.Sadallah S, Eken C, Schifferli JA. Ectosomes as modulators of inflammation and immunity. Clinical and Experimental Immunology. 2011;163(1):26–32. doi: 10.1111/j.1365-2249.2010.04271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Antonucci F, Turola E, Riganti L, et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO Journal. 2012;31(5):1231–1240. doi: 10.1038/emboj.2011.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Al-Nedawi K, Meehan B, Rak J. Microvesicles: messengers and mediators of tumor progression. Cell Cycle. 2009;8(13):2014–2018. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- 76.Bianco F, Perrotta C, Novellino L, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO Journal. 2009;28(8):1043–1054. doi: 10.1038/emboj.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kwaan HC, Magalhães Rego E. Role of microparticles in the hemostatic dysfunction in acute promyelocytic leukemia. Seminars in Thrombosis and Hemostasis. 2010;36(8):917–924. doi: 10.1055/s-0030-1267045. [DOI] [PubMed] [Google Scholar]
- 78.Siao C-J, Tsirka SE. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. Journal of Neuroscience. 2002;22(9):3352–3358. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hedhli N, Falcone DJ, Huang B, et al. The annexin A2/S100A10 system in health and disease: emerging paradigms. Journal of Biomedicine and Biotechnology. 2012;2012:13 pages. doi: 10.1155/2012/406273.406273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gauthier-Kemper A, Weissmann C, Golovyashkina N, et al. The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2-dependent manner. Journal of Cell Biology. 2011;192(4):647–661. doi: 10.1083/jcb.201007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ning L, Wang C, Ding X, Zhang Y, Wang X, Yue S. Functional interaction of TRPV4 channel protein with annexin A2 in DRG. Neurological Research. 2012;34:685–693. doi: 10.1179/1743132812Y.0000000065. [DOI] [PubMed] [Google Scholar]
- 82.Warner-Schmidt JL, Schmidt EF, Marshall JJ, et al. Cholinergic interneurons in the nucleus accumbens regulate depression-like behavior. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11360–11365. doi: 10.1073/pnas.1209293109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: micro-synapse interactions in the developing and mature CNS. Glia. 2013;61:24–36. doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]