

Abstract
Resistance to the cytostatic activity of the antimalarial drug chloroquine (CQ) is becoming well understood, however, resistance to cytocidal effects of CQ is largely unexplored. We find that PfCRT mutations that almost fully recapitulate P. falciparum cytostatic CQ resistance (CQRCS) as quantified by CQ IC50 shift, account for only 10–20% of cytocidal CQR (CQRCC) as quantified by CQ LD50 shift. Quantitative trait loci (QTL) analysis of the progeny of a chloroquine sensitive (CQS; strain HB3)×chloroquine resistant (CQR; strain Dd2) genetic cross identifies distinct genetic architectures for CQRCS vs CQRCC phenotypes, including identification of novel interacting chromosomal loci that influence CQ LD50. Candidate genes in these loci are consistent with a role for autophagy in CQRCC, leading us to directly examine the autophagy pathway in intraerythrocytic CQR parasites. Indirect immunofluorescence of RBC infected with synchronized CQS vs CQR trophozoite stage parasites reveals differences in the distribution of the autophagy marker protein PfATG8 coinciding with CQRCC. Taken together, the data show that an unusual autophagy – like process is either activated or inhibited for intraerythrocytic trophozoite parasites at LD50 doses (but not IC50 doses) of CQ, that the pathway is altered in CQR P. falciparum, and that it may contribute along with mutations in PfCRT to confer the CQRCC phenotype.
Introduction
Resistance to quinoline and antifolate antimalarial drugs, as well as emerging tolerance to artemisinin – based drugs [1] threatens the health of over half of the world’s population. Understanding the molecular details of P. falciparum and P. vivax antimalarial drug resistance phenomena facilitates surveillance of resistance and rapid development of more effective treatment. Two approaches for monitoring antimalarial drug resistance for P. falciparum malaria are available. One is analysis of clinical data to assess efficacy of specific treatments, the second is in vitro analysis of parasite strains or patient isolates to quantify their susceptibility to specific drugs. With the discovery of key genetic mutations in P. falciparum that confer resistance to either antifolate [2], [3] or quinoline – based [4] antimalarial drugs, rapid field-based surveillance of the geographic spread of existing drug resistant malaria is now possible, as is personalized delivery of second tier drug therapy to patients infected with a specific drug resistant strain.
Many antimicrobial drugs are both cytostatic and cytocidal, including quinoline antimalarial drugs such as CQ [5]–[7]. That is, under certain conditions a drug slows the rate of cell growth or impairs cell division such that the rate of proliferation of a mass population of the microbe is reduced, and under other conditions the drug kills the microbial cell. Often, cytocidal (cell kill) activity requires higher dose of drug, longer drug exposure time, or both. Cytostatic potency is usually quantified via IC50 values (the dose of drug at which growth is inhibited by 50% relative to control), whereas cytocidal potency is quantified via LD50 values (the dose of drug that kills 50% of a microbial population).
However, to date, all laboratory based quantification of antimalarial drug potency, and hence quantification of all antimalarial drug resistance phenomena, has been done with IC50 values alone. Again, IC50 assays quantify the concentration of drug required to inhibit proliferation of parasite populations by 50%. Drug IC50 for P. falciparum are typically measured in red blood cell culture suspensions in the continuous presence of serially diluted concentrations of the drug. Such quantification has proved critical for defining the genetics and biochemistry behind resistance to the cytostatic effects of CQ (CQRCS) [4], [8]–[11] and for identifying new drug leads with excellent cytostatic potential vs CQR malaria [12]. It is sometimes assumed that IC50 values measure the “cell kill” effect of a drug. Although this can be true in specific cases, in many others this is not the case. Use of IC50 values alone can over-estimate the cytocidal activity of a drug and can under-estimate potential parasite survival in the presence of higher (clinically relevant) levels of the drug. Since laboratory CQ IC50 are in the 10−9–10−8 M range, but peak plasma levels of CQ in patients are ∼1000 times higher (10−6–10−5 M; see [13]–[15]), clarification of these points is essential for fully understanding CQR.
That is, although the mechanism of CQRCS is becoming clear, much less is known about resistance to the cytocidal effects of CQ (CQRCC) (or cytocidal resistance vs any other antimalarial drug for that matter). This is a critical piece of missing information, given that parasite survival determines the rate of adaptation to selection by drugs. Only recently has it been possible to efficiently and reproducibly quantify LD50 for some antimalarial drugs, and rapid quantification of LD50 differences for drug sensitive vs drug resistant parasites are found in only one paper to our knowledge [6]. Formally, without additional information the ratio of drug IC50 values for drug sensitive vs drug resistant parasites expresses the degree of cytostatic resistance, whereas LD50 ratios express the degree of cytocidal resistance [6].
For any drug (anti-tumor, anti-bacterial, anti-fungal, anti-parasitic) it is theoretically possible that the molecular mechanisms controlling cytostatic and cytocidal activities could overlap. If this is the case, then the molecular mechanisms of resistance to those two distinct layers of drug pharmacology would also presumably overlap. In other cases, particularly when significantly different concentrations of drug are needed to kill the relevant cell vs merely slow its growth, molecular targets for the two layers of drug activity may differ, and then the mechanisms of cytostatic vs cytocidal resistance could be distinct. This has been observed in some examples of multidrug resistance, for example, overexpression of P- glycoprotein and other plasma membrane events can be particularly relevant for antitumor drug cytostatic resistance, whereas altered induction or regulation of apoptosis (programmed cell death) is particularly important in antitumor drug cytocidal resistance [16].
Perhaps relatedly, we recently reported that although decreased CQ accumulation within CQR P. falciparum is clearly related to elevated CQ IC50, it is not necessarily relevant for elevated CQ LD50 [17]. Surprisingly, much higher concentrations of CQ or fluorescent NBD - CQ can be found within parasites exhibiting cytocidal CQ resistance, even though reduced drug uptake is generally accepted to be the principle basis of CQR [17]. We have also recently found that, for both 4 amino quinolines similar to CQ and quinoline methanols similar to quinine (QN), IC50 is correlated with the ability of the drugs to inhibit hemozoin crystallization under close to physiologic conditions, but LD50 for the same drugs is not [18], [19]. Patterns of IC50 vs LD50 for a variety of quinoline drugs also suggest that the mechanisms for cytostatic vs cytocidal CQ resistance in P. falciparum are not entirely the same [6]. Taken together these data suggest that the cellular targets relevant for quinoline antimalarial drug cytocidal activities may differ from targets for cytostatic activities. Drug DV localization and drug/heme binding is the likely basis of CQ cytostatic pharmacology, but perhaps not the entire basis of CQ cytocidal pharmacology [17]–[19]. Since drug resistance is due to disruption of drug/drug target interactions, then different targets for cytostatic vs cytocidal effects predict distinct mechanisms of cytostatic vs cytocidal resistance, unless resistance is merely due to increased catabolism of the drug (which is not the case for CQR or QNR in P. falciparum).
With respect to P. falciparum CQRCS, elevated CQ IC50 and reduced parasite CQ accumulation are well correlated with mutations in the DV membrane CQ transporter PfCRT [11], suggesting that CQRCS is due to decreased drug accessibility to heme targets within the parasite DV [20]. However, resistance to the cytocidal effects of CQ is predicted to include alterations in additional targets, access to these additional targets [17], and/or to encompass mutations in key regulators of P. falciparum cell death pathways as recently hypothesized [21]. With regard to this last point, being a single celled organism, and due to the lack of caspase genes and other genes that encode key apoptosis regulators within the P. falciparum genome, it is questionable whether the canonical apoptosis pathway is the cause of drug - induced cell death for the malarial parasite [21]. Some evidence for an apoptotic - like cell death pathway for P. falciparum involving metacaspases has been presented [22]–[25] but there is disagreement on how relevant these observations are for P. falciparum death via different drugs [26]. More importantly, no molecular alterations in apoptosis have been found for CQR malaria.
These points led us to rank the progeny of the HB3 (CQS) × Dd2 (CQR) P. falciparum cross for CQ LD50 and to perform LD50– directed QTL analysis. Progeny of this genetic cross have proven invaluable to analysis of CQR phenomena [4], [8], [10], [27]. By quantifying CQ IC50 values for these progeny, a single locus on chr7 was previously identified as controlling the difference between CQR and CQS strain CQ IC50 [4]. Subsequent sequencing, in vitro drug pressure, and transfection results showed that multiple amino acid substitution mutations within a single gene in the chr7 locus, pfcrt, causes the large shift in IC50 values that has historically defined CQR and CQS status [4], [28], [29]. Allelic exchange experiments that directly replaced the “wild type” CQS associated pfcrt allele with mutant CQR associated pfcrt resulted in elevated CQ IC50 without the need to condition or select cells with CQ [30]. The degree to which CQ IC50 was elevated for these allelic exchange transfectants was very similar to that seen for highly drug selected CQR strains (70%–90% of the corresponding strain IC50 shift, see [30]), suggesting that the presence of mutant PfCRT protein was in-and-of-itself sufficient (or nearly sufficient) for conversion to a CQR phenotype. However, subsequent QTL analyses suggested that additional genetic components, such as inheritance of different chr5 loci containing mutations and varying copies of pfmdr1, may combine with PfCRT mutations in various isolates to confer the range of CQ IC50 and variable IC50 patterns for different drugs now known to exist across the globe [8]–[10], [27]. In contrast, QTL analysis in this paper shows a complete lack of the key chr5 locus previously identified for CQRCS, and identifies additional and unique genomic loci specific to CQRCC. Examination of genes in these loci suggests candidate pathways that may contribute to CQRCC. Relatedly, LD50 analysis of pfcrt allelic exchange progeny further supports our overall conclusion that although some PfCRT mutations in and of themselves confer nearly complete resistance to CQ cytostatic effects as defined by IC50 shift (see [30]), they are less important for cytocidal CQ resistance (CQRCC) as defined by LD50 shift. Using antibodies to the autophagy indicator protein ATG8 and high - resolution fluorescence microscopy, we find that a drug-induced autophagy-like cascade is dysregulated in CQRCC P. falciparum. Taken together, and considered alongside additional recent work with the related parasite T. gondii [31], our data suggest that a dysregulated autophagy-like process, combined with PfCRT mutations, promotes elevated CQ LD50 in CQR P. falciparum.
Materials and Methods
Cell Culture
P. falciparum strains HB3, Dd2, and their genetic cross progeny were acquired from the Malaria Research and Reference Reagent Resource Center (Manassas, VA), while GC03 transfectants (see [30]) were a kind gift from DA Fidock (Columbia University, New York, NY). All cultures were grown in complete media at 37°C as described [6] using type O+ human blood and human serum. Parasites were highly synchronized 3 times prior to experiments at 0, 10, and 52 hours using a solution of 5% sorbitol as described [7]. Parasites were incubated at 37°C/5%CO2 in either complete, starvation, or CQ containing media for 6 hours. When quantification of PfATG8 puncta was desired, CQ pulse was done for 6 hours using highly synchronized parasites at the mid trophozoite stage, followed by washing to remove drug as described [6].
IC50 and LD50 Quantification
Our assay for IC50 has been published previously [32] and has been used extensively and validated by many other laboratories. Our semi-high-throughput assay for LD50 quantification has recently been published elsewhere [6]. In brief, for LD50 quantification asynchronous P. falciparum cultures at 2% hematocrit and 2% parasitemia were treated with CQ in bolus fashion for 6 hr and then the drug was completely washed away [6]. After 48 hr of further incubation at 37°C under 5% CO2, Sybr Green I was added and fluorescence was measured at excitation and emission wavelengths of 485 nm and 538 nm, respectively [6]. For IC50 quantification initial parasitemia is reduced to 0.5% and cells remain in the constant presence of lower levels of drug [32]. In previous work [6] we defined differences in LD50 for synchronized vs asynchronous culture and vs other variables. LD50 and IC50 data for individual assays (each assay performed with triplicate plating of parasites treated at a given dose) were fit to a sigmoidal function using SigmaPlot 9.0 (San Jose, CA), and IC50 or LD50 values were calculated from at least three individual assays (at least 9 determinations in total) and averaged. Outgrowth in the LD50 assay 48 hr after drug is washed away represents expansion of live cells that survived cytocidal doseages of drug, as supported by staining for outgrowth after different incubation times (see [6] for detailed discussion of this point and related issues). CQ IC50 and LD50 values for laboratory strains analyzed in this paper are found in Table 1, and CQ LD50 values for HB3×Dd2 cross progeny can be found in Table S1.
Table 1. Average CQ IC50 and CQ LD50 (+/− [S.E.M.]; at least 3 separate assays in each case, each assay done in triplicate for ≥9 determinations each) for strains GC03 (CQS), Dd2 (CQR), 7G8 (CQR), C2GC03 (control CQS transfectant), C4Dd2 (CQS strain GC03 after allelic exchange with CQR Dd2 pfcrt allele), and C67G8 (CQS strain GC03 after allelic exchange with CQR 7G8 pfcrt allele).
| Strain | CQ IC50 (nM +/− [SEM]) | CQ LD50 (µM +/− [SEM]) |
| GC03 | 22.1 [1.7] | .0531 [0.005] |
| Dd2 | 206 [26.3] | 15.7 [1.82] |
| 7G8 | 161 [14.1] | 3.99 [0.62] |
| C2GC03 | 24.4 [1.9] | .096 [.008] |
| C4Dd2 | 187 [21.0] (90%) | 1.10 [0.082] (7%) |
| C67G8 | 128 [12.8] (79%) | 0.91 [0.067] (23%) |
Also shown for C4Dd2 and C67G8 are (% IC50 or % LD50, relative to Dd2 or 7G8 values, respectively). See [30] for description of the allelic exchange transfectants. Note IC50 assays are done in the continuous presence of low dose CQ for >48 hrs [33] whereas LD50 assays require higher doses of CQ given as a 6 hr bolus [6].
QTL Analysis
Using mean LD50 for each of the progeny of the HB3×Dd2 cross, genome-wide scans were run using Pseudomarker 2.04 to detect quantitative trait loci (QTL) associated with the drug response. Genome-wide significance thresholds which correct for multiple testing errors were determined by permutation testing (n = 1000 permutations). The strength of the association between a given locus and the trait (LD50) is expressed as a logarithm of odds (LOD) score. Loci that exceeded the 99th percentile (p<0.01), the 95th percentile (p<0.05), and the 37th percentile (p<0.63) were identified respectively as highly significant, significant, and suggestive QTL. Two-dimensional linear regression genome scans were run to test for potential loci interactions and joint LOD scores were calculated to identify significance interactions.
Candidate genes within QTL loci were selected as described [33]. In brief, candidate genes were selected based on four selection criteria: (1) genomic position, (2) structural polymorphisms, (3) correlation between LD50 and expression phenotypes for each parasite, and (4) gene annotations and enrichment analysis. PlasmoDB version 9.3 was used for SNP density scoring (CDS), and gene annotations. Gene enrichment analysis for biological processes and molecular functions was performed using DAVID [34]. Expression phenotypes for genes within our loci were taken from [35]. Permutation testing (n = 1000) confirmed significance of GO enriched terms within the loci of interest.
Preparation of Affinity Purified ATG8 IgG and ATG8 Monoclonal Antibody 2K19
A RACE validated cDNA encoding Toxoplasma gondii TgATG8 (Toxo.DB accession number TGGT1_003400) was used as template to amplify the full length coding region using the primers (CACCATGCCATCGATTCGCGACGAAGTGTCC and TTACCCCAGAGTGTTCTCTGAAGAGTATTCCA CGTACA). The amplicon was subcloned into pET100 establishing an in frame N-terminal hexa-His tag. Following expression in E. coli, the recombinant His-TgATG8 was purified on Ni-NTA magnetic beads (Invitrogen) and used as the immunogen to immunize a single rabbit (Cocalico, Reamstown, PA). The resulting antiserum was affinity purified using the bead immobilized crosslinked antigen and eluted using low pH.
Generation of a monoclonal antibody was contracted to AbMART.com (Shanghai, China) and accomplished using a synthetic polyprotein containing 6 tandemly arrayed epitopes. Epitopes were selected on the basis of the highest homology between TgATG8 and PfATG8. Supernatants from clones yielding >3 fold signal in an ELISA at 1∶128K dilution were screened by IFA on both Toxoplasma and Plasmodium. Multiple clones recognizing the epitope HRIRAKYPNR in Toxoplasma (HKIRSKYPNR in Plasmodium) gave excellent reactivity in both organisms. The clone 2K19 was selected for detailed work as it was found to be an IgG1 isotype (data not shown) and gave the best defined signal in both organisms.
Immunohistochemistry
For starvation treatments, cells at the mid trophozoite stage were pelleted and resuspended in HBS supplemented with 0.1 mg mL−1 hypoxanthine, 25 mM HEPES (pH 7.3), and 20 mM sodium bicarbonate. Cells were gassed and incubated at 37°C for a desired interval (typically 6 hours) before fixation. For CQ treatments, highly synchronized mid stage trophozoites were treated as described [6], [7] using drug concentrations noted in the text. Resultant cell pellets were resuspended in complete media and treated as below.
Cells were washed 3 times with 25 mM HEPES pH 7.3, fixed with 4% formaldehyde/0.0075% glutaraldehyde in PBS for 30 minutes, permeabilized with 0.1% Triton X-100 for 10 minutes, reduced with 0.3 mg mL−1 sodium triacetoxyborohydride for 10 minutes, blocked with 5% goat serum for 1 hour, and sequentially treated with antibodies (1∶500) diluted in 5% goat serum/PBS Tween-20 with PBS washes in between; antibody treatments lasted 1 hour at 37°C in the dark. For experiments involving mouse monoclonal 2K19, the primary antibody solution was prepared at 1∶500 and the secondary (typically goat anti mouse DyLight488) at 1∶500. For experiments involving antisera or purified IgG from rat or rabbit, primary solutions were prepared at 1∶500 and secondary solutions (goat anti rat AlexaFluor594 and goat anti rabbit DyLight488 or DyLight649) at 1∶500. Cells were attached to #1.5 coverslips and mounted using “Fluorogel” mounting media. Samples were imaged using a customized Perkin – Elmer spinning disk confocal microscope with 405 and 491 nm laser lines, typically at 200 ms exposure and 35% laser power [36].
Cell Fluorescence Data Analysis
Images were iteratively deconvolved using a point spread function obtained under identical imaging conditions (via doping one sample with fluorescent beads) and running multiple iterations in AutoQuantX2 [36]. Images were further processed and overlayed using Imaris 7.4.2 software. Using the “spots” routine in Imaris 7.4.2, puncta were defined and distances were measured from each spot to a single point within the DV as defined by the center of hemozoin optical density (see [36] and Scheme S1). These distances were exported to Excel and the resulting data were plotted as number of puncta vs distance from hemozoin.
Western Blot
Western blots were done as previously described [7] with slight modification. Fractionation of synchronized iRBC was as described [7]. SDS-PAGE gels (15% acrylamide) were pre loaded with lysed trophozoite-infected RBCs, electrophoresed and transferred to nitrocellulose over night at 4°C. Blots were blocked with 10% dry milk (Biorad)/PBS, washed×3 with PBS/0.1% Tween-20, labeled with rabbit anti-ATG8 antiserum (1∶10,000), washed again, and incubated with anti-rabbit HRP secondary antibody (1∶5000).
Results
Using a more rapid SybrGreen assay [32] in place of traditional 3H-hypoxanthine incorporation, our quantification of CQ IC50 for pfcrt transfectants agrees with that published previously (Table 1). Clones C4Dd2 and C67G8 [30] show approximately 8– fold and 6– fold shifted CQ IC50, relative to control transfectants (C2GC03) or the parental CQS strain GCO3 (Table 1; e.g. 187 nM/24 nM, C4Dd2 vs C2GCO3). That is, cytostatic CQ resistance for these clones is very close to that seen for laboratory strains Dd2 and 7G8 as noted earlier [30], suggesting that the Dd2 mutant PfCRT isoform and the 7G8 mutant PfCRT isoform are necessary and sufficient (or nearly sufficient) for the IC50 quantified CQR phenotypes in these widely studied drug selected strains. As hypothesized previously [30], it is perhaps possible that somewhat lower expression of PfCRT in the allelic exchange transfectants relative to the laboratory strains might be responsible for the measured IC50 shifts of 80% –90% of that observed in strains Dd2 and 7G8 (Table 1 and see also [37]).
Regardless, recently we showed that when expressed as a ratio of LD50 (dose required to kill 50% of parasites), “cytocidal” CQR was not the same as “cytostatic” CQR defined by IC50 ratios [6]. When CQ LD50 are quantified for CQR clones C4Dd2 and C67G8 as described [6] and the ratio vs control clone C2GCO3 calculated, we find that these clones are 10– fold and 8– fold cytocidal CQR (“CQRCC”), whereas strains Dd2 and 7G8 are 130– fold and 35– fold CQRCC, respectively (Table 1 and see also [6]). That is, in contrast to exhibiting 80%–90% of the CQ IC50 shifts, the allelic exchange transfectants exhibit only 7%–23% of the CQ LD50 shifts for laboratory strains harboring the same PfCRT mutant isoform (Table 1). This indicates that although mutant PfCRT protein is indeed responsible for most of the CQRCS phenotype (measured by CQ IC50), it makes a smaller contribution to the CQRCC phenotype (measured by CQ LD50) observed in the same strains.
Thus, we leveraged a QTL mapping approach within an available genetic cross to compare genetic profiles of the two phenotypes and to search for additional genetic loci contributing to CQRCC. We quantified CQ LD50 for progeny of the HB3 (CQS) × Dd2 (CQR) cross using our recently described high throughput assay ([6]; see Table S1 for CQ LD50 values). We were able to identify three major QTL loci associated with LD50. First, similar to CQ IC50– directed QTL [4] the chr7 locus harboring pfcrt segregates with the elevated LD50 phenotype (not shown, see [4]). The LOD score (∼21) is lower than the LOD score when IC50 is the measured phenotype (∼40; see [4], [10]). We emphasize that the magnitude of the LOD score reveals how well a locus segregates with a phenotype across a given population (in this case, progeny from the HB3 × Dd2 cross), but does not define the relative biochemical contribution of proteins encoded by that locus to the phenotype in question. Behavior of pfcrt transfectants that isolate PfCRT mutations in a common genetic background shows that the biochemical contribution of mutant PfCRT to elevated CQ LD50 is lower relative to its contribution to elevated CQ IC50 (Table 1), nonetheless, PfCRT mutations remain a prominent contribution to the mechanism of elevated LD50.
Second, two new chromosomal loci, not previously associated with any CQR phenomena, are associated with elevated CQ LD50 in CQR progeny of the HB3 × Dd2 cross (Fig. 1). One is a novel contribution from chr6 (0 cM –17.3 cM) that can increase CQ LD50 for CQR progeny of the cross (Fig. 1B). Furthermore, this locus interacts with chr8 (77.5 cM) with significant additive effects (Fig. 1E). In addition to defining a genetic architecture for CQ LD50 that is distinct from that previously defined for CQ IC50 (Fig. 1C), the loci contain genes that may contribute to CQRCC but that are not involved in CQRCS. In contrast, QTL scans of IC50 for the same CQR progeny identify a segment of chr5 (carrying the pfmdr1 amplicon) as contributing to elevated IC50 (Fig. 1A, [10]), and as described previously [10], pairwise interaction between the chr5 segment and the chr7 locus is associated with elevated IC50 in the CQR progeny (Fig. 1D). Results from many studies show that the relevant gene in this segment of chr5 is likely pfmdr1 [38]–[41]. However, importantly, LD50 - directed QTL analysis does not identify this chr5 fragment (Fig. 1C) or the chr5×chr7 pairwise interaction. Instead, it isolates a fragment of chr6 (Fig. 1B), and defines a chr6 × chr8 pairwise interaction relevant for elevated LD50 (Fig. 1E). In sum, these data force us to conclude that the LD50 and IC50 phenotypes share a critical feature (mutant PfCRT found on chr7) but are otherwise genetically distinct.
Figure 1. LD50 vs IC50 directed QTL analyses for CQR HB3 × Dd2 cross progeny.

A) IC50 QTL scan for CQR progeny shows a peak on chr5 (asterix) that encompasses pfmdr1 as previously described [10]. Notably, the chr6 locus that is pertinent for the LD50 scan (see Fig. 1B) does not pass the suggestive threshold on this IC50 scan. B) LD50 QTL scan for CQR progeny shows a peak on chr6 (asterix; L.O.D. = 2.5, passing the suggestive threshold). The locus that includes pfmdr1 does not pass the suggestive threshold for this scan (see also Fig. 1C). C) To more clearly highlight the differences in genetic architecture for LD50 vs IC50 phenotypes, an overlay of the two QTL scans is shown. The CQR progeny LD50 QTL scan is shown in blue, while the CQR progeny IC50 QTL scan is in black. The overlay shows quite clearly that the pfmdr1 locus does not factor at all into the LD50 phenotype, and that the chr6 locus does not factor at all into the IC50 phenotype. Thus the IC50 and LD50 phenotypes are genetically distinct. D) Similarly, the interaction locus between chr6 & 8 (see text) does not appear on a IC50 pair-wise scan. However, additive effects between chr5 and chr7 loci are seen, as previously reported [10]. E) Pair-wise scan of the CQR progeny shows that chr6 and chr8 loci (circle) have additive effects on LD50 (L.O.D. = 4.3).
Our interpretation is that the CQRCC and CQRCS mechanisms overlap (each requires mutant PfCRT), but that the relative contribution of PfCRT to CQRCC is less than, or mechanistically distinct from, its contribution to CQRCS (Table 1) and that high level CQRCC requires additional factors (Fig. 1B, 1E). The question then becomes how might genes in chr7, chr6, and chr8 loci act together to elevate CQ LD50 up to 250 fold (Table S1; note CQ LD50 = 32 µM for 3BA6), when Dd2 mutant PfCRT in and of itself only appears to elevate CQ LD50 10 fold (Table 1)?
Inspection of the chr6 and chr8 loci reveals multiple genes involved in processes linked to “vesicle traffic”, “proteasome function/proteolysis” and “lipid metabolism” (Table S2 & S3). For example, at least 6 genes within the chr8 locus encode proteins putatively involved in the proteasome pathway (Table S3). Candidate genes were also ranked based on the sequence similarity of strain HB3 (CQS) and strain Dd2 (CQR), and the correlation of expression levels with the LD50 phenotype (see Tables S2 & S3). Thus we used four previously vetted methods [33] to identify genes or pathways that are most likely relevant to LD50. Interesting genes within the chr6 and chr8 loci that have high CDS scores include putative orthologues of autophagy genes ATG11 and ATG14, several key metabolic regulators, multiple kinases, and a putative E3 ubiquitin ligase (Table S2 & S3). Also of note are several genes in the chr6 locus linked to response to oxidative stress. Due to the nature of chromosomal fragment inheritances in the HB3 × Dd2 cross, the chr6 × chr8 additive effect defines a much smaller region of the chr6 segment. This segment harbors only 20 genes, with 7 of those encoding proteins involved in lipid metabolism (see Discussion).
Elevated LD50 indicates resistance to cell death. Cell death is often mediated by signal transduction that controls a programmed cell death (PCD) pathway [21]. Importantly then, we find that the chr6 and chr8 loci do not harbor any candidate Pf metacaspases [23] or other molecules that typically regulate apoptotic PCD. That is, we find no genetic evidence from the HB3 × Dd2 cross for atypical apoptosis related to CQRCC. Autophagy (“self eating” upon starvation or stress) is an alternate pathway that has been linked to cell death for several cell types [42]–[44], including the related apicomplexan parasite T. gondii [31], [45]. It is an orchestrated, vesicle mediated, proteolysis/degradative pathway that is distinct from apoptosis. Since apoptosis genes were not found in chr6 or chr8 loci, and since “vesicle traffic”, “proteasome/proteolysis”, “lipid metabolism”, “oxidative stress” and autophagy pathways often overlap mechanistically (see Discussion), we wondered if altered autophagy might be related to LD50.
However, no experiments to our knowledge have been done to test if autophagy occurs in intraerythrocytic P. falciparum. The universal stimulus for autophagy is starvation, which also unambiguously induces cell death. An unambiguous feature of induction of autophagy in eukaryotes is redistribution of ATG8 protein (called LcIII in mammals) from a more localized and diffuse pattern to a more widely disbursed, more punctate pattern that defines the sites of autophagosome formation and/or recruitment of autophagosomal “cargo” [46]. Specific ATG8 antisera raised vs. T. gondii ATG8 protein (anti TgATG8) show excellent cross reactivity vs P. falciparum ATG8 because PfATG8 is ∼70% identical to TgATG8 [21]. We easily identify a PfATG8 doublet at predicted masses of 15 & 17 kDa in immunoblots of fractionated parasites (Fig. 2D) that is consistent with well known de-lipidated and lipidated forms of ATG8 [46]. IFA analysis using ATG8 antiserum suggests an autophagy-like process is active in intraerythrocytic P. falciparum trophozoites (Fig. 2,3). As expected, control iRBC trophozoite parasites grown in complete media show a cytosolic PfATG8 distribution (Fig. 2A, green) that appears somewhat punctate, perhaps due to at least partial localization to the apicoplast (see below and [47]). However, notably, when highly synchronized trophozoites are placed in starvation medium for 6 hrs, PfATG8 is redistributed in a much more expanded punctate fashion (Fig. 2B, green), as recently reported [21]. Closer inspection reveals puncta at the parasite periphery, possibly near the RBC membrane. Co – staining with a marker for Maurer’s cleft (anti - PfREX1, red) shows that some PfATG8 appears to be routed to very near Maurer’s cleft (MC) (yellow dots, Fig. 2B middle). The well - characterized inhibitor of autophagy, 3-methyl adenine (3-MA) partially reverses the starvation induced PfATG8 puncta redistribution (Fig. 2C), similar to what has recently been found for T. gondii [31]. Affinity purified IgG from the antisera as well as monoclonal antibody 2K19 raised against a highly conserved Apicomplexan ATG8 motif (see methods) yield results similar to polyclonal TgATG8 antisera (Fig. 3). Another recent report [47] presents data consistent with some localization of PfATG8 to the parasite apicoplast for control schizonts growing in normal media. We also obtain data consistent with partial (but not exclusive) localization of PfATG8 to the apicoplast for control late trophozoites/early schizonts, by co – staining for apicoplast – specific PfACP protein (see Fig. S1). We note that the trophozoite (feeding) and schizont (nuclear division/parasite replication) stages of parasite development would be expected to utilize autophagy machinery in different ways [48], [49] and that further study of PfATG8 in trophozoites vs schizonts is warranted.
Figure 2. PfATG8– positive puncta.

Shown are puncta for (A) control HB3 iRBC grown under normal culture conditions (B) HB3 iRBC grown for 6 hours under starvation conditions (see Methods) and (C) HB3 iRBC grown under starvation conditions plus the autophagy inhibitor 3 methyl adenine (3 MA). Shown are transmittance (left), immunofluorescence vs antiPfREX1 (Maurer’s cleft marker; red; second column), immunofluorescence vs antiTgATG8 (cross reacts with PfATG8; green, third column) and overlays (right). Bar = 5 µm. Also shown (D) are western blot data for iRBC harboring HB3 (CQS) and Dd2 (CQR) trophozoites grown under control culture conditions. Two separate gels for two independent sets of samples (two iRBC isolations for each culture) are shown. We note our data show a clear doublet at 15 and 17 kDa, similar to all other studies of eukaryotic ATG8 protein of which we are aware except one [47], which resolves only a single band instead of the usual doublet with a polyclonal antisera raised against a recombinant GST-PfATG8 fusion. We suggest three possible reasons for the discrepancy: 1) we use higher density [15% acrylate] gels relative to [47] in order to resolve the low mass doublet, 2) we do not solubilize parasites with saponin as in [47] which would release de - lipidated ATG8 into wash supernatant, 3) perhaps abundance of one PfATG8 species (presumably de lipidated) is higher in trophozoites relative to schizonts examined in [47].
Figure 3. Comparison of antisera vs monoclonal Ab staining.

Shown are results for TgATG8 antisera (bottom row) vs staining using a monoclonal antibody raised vs a highly conserved Apicomplexan ATG8 epitope (top row, see methods). Panels A,G,D,J are transmittance, B,H,E,K are ATG8 fluorescence, and C,I,F,L are overlay, respectively. Left side is control intraerythrocytic HB3 P. falciparum; right side is HB3 starved for 6 hrs as described in methods. Bar = 5 µm.
We next tested if cytocidal levels of CQ induced similar PfATG8 redistribution. Indeed, CQS parasites show similar extensively distributed PfATG8 puncta when they are treated at 2×LD50 cytocidal dose of CQ (Fig. 4A), but not when they are treated with 2×IC50 cytostatic dose (Fig. S2). Thus the autophagy – like cascade induced by starvation is also involved in the response to cytocidal levels of CQ, but not response to cytostatic levels. When CQR parasites are treated with the same absolute dose (2×CQS LD50 dose; 250 nM), we do not observe the punctate redistribution of PfATG8 (see below). However, when CQR parasites are treated with 2×CQR parasite LD50 dose (e.g. a similar effective pharmacologic dose, ∼32 µM for strain Dd2, see [6]) some PfATG8 redistribution is observed (Fig. 4B), but the ATG8 response appears somewhat muted.
Figure 4. PfATG8 (green) re distribution, CQ vs time.

CQS (strain HB3; top panel A) and CQR (strain Dd2; bottom panel B) parasites were grown in control media (left column) or in media plus 2×LD50 dose of CQ (250 nM for HB3 Fig. 4A, 30 µM for Dd2, Fig. 4B) for 2, 4 or 8 hrs (column 2,3,4 respectively). The top row in panels A and B is transmittance, the second (green) is staining for PfATG8, the third is DAPI to visualize the single trophozoite nucleus, and the bottom row for each panel presents the overlay. Fluorescence acquired at 35% power, 200 ms, 642 nm; emission 700/75 nm dichroic, 690 nm cut-off. Bar = 5 µm.
To quantify this behavior, we devised a method based on spinning disk confocal microscopy and 3D Imaris rendering of z stacks to plot radial distributions of PfATG8 puncta relative to hemozoin optical density (Fig. 5). Essentially, very optically dense hemozoin within the DV is used to define a “center” point of reference for the iRBC parasite (Fig. 5 left) and distinct, clearly defined spots of ATG8 fluorescence (Fig. 5 middle, see also Scheme S1) are then measured for their relative distance (x,y,z) from the center of hemozoin optical density (white lines, Fig. 5 right). Using these methods, we quantified PfATG8 puncta abundance at >3.5 µm for CQS HB3, CQR Dd2 and C4Dd2 parasites +/− starvation and across a range of bolus CQ dosages (Fig. 6). These puncta distributions show that starvation produces similar number and similar radially distributed patterns of peripheral PfATG8 puncta for CQS and CQR parasites (compare far left and far right bars in each panel Fig. 6), but that 2×CQS LD50 dose of CQ (250 nM) only produces high numbers of distal radially - distributed puncta for CQS strain HB3 (Fig. 6A top), not for CQR strain Dd2 (Fig. 6B middle). C4Dd2 transfectants, wherein CQR is mediated solely by allelic exchange with CQR associated mutant pfcrt, show PfATG8 behavior that is intermediate relative to CQS HB3 and CQR Dd2 (Fig. 6C bottom), and again, Dd2 shows a muted ATG8 response (hashed bars indicate 2×LD50 dose for each strain).
Figure 5. Semi automated computational method for quantifying the distribution of ATG8 puncta relative to hemozoin crystals within the DV.

(A) Hemozoin within the DV is detected by transmittance, the outline of <10% transmittance is defined (labeled “DV”, left panel) and the center of this outline then defined by voxel analysis using Imaris software. (B) The center of distinct ATG8 puncta (bright green dots) are labeled using the option ‘Add new measurement points’ in the Imaris “spots” subroutine. (C) Distances from the hemozoin center to the ATG8 puncta centers are then computed (white lines) and data collated in excel.
Figure 6. Quantified PfATG8 puncta distribution for synchronized trophozoite parasites.
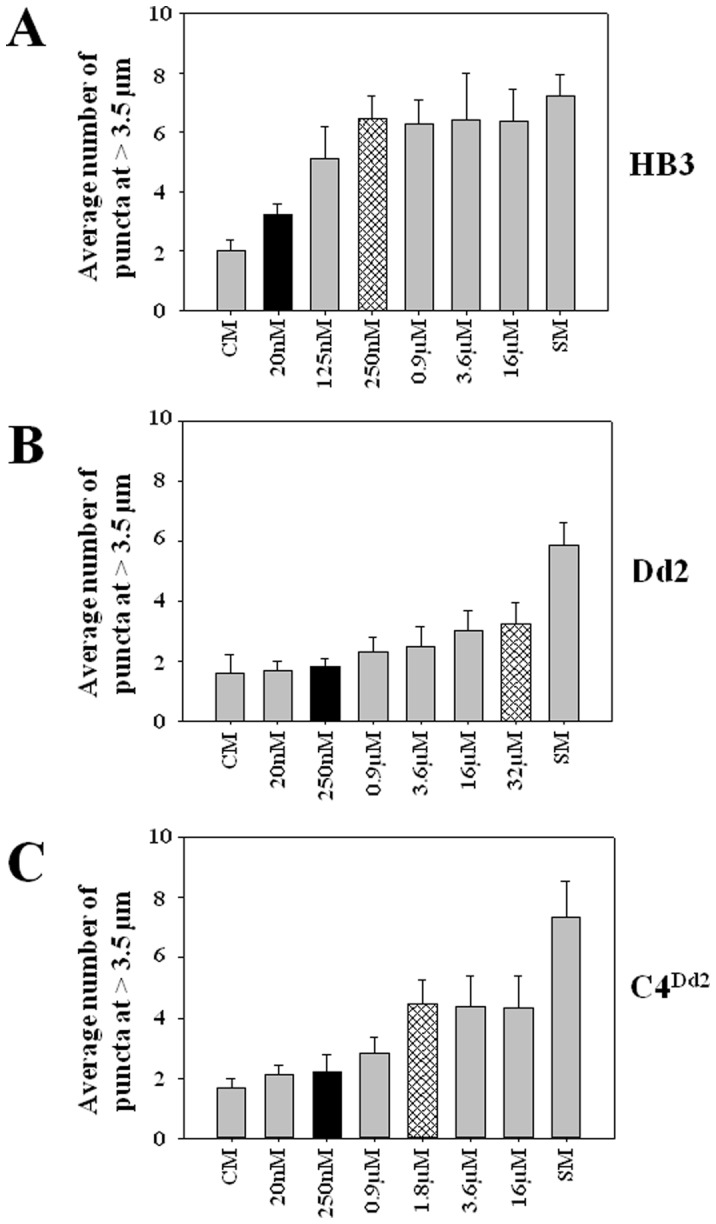
Shown are CQS HB3 (top) CQR Dd2 (middle) and transfectant C4Dd2 (bottom) under different conditions. Far left, “CM” = control culture conditions, far right, “SM” = iRBC in starvation media for 6 hr. In between are puncta quantified for iRBC treated for 6 hrs with the indicated [CQ]. Black bars in each panel denote 2×IC50 [CQ] for the strain, hashed bars denote 2×LD50 [CQ] for the strain. Data are the average of at least 20 iRBC, +/− s.d., and puncta that are ≥3.5 µm from DV hemozoin optical density are plotted. The three phenotypes are distinct, as evidenced by statistical comparison of results upon 250 nM and 3.6 µM treatments common to all three strains; for 3.6 µM data, HB3 vs Dd2, HB3 vs C4Dd2, and Dd2 vs C4Dd2, P value <0.05 in each case. For 250 nM data, HB3 vs Dd2 or HB3 vs C4Dd2, P value <0.05, but Dd2 vs C4Dd2>0.05. That is, as proposed in the text, C4Dd2 is intermediate relative to HB3 and Dd2: both Dd2 and C4Dd2 differ from HB3, but C4Dd2 shows behavior similar to Dd2 at lower dose CQ, but different behavior vs Dd2 at higher dose.
We further tested the association between peripherally distributed PfATG8– positive puncta and CQR status by examining 4 other well-characterized strains, two CQS (strains 3D7 and Sudan 106) and two CQR (strains FCB and 7G8). Upon starvation for 6 hr, all 4 strains distribute PfATG8– positive puncta in a peripheral fashion similar to starved HB3, Dd2, and C4Dd2 (Fig. 7). However, as shown in Fig. 7, only the CQR strains show no increased peripheral puncta at 250 nM CQ (2×CQS LD50) whereas both CQS strains do. Also, even though 2×CQR LD50 (32 µM) induces formation of peripheral puncta for the CQR strains, similar to CQR strain Dd2 (Fig. 6), the numbers are reduced relative to 2×CQS LD50 treated CQS. To summarize, 3 CQR laboratory strains behave similarly, and are distinct from all 3 CQS strains examined. In terms of statistical significance of these data, P<0.01 when puncta produced upon 250 nM CQ exposure for any CQS strain (HB3, 3D7, S106) is compared vs 250 nM CQ puncta data for any CQR strain (Dd2, FCB, 7G8). However, P values are >0.4 when any two CQR or any two CQS strains treated with the same dose are compared. That is, no CQR strains are significantly different from each other, no CQS strains are significantly different from each other, but all CQR strains show statistically significant differences in PfATG8 redistribution vs all CQS strains. This suggests that redistribution of PfATG can serve as a marker to quantify sensitivity to cytocidal CQ.
Figure 7. Quantified PfATG8 puncta at ≥3.5 µm from hemozoin for synchronized trophozoites.

Two additional CQR and two additional CQS strains are analyzed. Shown are puncta counts for at least 20+/− s.d. grown under control conditions (“CM”, left side each panel), upon starvation (SM; far right, each panel) and upon dosing for 6 hrs with either 2×IC50 or 2×LD50 concentrations of CQ (50 nM and 250 nM for CQS strains, and 250 nM and 32 µM for CQR strains, respectively).
We next inventoried sequences of all P. falciparum orthologues of autophagy pathway proteins that we can identify at this time (see also [50]) using available genomes, and note that a number of putative orthologues of key autophagy proteins are mutated in Dd2 (CQR) vs HB3 and 3D7 (CQS) parasites (see Discussion). For example, a recently identified P. falciparum PI3’ kinase (PF3D7_0515300; [51]) appears to us to be a Vps34 orthologue, based on its very high identity to ScVps34 (E-value = 1×10−73; homology index score = 567). A similar conclusion was also reached by Kitamura et al. [47]. During autophagy in other eukaryotic cells, Vps34 produces copious PI3P at the developing autophagosome, where ATG8 protein also localizes. PfVps34 from CQR strain Dd2 shows point mutations and deletions relative to CQS strains HB3 and 3D7 that could conceivably affect function or regulation, as described in Discussion. Quite interestingly then, this enzyme has recently been localized to near Maurer’s cleft organelles [51], very similar to the PfATG8 relocalization upon starvation that is reported here. It remains to be determined if these additional autophagy gene mutations uniformly segregate with CQRCC phenomena for geographically distinct isolates of CQR P. falciparum. Importantly, LD50 quantification for these isolates will need to be performed, which will require the ability to culture these isolates over long time periods.
Discussion
Data in this paper reveal three important concepts:
Mutant PfCRT protein, while responsible for the majority of the shift in CQ IC50 that has traditionally characterized CQR P. falciparum strains Dd2 and 7G8 ([30], and present data), is only responsible for 10–20% of the shift in CQ LD50 for the same strains of CQR parasites. PfCRT is thought to confer CQRCS by reducing access of CQ to DV – localized heme target and thereby reduce the ability of the drug to inhibit heme – hemozoin biomineralization [11], [29]. However, recent data [17] shows that DV levels of CQ are not necessarily correlated with CQ LD50. In addition, other recent work [18], [19] shows that the ability of CQ or QN analogues to inhibit hemozoin formation correlates with their IC50 activity, but not with their LD50 activity. Thus our observations are not as surprising as they might initially appear, and one important implication is that targets in addition to inhibition of hemozoin crystallization are likely relevant for the cytocidal activity of quinoline antimalarial drugs.
LD50– directed Quantitative trait loci (QTL) analysis in the well-characterized HB3 × Dd2 genetic cross shows that the chr5 segment previously identified as modulating PfCRT mediated CQRCS does not contribute to CQRCC. Other distinct differences in genetic architecture include two novel loci, on chr6 and chr8, associated with elevated LD50. These loci are enriched for genes that encode proteins linked to proteosome and autophagy pathways, but no genes encoding metacaspases or other proteins that regulate apoptosis are found in the loci (Tables S2– S7). A number of recent observations suggest interesting cross talk between proteasome and autophagy pathways (e.g. [52], [53]), thus enrichment for both processes in the LD50 loci is intriguing.
Staining for a definitive marker of autophagy (ATG8 protein puncta) shows cell death from LD50 CQ treatment is linked to an autophagy – like process and that this process is altered in CQR parasites. Interestingly, we find that starvation and CQ – induced death lead to accumulation of PfATG8– positive puncta to near Maurer’s clefts (MC). These poorly understood organelles are known to be involved in export of proteins from the parasite to the red cell plasma membrane but they are also believed to have several other functions, including vesicle fusion [54].
To our knowledge, only three studies have attempted to distinguish CQ cytostatic and cytocidal activities in vitro [5], [6], [55] but only recently have LD50 been quantified [6], allowing QTL analyses as presented here. All previous cellular quantification of antimalarial drug resistance phenomena of which we are aware has been solely via IC50 ratios. IC50 does not a priori define cytocidal (parasite kill) potency; formally it defines growth inhibition potency vs mass populations of parasites. It is certainly true that killing some parasites over time with a drug prevents growth seen in control cultures, but it can also be true that a drug in continuous culture does not kill parasites, but merely slows the cell cycle, changes multiplicity of schizogony, or has other effects that yield IC50 of a given value for mass populations of parasites [7]. CQ appears to exhibit both dose dependent (“Cmax”) ([7] and this work) and time dependent (“TMIC”) [56] parasite killing. In studies that report IC90 data there may be greater “mixing” between CQ cytostatic and cytocidal effects relative to studies that report IC50 data. In this paper, we have made the first attempt to our knowledge to more fully separate CQ IC50 and LD50 phenomena for progeny of the HB3×Dd2 cross.
We previously found that some strains are much more CQRCC than CQRCS [6], and that the rank order of LD50 and IC50 for different drugs varies (meaning the drugs to which a parasite shows the highest IC50 are not necessarily the drugs to which it shows the highest LD50 [6]). Plotting CQ IC50 [4] vs CQ LD50 [Table S1] for HB3×Dd2 progeny yields poor correlation (r2<0.4; not shown). Also, a recent study shows that CQ transport ability for 13 mutant PfCRT isoforms found in 13 different CQR strains does not correlate with CQ IC50 for those strains [57]. Considering these observations along with the current data suggests that parasite resistance to cytocidal effects of CQ is influenced by additional genetic or physiological events, along with PfCRT mutations. Our initial analysis suggests these events include alterations in a novel pathway showing some similarities to autophagy.
In all eukaryotic cell types examined, the re distribution of ATG8 protein to more widely disbursed puncta marks the induction of autophagy by starvation [46] . In all other examples, the membranes to which ATG8 is routed are synthesizing double membraned autophagosomes and copius phosphatidyl inositol 3′ phosphate (PI3P) via Vps34. It is striking then that previous work completely unrelated to the present study places PfVps34 near MC, similar to our localization for some re – routed PfATG8 [51]. In all eukaryotic cell types examined, autophagosomes containing high levels of ATG8 and PI3P lipid then engulf cytosolic or organellar targets, fuse with lysosomes, and the contents are then degraded, serving as nutrient pools that temporarily keep the starving cell alive. In the case of P. falciparum, the parasite trophozoite undergoes heightened accumulation of PfATG8– associated vesicles at or near the MC upon starvation and cytocidal CQ treatment. The parasite trophozoite does not appear to engulf and degrade its sole mitochondrion to provide additional food upon starvation. Indeed, starvation induced mitochondrial fragmentation by autophagy in T. gondii causes cell death [31]. Instead, the unique properties of the RBC, which is devoid of de novo biosynthetic activity as a host cell, necessitates enhanced endocytosis to acquire extracellular food. We suggest that under starvation pressure the parasite up regulates additional endocytosis from the red cell cytosol using (at least in part) the vesicle formation and fusion functions of encoded autophagy machinery. This might be consistent with PfATG8 vesicles eventually interacting with the parasite DV, analogous to ATG8 positive vesicle fusion to lysosomes in other eukaryotes after they recruit nutritional “cargo”.
In yeast and higher eukaryotes, membrane association of ATG8 is mediated by lipidation. The terminal G residue of ATG8 that becomes lipidylated is blocked, necessitating proteolytic cleavage by ATG4 as a prelude to membrane association [46]. Notably however, P. falciparum PfATG8 terminates with G, thus ATG4 proteolysis to reveal G as in other species is absent, suggestive of constitutive membrane association [49]. In reality however, PfATG8 exists in both the unlipidated and lipidated forms (Fig. 2D) suggesting mechanisms other than ATG4 regulate PfATG8 dynamics. We propose that a low level of constitutively activated autophagy is present in iRBC parasites, and that CQR parasites have developed resistance to CQ induced perturbations in autophagy. Accumulation of PfATG8 puncta upon toxic CQ treatment is consistent with either upregulation of puncta formation or inhibition of autophagosome fusion, so CQR parasites could in theory have perturbations in either (or both) steps of the autophagy pathway.
Interestingly then, CQ is a known potent inhibitor of autophagy in other cell types. Its diprotic weak base character promotes profound accumulation in acidic compartments such as lysosomes, autophagosomes, and vacuoles. At doses that correspond to these LD50 CQ is known to block the fusion of autophagosomes with lysosomes/vacuoles and also raises the pH of these compartments, thereby inhibiting processes that require acidic pH (e.g. intra lysosomal proteolysis ([42], [44] and references within)). A few molecular possibilities specific to P. falciparum are that LD50 dosages of CQ (i) block the fusion of endocytic vesicles carrying hemoglobin to the DV (in fact, a somewhat overlooked paper [58] shows that CQ LD50 doses do indeed lead to a buildup of undigested Hb trapped within arrested parasite vesicles), (ii) inhibit falcipain and plasmepsin activity by raising the pH in endocytic vesicles and/or the DV, (iii) inhibit fusion of autophagosomes and/or other vesicles with their target organelles. Other lysosomotropic agents would be expected to mimic this CQ pharmacology. Interestingly then, certain alkaloids that inhibit autophagy also show antimalarial activity [59], [60]. One example is voacamine, a tertiary alkaloid isolated from Peschiera fuchsiaefolia stem bark that shows good antimalarial activity (238 ng/mL vs strain D6 and 290 ng/mL vs strain W2), and which has also been reported to chemosensitize MDR cancer cells in an autophagy-dependent manner [61]. Overall, since P. falciparum has been subjected to decades of cytocidal CQ selective pressure, it is logical that the parasites would evolve resistance to CQ autophagy inhibition.
With regard to the starvation effects that we observe, work in the Goldberg laboratory has shown that P. falciparum meets its amino acid requirements by a combination of hemoglobin degradation and uptake of free amino acids from the medium [62]–[64]. When some extracellular amino acids are removed, the parasite responds by up – regulating additional hemoglobin transport and degradation; hemoglobin, however, lacks the essential amino acid Ile, so parasite survival is conditional under these circumstances [63]. Conversely, if the hemoglobin pathway is inhibited, the parasite survives by acquiring additional amino acids from the extracellular medium. If Ile is withdrawn the parasite can enter a hibernatory state [64]. These observations suggest that (i) malaria parasites are able to sense amino acid levels in the medium and (ii) they possess a system that can respond to the lack of some extracellular amino acids by regulating intracellular transport to the vacuole. During starvation induced autophagy, other eukaryotic cells respond to low amino acid levels in the medium by trapping cytosolic material in transport vesicles, which will eventually fuse and release cargo into a lysosome or vacuole to then be digested to amino acids. Although for intraerythrocytic P. falciparum the “cargo” is presumably within the host cell cytosol, starvation induced autophagy reported here could be somewhat reminiscent of elevated hemoglobin endocytosis in P. falciparum.
We wondered if autophagy genes in the identified chr6/chr8 loci might be hinting at mutations in other Pf autophagy gene (PfATG gene) orthologues for CQR parasites. A partial inventory of PfATG gene orthologues has been published [50] but does not include important co factors such as Vps34, Pex14, Vps15, etc. We re – queried the Pf genome with a more complete set of 42 autophagy related genes. Remarkably, a number of candidate orthologues (16 of 42) lie within QTL loci previously associated with drug resistance phenomena, or within “eQTL” gene sets that are up/down regulated in trans by resistance associated eQTL [35]. Also, after alignment of Dd2 (CQR) vs HB3 and 3D7 (CQS) alleles via Broad Institute data (http://www.broadinstitute.org/), we find that many candidate PfATG gene orthologues are mutated in the Dd2 CQR genome relative to 3D7 or HB3 CQS. One in particular is candidate PfVps34 (PF3D7_0515300; E-value 10−73 and homology index score 567 relative to ScVps34) which encodes K at codon 423 for Dd2 in place of Q found for HB3 and 3D7, deletion of a N residue found at position 578 for HB3 and 3D7, and a deletion of 24 codons relative to HB3 and 3D7 (from codon 931 to 955). Interestingly, this deletion is within what we predict is a C2 regulatory domain that presumably binds Ca2+; a similar C2 domain prediction for this protein has also been made by two other groups [51], [65]. Since a clear TOR orthologue is missing within the P. falciparum genome [50] altered regulation via mutant PfVps34 is one possibility for the altered cascade seen in CQR strain Dd2.
Regardless, we predict that multiple routes to dysregulated autophagy will be found for various strains and isolates of CQRCC P. falciparum, analogous to how multiple routes to dysregulated programmed cell death are found in various multidrug resistant tumor cell lines [16]. At LD50 doses, via its well known lysosomotropic behavior [58], [66] CQ will inhibit vesicle formation and vesicle fusion, two processes that are essential to autophagy. Since these processes are controlled by a number of proteins, many mutations are possible for altering CQ response.
In sum, we find that mutant PfCRT protein confers the majority of CQRCS as defined by CQ IC50 shift, but only partial CQRCC as defined by LD50 shift. Not surprisingly then, our data also show that distinct genetic architecture is associated with CQRCS vs CQRCC, that a specialized P. falciparum autophagy cascade is induced by LD50 doses of CQ but not IC50 doses of the drug, and that altered regulation of this unique autophagy – like process is present in CQRCC parasites. Consistent with a clear role for autophagy in cell death for other apicomplexa [31], [45], altered PfATG8 redistribution indicative of a novel autophagy – like cascade appears to be associated with CQRCC for intraerythrocytic P. falciparum trophozoites. Elucidating additional molecular events controlling perturbation of autophagy signaling for CQR P. falciparum, potentially involving at least two dozen autophagy gene orthologues [50], should prove to be a fertile area of future research. Among additional data needed to elucidate the pathway further are precise measurements of ATG8 puncta flux, meaning the rate of ATG8 vesicle production vs presumed fusion with either MC and perhaps the DV.
Finally, regardless the exact role of the novel autophagy – like process for iRBC trophozoites that we describe here, we propose that CQRCC requires prior acquisition of CQRCS, meaning that parasites must first be able to survive lower IC50 dose via PfCRT mutations, before they can survive higher LD50 dose via acquiring additional autophagy mutations. Precise measurements of IC50 vs LD50 phenomena among geographically diverse isolates of CQR P. falciparum will further test this hypothesis. Also, since PvCRT mutations have not been found for CQR P. vivax, it should prove interesting to investigate the status of genes in the chr6 and chr8 loci for CQR P. vivax isolates, as well as the formation of PvATG8 positive structures in response to CQ treatment.
Supporting Information
Late trophozoite/early schizont stained with anti-ATG8 (green) and anti-apicoplast (red) antibodies. Some overlap between ATG8 and apicoplast localized ACP (D) suggests partial (but not complete, ∼25%) co – localization of PfATG8 and apicoplast as previously suggested [46]. Scale bar = 5 µm.
(DOC)
CQS (HB3, A) and CQR (Dd2, B) parasites treated with IC50 (top row each panel) and 2× IC50 (bottom row) doses of CQ (10, 20 nM and 125, 250 nM, respectively) for 48 hr. Parasites were then stained for ATG8 (green) and with DAPI (blue), as described in Methods. Very few peripherally disposed PfATG8 puncta are observed, supporting the conclusion that puncta formation is associated with LD50, but not IC50 doses of CQ. Scale bar = 5 µm.
(DOC)
LD50 values for the HB3×Dd2 cross progeny.
(DOC)
All genes within the chr6 LD50 locus.
(DOC)
All genes within the LD50 chr6×chr8 interaction.
(DOC)
GO enriched molecular functions for the chr6 LD50 locus.
(DOC)
GO enriched biological processes for the LD50 chr6 locus.
(DOC)
GO enriched molecular functions for the LD50 chr6×chr8 interaction.
(DOC)
GO enriched biological processes for the LD50 chr6×chr8 interaction.
(DOC)
Cartoon representation of 3D puncta quantification method used in the present work. Hemozoin density (red “x”) is used as the origin, and distances toPfATG8 positive puncta are defined for the reconstructed confocal z stack of images, relative to hemozoin, using 3D Cartesian (x,y,z) coordinates. The cartoon shows an abbreviated depiction of 3 SDCM “slices”, but as described in methods the z stack data set for each cell is a fully assembled, iteratively deconvolved 3D image constructed from approximately 15–20 z slices (see [35] for additional detail).
(DOC)
Acknowledgments
We thank Dr. Don Gardiner (Malaria Biology Laboratory Queensland Institute of Medical Research) for anti PfREX-1 antibody, Dr. Sean Prigge (Johns Hopkins University) for rat anti PfACP antibody, Ms. Chardai Gray and Lauren Jacobs for help with bioinformatics, and Mr. Paul Callaghan, Ms. Katy Sherlach and Dr. Nick Baro for helpful discussions. The following Plasmodium falciparum strains were obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH: HB3, Dd2, 7G8, and the progeny of the HB3×Dd2 cross, deposited by Drs. TE Wellems, D Walliker, DE Kyle, and TE Wellems, respectively. We also thank Dr. David A. Fidock (Columbia University, New York, NY) for providing C2GC03, C4Dd2, and C67G8.
Funding Statement
This work was supported by National Institutes of Health (NIH) grants: NIH RO1 AI045957 (to PDR); NIH RO1 AI049367 (to APS); NIH RO1 AI071121 (to MTF); and Kentucky Science and Education Foundation grant: KSEF-2624-RDE015 (to APS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cheeseman IH, Miller BA, Nair S, Nkhoma S, Tan A, et al. (2012) A major genome region underlying artemisinin resistance in malaria. Science 336: 79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plowe CV, Cortese JF, Djimde A, Nwanyanwu OC, Watkins WM, et al. (1997) Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase and epidemiologic patterns of pyrimethamine-sulfadoxine use and resistance. J Infect Dis. 176: 1590–1596. [DOI] [PubMed] [Google Scholar]
- 3. Wang P, Read M, Sims PF, Hyde JE (1997) Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization. Mol Microbiol. 23: 979–86. [DOI] [PubMed] [Google Scholar]
- 4. Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, et al. (2000) Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 4: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Young RD, Rathod PK (1993) Clonal viability measurements on Plasmodium falciparum to assess in vitro schizonticidal activity of leupeptin, chloroquine, and 5-fluoroorotate. Antimicrob Agents Chemother. 37: 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paguio MF, Bogle KL, Roepe PD (2011) Plasmodium falciparum resistance to cytocidal versus cytostatic effects of chloroquine. Mol Biochem Parasitol. 178: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gligorijevic B, Purdy K, Elliott DA, Cooper RA, Roepe PD (2008) Stage independent chloroquine resistance and chloroquine toxicity revealed via spinning disk confocal microscopy. Mol Biochem Parasitol. 159: 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferdig MT, Cooper RA, Mu J, Deng B, Joy DA, et al. (2004) Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol. 52: 985–997. [DOI] [PubMed] [Google Scholar]
- 9. Sá JM, Twu O, Hayton K, Reyes S, Fay MP, et al. (2009) Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci U S A. 106: 18883–18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patel JJ, Thacker D, Tan JC, Pleeter P, Checkley L, et al. (2010) Chloroquine susceptibility and reversibility in a Plasmodium falciparum genetic cross Mol Microbiol. 78: 770–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roepe PD (2011) PfCRT-Mediated Drug Transport in Malarial Parasites. Biochemistry. 50: 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mäser P, Wittlin S, Rottmann M, Wenzler T, Kaiser M, et al. (2012) Antiparasitic agents: new drugs on the horizon. Curr Opin Pharmacol. 12: 562–566. [DOI] [PubMed] [Google Scholar]
- 13. Salako LA, Aderounmu AF, Walker O (1987) Influence of route of administration on the pharmaco-kinetics ofchloroquine and desethylchloroquine. Bull World Health Organ. 65: 47–50. [PMC free article] [PubMed] [Google Scholar]
- 14. Hodel EM, Zanolari B, Mercier T, Biollaz J, Keiser J, et al. (2009) A single LC-tandem mass spectrometry method for the simultaneous determination of 14 antimalarial drugs and their metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 877: 867–886. [DOI] [PubMed] [Google Scholar]
- 15. Khalil IF, Alifrangis M, Recke C, Hoegberg LC, Ronn A, et al. (2011) Development of ELISA-based methods to measure the anti-malarial drug chloroquine in plasma and in pharmaceutical formulations. Malar J. 10: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baguley BC (2010) Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 46: 308–316. [DOI] [PubMed] [Google Scholar]
- 17. Cabrera M, Paguio MF, Xie C, Roepe PD (2009) Reduced digestive vacuolar accumulation of chloroquine is not linked to resistance to chloroquine toxicity. Biochemistry 48: 11152–11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorka AP, Alumasa JN, Sherlach KS, Jacobs LM, Nickley KB, et al. (2013a) Cytostatic vs Cytocidal Activities of Chloroquine Analogues and Inhibition of Hemozoin Crystal Growth. Antimicro. Agents and Chemotherapy. 57: 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorka AP, Sherlach KS, de Dios AC, Roepe PD (2013b) Relative to Quinine and Quinidine, their 9-epimers exhibit decreased cytostatic activity and altered heme binding, but similar cytocidal activity vs P. falciparum. Antimicro. Agents and Chemotherapy. 57: 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorka AP, de Dios A, Roepe PD (2013c) Quinoline Drug-Heme Interactions and Implications for Antimalarial Cytostatic versus Cytocidal Activities. J Med Chem. 2013 Apr 29. [Epub ahead of print, PMID: 23586757] [DOI] [PubMed]
- 21. Sinai AP, Roepe PD (2012) Autophagy in Apicomplexa: a life sustaining death mechanism? Trends Parasitol. 28: 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Totino PR, Daniel-Ribeiro CT, Corte-Real S, de Fátima Ferreira-da-Cruz M (2008) Plasmodium falciparum: erythrocytic stages die by autophagic-like cell death under drug pressure. Exp Parasitol. 118: 478–86. [DOI] [PubMed] [Google Scholar]
- 23. Meslin B, Beavogui AH, Fasel N, Picot S (2011) Plasmodium falciparum metacaspase PfMCA-1 triggers a z-VAD-fmk inhibitable protease to promote cell death. PLoS One. 6: e23867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suradji EW, Hatabu T, Kobayashi K, Yamazaki C, Abdulah R, et al. (2011) Selenium-induced apoptosis-like cell death in Plasmodium falciparum. Parasitology. 19: 1–11. [DOI] [PubMed] [Google Scholar]
- 25. Ch’ng JH, Kotturi SR, Chong AG, Lear MJ, Tan KS (2010) A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis. 1: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nyakeriga AM, Perlmann H, Hagstedt M, Berzins K, Troye-Blomberg M, et al. (2006) Drug-induced death of the asexual blood stages of Plasmodium falciparum occurs without typical signs of apoptosis. Microbes Infect. 8: 1560–1568. [DOI] [PubMed] [Google Scholar]
- 27. Sanchez CP, Mayer S, Nurhasanah A, Stein WD, Lanzer M (2011) Genetic linkage analyses redefine the roles of PfCRT and PfMDR1 in drug accumulation and susceptibility in Plasmodium falciparum. Mol Microbiol. 82: 865–878. [DOI] [PubMed] [Google Scholar]
- 28. Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, et al. (2002) Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 61: 35–42. [DOI] [PubMed] [Google Scholar]
- 29.Ecker A, Lehane AM, Clain J, Fidock DA (2012) PfCRT and its role in antimalarial drug resistance. Trends Parasitol. pii: S1471–4922. [DOI] [PMC free article] [PubMed]
- 30. Sidhu AB, Verdier-Pinard D, Fidock DA (2002) Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 298: 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghosh D, Walton JL, Roepe PD, Sinai AP (2012) Establishment of autophagy as a cell death mechanism in Toxoplasma gondii Cell Microbiol. 4: 589–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, et al. (2004) Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob Agents Chemother. 48: 1807–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reilly Ayala H, Wacker MA, Siwo G, Ferdig MT (2011) Quantitative trait loci mapping reveals candidate pathways regulating cell cycle duration in Plasmodium falciparum. BMC Genomics. 11: 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 4(1): 44–57. [DOI] [PubMed] [Google Scholar]
- 35. Gonzales JM, Patel JJ, Ponmee N, Jiang L, Tan A, et al. (2008) Regulatory hotspots in the malaria parasite genome dictate transcriptional variation. PLoS Biol. 30 6(9): e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gligorijevic B, McAllister R, Urbach JS, Roepe PD (2006) Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 1. Quantification of hemozoin development for drug sensitive versus resistant malaria. Biochemistry. 45: 12400–12410. [DOI] [PubMed] [Google Scholar]
- 37. Waller KL, Muhle RA, Ursos LM, Horrocks P, Verdier-Pinard D, et al. (2003) Chloroquine resistance modulated in vitro by expression levels of the Plasmodium falciparum chloroquine resistance transporter. J. Biol. Chem. 278: 33593–33601. [DOI] [PubMed] [Google Scholar]
- 38. Wilson CM, Serrano AE, Wasley A, Bogenschutz MP, et al. (1989) Amplification of a gene related to mammalian mdr genes in drug-resistant Plasmodium falciparum. Science. 244: 1184–1186. [DOI] [PubMed] [Google Scholar]
- 39. Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF (2000) Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 403: 906–9. [DOI] [PubMed] [Google Scholar]
- 40. Amoah LE, Lekostaj JK, Roepe PD (2007) Heterologous expression and ATPase activity of mutant versus wild type PfMDR1 protein. Biochemistry 46: 6060–6073. [DOI] [PubMed] [Google Scholar]
- 41. Pleeter P, Lekostaj JK, Roepe PD (2010) Purified Plasmodium falciparum multi-drug resistance protein (PfMDR 1) binds a high affinity chloroquine analogue. Mol Biochem Parasitol. 173: 158–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Notte A, Leclere L, Michiels C (2011) Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem Pharmacol. 82: 427–434. [DOI] [PubMed] [Google Scholar]
- 43. Platini F, Pérez-Tomás R, Ambrosio S, Tessitore L (2010) Understanding autophagy in cell death control. Curr Pharm Des. 16: 101–13. [DOI] [PubMed] [Google Scholar]
- 44. Gozuacik G, Kimchi K (2007) Autophagy and Cell death Curr. Topics in Dev Biol 78: 217–245. [DOI] [PubMed] [Google Scholar]
- 45. Lavine MD, Arrizabalaga G (2012) Analysis of monensin sensitivity in Toxoplasma gondii reveals autophagy as a mechanism for drug induced death. PLoS One. 7(7): e42107 10.1371/journal.pone.0042107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) ATG8: An Autophagy – Related Ubiquitin – Like Protein Family. Genome Biology 12: 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kitamura K, Kishi-Itakura C, Tsuboi T, Sato S, Kita K, et al. (2012) Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLoS One 7 8: e42977 doi:10.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsui A, Kamada Y, Matsuura A (2013) The role of autophagy in genome stability through suppression of abnormal mitosis under starvation. PLoS Genet. 9(1): e1003245 10.1371/journal.pgen.1003245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dotiwala F, Eapen VV, Harrison JC, Arbel-Eden A, Ranade V, et al. (2013) DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc Natl Acad Sci U S A. 110: E41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brennand A, Gualdrón-López M, Coppens I, Rigden DJ, Ginger ML, et al. (2011) Autophagy in parasitic protists: unique features and drug targets. Mol Biochem Parasitol. 177: 83–99. [DOI] [PubMed] [Google Scholar]
- 51. Vaid A, Ranjan R, Smythe WA, Hoppe HC, Sharma P (2010) PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood. 115: 2500–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, et al. (2012) Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A. 109(34): 13561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin M, Chandramani-Shivalingappa P, Jin H, Ghosh A, Anantharam V, et al. (2012) Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction andautophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience. 17 210: 308–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lanzer M, Wickert H, Krohne G, Vincensini L, Braun Breton C (2006) Maurer’s clefts: A novel multi-functional organelle in the cytoplasm of Plasmodium falciparum-infected erythrocytes. Intl. J. Parasitol. 36: 23–36. [DOI] [PubMed] [Google Scholar]
- 55. Richards WH, Maples BK (1979) Studies on Plasmodium falciparum in continuous cultivation. I. The effect of chloroquine and pyrimethamine on parasite growth and viability. Ann Trop Med Parasitol. 73: 99–108. [PubMed] [Google Scholar]
- 56.Bakshi RP, Nenortas E, Tripathi AK, Sullivan DJ, Shapiro TA (2013) Model System to Define Pharmacokinetic Requirements for Antimalarial Drug Efficacy. Science (Translational Medicine). In the press. [DOI] [PMC free article] [PubMed]
- 57. Baro NK, Callaghan PS, Roepe PD (2013) Function of resistance conferring Plasmodium falciparum chloroquine resistance transporter isoforms. Biochemistry. 52: 4242–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roberts L, Egan TJ, Joiner KA, Hoppe HC (2008) Differential effects of quinoline antimalarials on endocytosis in Plasmodium falciparum. Antimicrob Agents Chemother. 52: 1840–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Federici E, Palazzino G, Nicoletti M, Galeffi C (1999) Antiplasmodial activity of the alkaloids of Peschiera fuchsiaefolia. 66: 93–95. [DOI] [PubMed] [Google Scholar]
- 60. Ramanitrahasimbola D, Rasoanaivo P, Ratsimamanga-Urverg S, Federici E, Palazzino G, et al. (2001) Biological activities of the plant-derived bisindole voacamine with reference to malaria. Phythother. Res. 15: 30–33. [DOI] [PubMed] [Google Scholar]
- 61. Meschini S, Condello M, Marra M, Formisano G, Federici E, et al. (2007) Autophagy-mediated chemosensitizing effect of the plant alkaloid voacamine on multidrug resistant cells. Toxicol. 21: 197–203. [DOI] [PubMed] [Google Scholar]
- 62. Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE (2006) Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc. Natl. Acad. Sci. 103: 8840–8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Istvan ES, Dharia NV, Bop SE, Gluzman I, Winzeler EA, et al. (2011) Validation of isoleucine utilization targets in Plasmodium falciparum. Proc. Natl. Acad. Sci. 108: 1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Babbitt SE, Altenhofen L, Cobbold SA, Istvan ES, Fennell C, et al.. (2012) Plasmodium falciparum responds to amino acid starvation by entering into a hibernatory state. Proc. Natl. Acad. Sci. USA in press. (early edition doi 10.1073). [DOI] [PMC free article] [PubMed]
- 65. Tawk L, Chicanne G, Dubremetz JF, Richard V, Payrastre B, et al. (2010) Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryotic Cell 9: 1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, et al. (1974) Lysosomotropic agents. Biochem Pharmacol. 23: 2495–531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Late trophozoite/early schizont stained with anti-ATG8 (green) and anti-apicoplast (red) antibodies. Some overlap between ATG8 and apicoplast localized ACP (D) suggests partial (but not complete, ∼25%) co – localization of PfATG8 and apicoplast as previously suggested [46]. Scale bar = 5 µm.
(DOC)
CQS (HB3, A) and CQR (Dd2, B) parasites treated with IC50 (top row each panel) and 2× IC50 (bottom row) doses of CQ (10, 20 nM and 125, 250 nM, respectively) for 48 hr. Parasites were then stained for ATG8 (green) and with DAPI (blue), as described in Methods. Very few peripherally disposed PfATG8 puncta are observed, supporting the conclusion that puncta formation is associated with LD50, but not IC50 doses of CQ. Scale bar = 5 µm.
(DOC)
LD50 values for the HB3×Dd2 cross progeny.
(DOC)
All genes within the chr6 LD50 locus.
(DOC)
All genes within the LD50 chr6×chr8 interaction.
(DOC)
GO enriched molecular functions for the chr6 LD50 locus.
(DOC)
GO enriched biological processes for the LD50 chr6 locus.
(DOC)
GO enriched molecular functions for the LD50 chr6×chr8 interaction.
(DOC)
GO enriched biological processes for the LD50 chr6×chr8 interaction.
(DOC)
Cartoon representation of 3D puncta quantification method used in the present work. Hemozoin density (red “x”) is used as the origin, and distances toPfATG8 positive puncta are defined for the reconstructed confocal z stack of images, relative to hemozoin, using 3D Cartesian (x,y,z) coordinates. The cartoon shows an abbreviated depiction of 3 SDCM “slices”, but as described in methods the z stack data set for each cell is a fully assembled, iteratively deconvolved 3D image constructed from approximately 15–20 z slices (see [35] for additional detail).
(DOC)