

Abstract
Thrombospondin 1 (THBS1 or TSP-1) is a circulating glycoprotein highly expressed in hypertrophic visceral adipose tissues of humans and mice. High-fat diet (HFD) feeding induces the robust increase of circulating THBS1 in the early stages of HFD challenge. The loss of Thbs1 protects male mice from diet-induced weight gain and adipocyte hypertrophy. Hyperinsulinemic euglycemic clamp study has demonstrated that Thbs1-null mice are protected from HFD-induced insulin resistance. Tissue-specific glucose uptake study has revealed that the insulin-sensitive phenotype of Thbs1-null mice is mostly mediated by skeletal muscles. Further assessments of the muscle phenotype using RNA sequencing, quantitative PCR, and histological studies have demonstrated that Thbs1-null skeletal muscles are protected from the HFD-dependent induction of Col3a1 and Col6a1, coupled with a new collagen deposition. At the same time, the Thbs1-null mice display a better circadian rhythm and higher amplitude of energy expenditure with a browning phenotype in sc adipose tissues. These results suggest that THBS1, which circulates in response to a HFD, may induce insulin resistance and fibrotic tissue damage in skeletal muscles as well as the de-browning of sc adipose tissues in the early stages of a HFD challenge. Our study may shed new light on the pathogenic role played by a circulating extracellular matrix protein in the cross talk between adipose tissues and skeletal muscles during obesity progression.
White adipose tissue (WAT) is surrounded by the network of extracellular matrix (ECM) proteins (1, 2). The dynamic equilibrium between the synthesis and degradation of ECM proteins plays a key role in the regulation of adipose tissue size and function during development and obesity progression (3–5). The dysregulation of ECM remodeling may lead to adipose tissue fibrosis, which is the hallmark of proinflammatory tissue damage observed in insulin-resistant, obese humans (6). The diet-induced gene regulation in the pathway of ECM remodeling is observed within a few weeks of a high-fat diet (HFD) challenge (3); however, the functional consequence of diet-induced adipose ECM remodeling in the early stages of a HFD challenge has not been fully defined.
Among a cohort of ECM proteins, thrombospondin 1 (THBS1; also known as TSP-1) is a large adhesive ECM glycoprotein that contains multiple functional domains (7). THBS1 is expressed predominantly in visceral adipose tissues, and its expression is elevated in insulin-resistant, obese humans (8). THBS1 is one of a few genes highly expressed in the visceral adipose tissues of Otsuka Long-Evans Tokushima fatty rats, a disease model of visceral adiposity and insulin resistance (9). In a brief report, Voros and Lijnen (10) reported the lack of significant difference in weight gain after 15 weeks of a HFD (42% kcal fat) between wild-type and Thbs1-null mice. On the other hand, Li et al (11) demonstrated that Thbs1-null mice were protected from adipose tissue inflammation and insulin resistance when examined after 16 weeks of a HFD (60% kcal fat). Despite these reports, the temporospatial regulation of insulin sensitivity by THBS1 and the potential target organs that mediate THBS1 effects in vivo have not been fully defined.
We hypothesized that ECM proteins derived from expanding visceral adipose tissues in the early stages of obesity may contribute to the pathogenesis of insulin resistance. Herein we demonstrate that HFD acutely induces Thbs1 expression in visceral adipose tissues and increases the level of circulating THBS1 protein. We have discovered that Thbs1-null male mice are protected from the HFD-induced insulin resistance and fibrotic tissue remodeling of skeletal muscles. The metabolically beneficial muscle phenotype of Thbs1-null mice was observed in conjunction with the emerging uncoupling protein 1 (UCP1)-positive brown fat-like structures in subcutaneous adipose tissues. These results suggest that THBS1 may act as a diet-dependent metabolic switch that may trigger muscle fibrosis and insulin resistance as well as adipose tissue dysfunction in the very beginning of diet-induced obesity.
Materials and Methods
Mice
B6.129S2-Thbs1tm1Hyn/J (Thbs1-null) (12) and age-matched control C57BL/6J male mice were obtained from the Jackson Laboratory. Thbs1-null mice were backcrossed to C57BL/6J for eight generations and have been subsequently maintained at The Jackson Laboratory. Mice were fed a normal-fat diet with 10% total calories from fat (NFD; D12450B; Research Diets) or a HFD with 45% of total calories from fat (D12451; Research Diets). Diet manipulation was started at 8 weeks of age unless otherwise stated. Animals were housed in a specific pathogen-free facility of the University of Michigan with a 12-hour light, 12-hour dark cycle and given free access to food and water. All animal use was approved by the University Committee on Use and Care of Animals and was compliant with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals. The inguinal and epididymal white adipose tissues, interscapular brown adipose tissues, and the quadriceps femoris were isolated at the end of each study (at age 11, 23, or 28 weeks old). For experiments, approximately six to eight male mice were used for each group. A set of experiments was performed at least twice.
Immunostaining and Picosirius Red staining
Immunostaining was performed in either paraffin-embedded sections or whole-mount tissues. Whole-mount tissue staining was performed as described previously (13, 14). Anti-Galectin 3 (also known as MAC2) antibody was obtained from eBioscience. Paraffin-embedded sections were preincubated in permeabilization buffer (0.5% Triton X-100 in PBS) for 10 minutes and then in blocking buffer (5% normal goat serum in PBS) for 30 minutes at room temperature, followed by the incubation with primary and secondary antibodies. Paraffin-embedded sections were used for hematoxylin and eosin (H&E) staining. The UCP1 staining was performed as described previously (15). Anti-mouse UCP1 antibody (catalog number UCP11-A) was obtained from Alpha Diagnostic International. Collagen fibers in skeletal muscles were detected with Picosirius Red staining (16) and observed under a polarized light microscopy (Olympus IX71).
Microscopic imaging and morphometric analysis
Olympus IX71 was used to capture epifluorescent and H&E images. The average size of adipocytes was determined using Image J (National Institutes of Health), and the adipocyte number within each adipose tissue was estimated as previously described (17). Immunofluorescent images were pseudocolored with FluoView (Olympus). The percent of area positive for fibrillar collagen within the perimysium and endomysium of the quadriceps femoris was quantified using Image J. The preexisting collagen fibers found in perivascular areas and the epimysium were excluded from the analysis.
Cells
3T3-L1 mouse preadipocytes were obtained from American Type Culture Collection. Cells were maintained in high-glucose DMEM (Life Technologies supplemented with 10% newborn calf serum (Thermo Scientific and 1× penicillin-streptomycin (Life Technologies). Adipogenesis of 3T3-L1 cells was induced by adding the adipogenic cocktail at the final concentration of 1 μg/mL insulin, 0.5 mM 3-isobutyl-1-methylxanthine, and 0.25 μM dexamethasone (all from Sigma-Aldrich) (18). Three days after adding the adipogenic cocktail, the media were changed and cells were incubated for 3 days in DMEM with 10% fetal bovine serum and 1 μg/mL insulin, then in DMEM with 10% fetal bovine serum.
THBS1 Western blot
The serum (1 μL) was diluted in sodium dodecyl sulfate lysis buffer, denatured, and run in 5% SDS-PAGE. THBS1 was detected with Western blot in denatured condition (18) using rabbit anti-THBS1 antibody (Santa Cruz Biotechnology).
Body composition, energy expenditure, and respiratory quotient
Body composition was measured with a nuclear magnetic resonance analyzer (Minispec LF9011; Bruker AXS) at the Animal Phenotyping Core, University of Michigan. Food consumption, spontaneous cage activity, O2 consumption (VO2), CO2 production (VCO2), and respiratory exchange ratio were obtained using the Comprehensive Laboratory Monitoring System (Columbus Instruments).
Flow cytometry
Flow cytometry analysis of adipose tissue macrophages (ATMs) was performed as recently described (19). Briefly, after collagenase digestion, stromal-vascular cells (107/mL) were incubated in Fc Block (rat antimouse CD16/CD32; eBioscience) for 15 minutes on ice. Cells were stained with F4/80-PE, CD11b-PE-Cy7, CD11c-APCCy7, and CD301-APC (eBioscience) fluorescence-conjugated antibodies prior to analysis with FACSCanto II (BD Biosciences) and FlowJo (Tree Star, Inc) software. M1-like ATM was defined as CD11b+ F4/80+ CD11c+ CD301− cells and M2-like cells as CD11b+ F4/80+ CD11c− CD301+ cells.
Real-time PCR
Total RNA was isolated using an RNeasy kit (QIAGEN) according to the manufacturer's instruction. After cDNA synthesis using Superscript III (Invitrogen), real-time quantitative PCR amplification was performed in StepOnePlus (Applied Biosystems) using either SYBR Green primers or Taqman primers/probes (Integrated DNA Technologies). The quantity of 36B4 (Rplp0) was used as an internal control to obtain the relative expression levels of target genes. RNAs obtained from mouse inguinal and perigonadal adipose tissues were pooled and used as a standard RNA. The sequences of primers and probes are the following. Thbs1 (primers, ACT TCA CCT TTG CCA CCT C, AGA CTC TGG AAT GCG GTT G; probe, 56-FAM/ACA CAT ATG /ZEN/CAA CAG GAA CAG GAC ACC /3IABkFQ); Cd36 (primers, GCG ACA TGA TTA ATG GCA CAG, GAT CCG AAC ACA GCG TAG; probe, 56-FAM/CAA CAA AAG /ZEN/GTG GAA AGG AGG CTG C/3IABkFQ); Cd47 (primers, GGA TCA TAG CTC TAG CAG AAC T, GTC ACT TCC CTT CAC CTA TTC C; probe, 56-FAM/AGT TTG TCG/ZEN/CTT CCA ACC AGA GGA C/3IABkFQ); Col1a1 (primers, AGA GTC TAC ATG TCT AGG GTC; CTT CAG GGA TGT CTT CTT GG); Col3a1 (primers, CCT CTG GTT CTC CTG GTC TG, CCA CCT TCA CCC TTA TCT CC); Col4a1 (primers, CAT TGT GGA GTG TCA ACC TG, TCC TTT CTG ACC TTT CTG TCC); Col5a3 (primers, TGC TGT CCC TGC TCT ACT, GTC CTT ACA GAC TGA GTT CCA G; probe, 56-FAM/CTC TCC TTT/ZEN/CGC TCC CTT TTC TCC A/3IABkFQ); and Col6a1 (primers, AAG TTC TGT AGG CCA ATG CTC, CCA GAT GAG TGT GAG ATC CTG; probe, 56-FAM/AAA TGT GCT/ZEN/CCT GCT GTG AGT GC/3IABkFQ).
RNA sequencing and gene expression profiling
Illumina libraries were prepared at the University of Michigan DNA sequencing core (Bob Lyons, director) using TrueSeq RNA kit (Illumina) following the manufacture's protocol. Every four cDNA samples were indexed with adapter sequences and run on one lane of an Illumina HiSeq2000. The sequenced reads were aligned to the mouse genome (Ensembl m38.70, Genbank Assembly identification: GCA_000001635.3) using Bowtie2 version 2.0.6 (20), and gene expression was analyzed using Cufflinks version 2.0.2 (21). A P value corrected for an false discovery rate (q value) of .05 was considered statistically significant (22). Heat maps were generated in R, version 2.15.1, with the heatmap.2 function in the gplots package (version 2.11.0) using the hierarchical clustering method ward and the distance function euclidean. Gene ontology (GO) analysis was conducted using Database for Annotation, Visualization, and Integrated Discovery (23).
Hyperinsulinemic-euglycemic clamp study
Hyperinsulinemic-euglycemic clamp experiments were performed at the Vanderbilt-National Institute of Diabetes and Digestive and Kidney Diseases Mouse Metabolic Phenotyping Center (Nashville, Tennessee). All procedures were approved by the Vanderbilt University Animal Care and Use Committee. Mice were fed a HFD for 3 weeks beginning at 8 weeks of age. Catheters were implanted into a carotid artery and a jugular vein of mice for sampling and infusions, respectively, 5 days before the study as described (24). Insulin clamps were performed on mice fasted for 5 hours using a modification of the method described previously (25). [3-3H]-glucose was primed (2.4 μCi) and continuously infused for 90-minute equilibration and basal sampling periods (0.04 μCi/min). [3-3H]-glucose was mixed with the nonradioactive glucose infusate (infusate specific activity of 0.4 μCi/mg) during the 2-hour clamp period. Arterial glucose was clamped using a variable rate of glucose (plus trace [3-3H]-glucose) infusion, which was adjusted based on the measurement of blood glucose at 10-minute intervals. By mixing radioactive glucose with the nonradioactive glucose infused during a clamp, deviations in arterial glucose specific activity were minimized and steady state conditions were achieved.
The calculation of glucose kinetics was therefore more robust (26). Baseline blood or plasma variables were calculated as the mean of values obtained in blood samples collected at −15 and −5 minutes. At time zero, insulin infusion (2.5 mU/kg of body weight per minute) was started and continued for 120 minutes. Mice received heparinized saline-washed erythrocytes from donors at 5 μL/min to prevent a fall in hematocrit. Insulin clamps were validated by assessment of blood glucose over time. Blood was taken at 80–120 minutes for the determination of [3-3H]-glucose. Clamp insulin was determined at t = 120 minutes. At 120 minutes, 13 μCi of [14C]-2-deoxyglucose ([14C]2DG) was administered as an iv bolus. Blood was taken at 2–25 minutes for the determination of [14C]2DG. After the last sample, mice were anesthetized and tissues were freeze clamped for biochemical analysis. Plasma insulin was determined by ELISA. Radioactivity of [3-3H]-glucose and [14C]2DG in plasma samples and [14C]2DG-6-phosphate in tissue samples were determined by liquid scintillation counting. Glucose appearance and disappearance rates were determined using non-steady-state equations (27). Endogenous glucose appearance was determined by subtracting the glucose infusion rate from total glucose appearance. The glucose metabolic index was calculated as previously described (28).
Statistical analysis
All data are presented as mean ± SEM and analyzed with a two-tailed Student's t test and ANOVA. Harmonic analysis of ultradian energy expenditure was performed as described by others (29). Differences were considered significant for P < .05 (*, P < .05; **, P < .005; ***, P < .0005).
Results
HFD-dependent induction of THBS1 in epididymal WAT (eWAT) and in the circulation
When screening for ECM genes that are regulated during the adipocyte differentiation of 3T3-L1 cells, we identified Thbs1 as the most significantly down-regulated ECM gene in the beginning of adipogenesis (Figure 1A). Of note, however, the reduction of Thbs1 gene expression was transient, and mature adipocytes expressed higher levels of Thbs1 than differentiating adipocytes (Figure 1A). Given the potential role of Thbs1 associated with adipocyte differentiation, we next tested which fat depots expressed Thbs1. Thbs1 was highly expressed in eWAT relative to inguinal WAT (iWAT) and brown adipose tissues (Figure 1B). After a short-term, 3-week HFD feeding, Thbs1 expression was significantly increased in eWAT but not in iWAT (Figure 1C). In conjunction with the increased expression of Thbs1 in eWAT, the circulating THBS1 protein levels increased within 3 weeks of short-term HFD and stayed at elevated levels during a long-term HFD (Figure 1D). Underscoring the pleiotropic role of THBS1 as a circulating factor, its cognate receptors, CD36 and CD47, were equally expressed in both iWAT and eWAT (Figure 1E). No significant change in the expression of Cd36 or Cd47 was noted upon HFD (Figure 1E). Thbs1 expression in skeletal muscles (quadriceps femoris) was very low (0.96% ± 0.18% of eWAT Thbs1, n = 8), and the levels of Thbs1 expression in the skeletal muscles did not change in response to HFD (Figure 1F). Cd36 and Cd47 were both well expressed in skeletal muscles; however, no dietary effects on their expression were noted (Figure 1F).
Figure 1.

Thbs1 expression during adipogenesis and HFD-induced obesity. A, Thbs1 expression during the differentiation of 3T3-L1 cells into adipocytes (n = 3, mean ± SEM). The induction of aP2 (Fabp4) and Pparγ is shown for comparison. B, Thbs1 expression in the iWAT, eWAT, and brown adipose tissues isolated from 11-week-old C57BL/6J male mice, respectively (n = 4, mean ± SEM). C, Thbs1 expression in iWAT and eWAT isolated from 11-week-old male mice fed a NFD (open bar)or HFD (closed bar) for 3 weeks (n = 4, mean ± SEM). D, THBS1 protein levels in the sera collected from 11-week-old male mice fed a NFD or HFD for 3 weeks and 15 weeks. The staining with Ponceau S is shown as loading control (Po-S). Representative figures (n = 4 each) are shown. E, Cd36 and Cd47 expression in iWAT and eWAT isolated from male mice fed a NFD (open bar) or a HFD (closed bar) for 3 weeks (n = 8, mean ± SEM). F, The expression of Thbs1, Cd36, and Cd47 in the quadriceps femoris isolated from wild-type male mice fed a NFD or HFD for 3 weeks (n = 8, mean ± SEM). NFD (open bar), HFD (closed bar), *, P <.05.
Thbs1 deficiency prevents HFD-induced adipose tissue hypertrophy
To test the causal role of Thbs1 in diet-induced obesity, Thbs1-null and age-matched C57BL/6J mice were fed a NFD or HFD beginning at 8 weeks of age. Weight gain upon HFD feeding was significantly suppressed in Thbs1-null mice (Figure 2A), whereas no significant weight differences were observed between wild-type and Thbs1-null mice under NFD.
Figure 2.
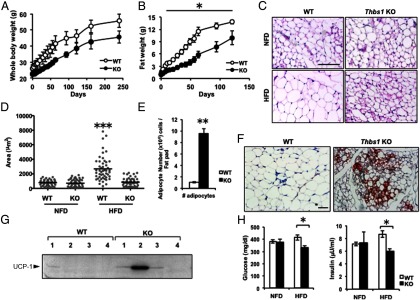
Thbs1-null mice are protected from HFD-induced obesity. A, Body weight change during a 45% HFD feeding. WT and Thbs1-null (KO) male mice, fed a 45% HFD beginning at age 8 weeks (n = 8 for each group). B, Fat mass assessed with a nuclear magnetic resonance body composition analyzer (n = 8). C, H&E staining of inguinal white adipose tissues. Upper panel, Eleven-week-old male mice fed normal fat diet (NFD). Lower panel, Eleven-week-old male mice fed a 45% fat diet (HFD) for 3 weeks. Scale, 100 μm. D, The morphometric quantification of adipocyte size in adipose tissue (square micrometers, n = 50 adipocytes per field). E, The number of adipocytes per tissue. Open bar, WT; closed bar, Thbs1 KO mice. F, UCP1-positive adipocytes within iWAT (brown); nuclei are counterstained (blue). G, Western blot of UCP1 in tissue lysates of inguinal adipose tissues (n = 4 for each group). H, The levels of serum glucose (left panel) and insulin (right panel) in mice fed a NFD or HFD for 3 weeks (n = 8, mean ± SEM). *, P < .05 compared with WT.
The lower body weight of Thbs1-null mice during HFD feeding was associated with decreased adiposity (Figure 2B). When fed a HFD, inguinal fat mass increased at the rate of 0.2 g/d (or 0.5% body weight per day) in wild-type mice, whereas the expansion rate was reduced by 60% in Thbs1-null mice (0.08 g/d or 0.2% body weight per day, Figure 2B). Adipocyte size increased 3-fold in wild-type mice in 3 weeks of the HFD, whereas the adipocyte size of Thbs1-null iWAT did not change within 3 weeks of HFD (Figure 2, C and D). Consistently, the increase in serum leptin levels after 3 weeks of HFD was suppressed by 50% in Thbs1-null mice (Supplemental Figure 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). Of note, the groups of small and multilocular adipocytes emerged in Thbs1-null iWAT upon the HFD challenge (Figure 2, C and F), and the number of adipocytes in the iWAT of Thbs1-null mice was 10 times higher than that of wild-type mice (Figure 2E). These multilocular adipocytes found in Thbs1-null mice were positive for UCP-1, suggesting the presence of mitochondria-rich, brown fat-like adipocytes in Thbs1-null iWAT (Figure 2F). Indeed, the mice with higher expression of UCP1 in iWAT were found selectively in Thbs1-null mice but not in wild-type mice (Figure 1G), even though the levels of UCP-1 expression in tissue lysates varied in the group (Figure 1G). Metabolically, Thbs1-deficient state rendered mice insulin sensitive as evidenced by the lower levels of insulin and glucose relative to controls (Figure 2F). In this early stage of HFD (3 weeks), we observed no significant difference in serum triglyceride content; however, Thbs1-null mice displayed lower total cholesterol levels (Supplemental Figure 2).
The protection from diet-induced weight gain in Thbs1-null mice was not due to decreased food intake or increased physical activity (Figure 3, A and B). To assess their basal metabolic rates, the oxygen consumption rates of Thbs1-null and wild-type mice were assessed in the third week of HFD. On average, oxygen consumption and respiratory quotient were elevated in Thbs1-null mice but not statistically different from wild-type mice (Figure 3, C and D). The harmonic analysis of oxygen consumption, however, demonstrates that Thbs1-null mice display a better fitting to a cosine curve with higher amplitude of VO2, pointing to the well-controlled circadian rhythm in their energy expenditure (R2, wild type, 0.29 ± 0.07 vs Thbs1-null mice, 0.37 ± 0.05, P = .01, n = 8 for each group). The peak amplitude of VO2 per lean mass was significantly higher in Thbs1-null mice than wild-type control (wild type, 584 ± 94 vs Thbs1-null, 738 ± 139 mL/kg·min, P = .02).
Figure 3.

Food consumption, physical activity, and oxygen consumption. A, Weekly food consumption of WT (open circle) and Thbs1-null male mice (KO; closed circle) during a 45% HFD challenge. B, Total physical activities of WT and KO male mice under a 45% HFD challenge. C, Oxygen consumption of WT and KO mice adjusted for lean mass (LBM). D, The respiratory quotient (RQ; VCO2 to VO2 ratio) of WT and KO mice during a 45% HFD feeding (n = 6, mean ± SEM).
Loss of Thbs1 prevents HFD-induced adipose tissue inflammation
Perigonadal adipose tissues (eWAT) of Thbs1-null mice fed a NFD showed no morphological differences from control mice under NFD (Figure 4A). HFD feeding induced the significant hypertrophy of eWAT in wild-type mice, whereas Thbs1-null mice were relatively protected from diet-induced hypertrophy (Figure 4A). Although the protection from adipocyte hypertrophy in eWAT was similar to that observed in iWAT (Figure 2), the Thbs1-null eWAT did not display the emergence of eosinophilic, brown fat-like adipocytes under a HFD challenge (Figure 4A).
Figure 4.

The loss of Thbs1 protects mice from HFD-induced adipose tissue inflammation. A, H&E staining of eWAT. Upper panel, Eleven-week-old male mice fed a NFD. Lower panel, Eleven-week-old male mice fed a 45% HFD for 3 weeks. Scale, 100 μm. B, HFD-induced crown-like structure is shown with Mac2 staining in eWAT after 28 weeks of HFD feeding. Scale, 200 μm. The higher magnification of WT crown-like structure is shown at the bottom. Scale, 50 μm. C, Gene expression of Emr1 encoding F4/80 in WT or KO eWAT after 28 weeks of a HFD (n = 4). D, Flow cytometry-based cell population analysis. The percentage of the parent cell population is shown for ATMs, M1-like ATMs (M1s), M2-like ATMs (M2s), and double-negative cells. E, The ratio of M1-like to M2-like ATMs in WT and KO mice at 3 and 21 weeks of a HFD (n = 4, mean ± SEM). *, P < .05; **, P < .005.
A long-term HFD challenge (20 weeks) induced the significant infiltration of M1-like ATMs and crown-like structures (CLSs) within the eWAT of wild-type mice (Figure 4B) (14). By contrast, Thbs1-null eWAT displayed only a few CLSs upon histological evaluation (Figure 4B). Consistent with the histological finding, the expression of a macrophage marker Emr1 (encoding F4/80) was significantly reduced in Thbs1-null eWAT (Figure 4C). This observation was further validated by the quantitation of M1-like (CD11b+ F4/80+, CD11c+ CD301−) and M2-like (CD11b+ F4/80+, CD11c− CD301+) ATMs using flow cytometry. The total number of ATMs in Thbs1-null mice was reduced by 25% relative to control [wild type (WT), 13.0% ± 1.1%; knockout (KO), 9.8% ± 0.76% of total vascular stromal cells, n = 4 per group, P = .04]. In addition, the M1-like ATM population in Thbs1-null adipose tissue was significantly reduced (WT, 57.0% ± 6.2%; KO, 32.1% ± 0.8%; P < .005). Nonetheless, the proportion of M2-like ATMs was significantly larger in KO ATMs (WT, 13.5% ± 4.1%; KO, 32.7% ± 2.6%; P < .005) (Figure 4D). Therefore, the ratio of M1 to M2 ATMs was substantially reduced in Thbs1-null mice after a prolonged HFD challenge (Figure 4E). Despite these adipose tissue phenotype, we were not able to see significant differences in the development of fatty liver between wild-type and Thbs1-null mice (Supplemental Figure 3).
Thbs1-dependent muscle insulin resistance induced by HFD challenge
To assess the role of THBS1 in regulating whole-body glucose metabolism particularly in the early stages of obesity, hyperinsulinemic euglycemic clamp studies were performed on Thbs1-null and wild-type mice. Throughout the clamp procedure, blood glucose levels were maintained at equivalent levels in the two groups (Figure 5A). Thbs1-null mice required a significantly higher rate of glucose infusion than WT mice (Figure 5B, P = .01). Consistently, the total glucose flux rate was significantly higher in the KO than the WT mice under hyperinsulinemic euglycemic conditions (Figure 5C; the rate of appearance Ra, WT, 34.4 ± 2.3 vs KO, 45.8 ± 3.2 mg/kg·min at 120 min, P = .01). By contrast, the insulin-dependent suppression of endogenous glucose production was not different between the groups, suggesting that Thbs1 does not regulate liver insulin sensitivity (Figure 5D, percentage suppression of the endogenous Ra, WT, 80.0% ± 6.8% vs KO, 75.9% ± 9.8%, P = .71).
Figure 5.

Hyperinsulinemic euglycemic clamp study. A, Blood glucose levels of WT and Thbs1-null (KO) mice during hyperinsulinemic euglycemic clamp studies. B, Glucose infusion rate (120 min) (mean ± SEM; WT, n = 10; KO, n = 6; P = .03 at 120 min). C, Glucose flux rate of WT and Thbs1-null (KO) mice under a hyperinsulinemic, euglycemic condition (P = .01 at 120 min). D, Suppression of endogenous glucose production during the study. *, P < .05.
Given the improved insulin-dependent glucose disposal observed in Thbs1-null mice (Figure 5), we assessed tissue-specific glucose uptake at the conclusion of the insulin clamp study after a bolus injection of [14C]-2-deoxyglucose. Glucose uptakes by the soleus, gastrocnemius, and vastus lateralis were significantly higher in Thbs1-null mice than WT mice (Figure 6A, P < .05). By contrast, the average glucose uptake by inguinal or eWAT was not statistically different between wild-type and Thbs1-null mice (Figure 6B; P = .15 and P = .25, respectively).
Figure 6.

Tissue glucose uptake. Tissue glucose uptake (Rg) in soleus, gastrocnemius, and vastus lateralis (A) and the iWAT and eWAT (B) [WT, n = 10 (open bars), and Thbs1-null (KO) mice, n = 6 (closed bars); mean ± SEM]. *, P < .05.
Thbs1-dependent fibrotic tissue remodeling in skeletal muscles under HFD challenge
To determine the molecular pathways underlying the metabolically beneficial phenotype of Thbs1-deficiency, we determined the gene expression profile of skeletal muscles in the early stages of HFD. Expression profiles were obtained from mice fed a HFD for 3 weeks because this short duration of HFD was sufficient to observe the metabolically beneficial effects of Thbs1 deficiency; however, this time point preceded a recognizable difference in body weight to be noted between Thbs1-null and wild-type. Therefore, we should be able to minimize the obesity-induced secondary effects on gene expression.
Quadriceps femoris muscles were isolated from wild-type and Thbs1-null mice fed a HFD for 3 weeks since 8 weeks of age because this muscle was found to display an increased glucose uptake in Thbs1-null mice (Figure 5A). Genome-wide transcriptome quantification was performed with RNA sequencing. The correlation coefficient (r) between the two groups was 0.998. Among 41 381 genes analyzed, 13 095 genes (32% total) were defined as expressed (fragments per kilobase of exon per million fragments mapped > 1.0). Among these genes, 552 (4.2% of expressed genes) were differentially expressed between the two groups (adjusted P < .05). The pathway analyses of these differentially expressed genes with GO and the Kyoto Encyclopedia of Genes and Genomes point to the enrichment of the genes in the pathways associated with ECM remodeling, cell adhesion, and collagen fibril organization (Figure 7A and Supplemental Table 1). Consistently, Thbs1-null muscles displayed a reduced gene expression in the pathway of collagen (Figure 7B).
Figure 7.
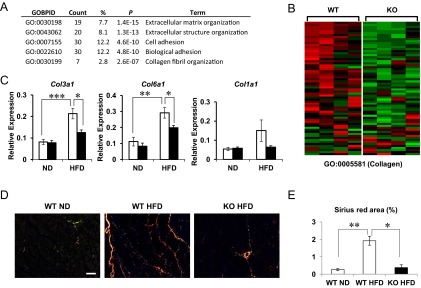
Thbs1-dependent skeletal muscle gene expression. A, The list of Gene Ontology Biological Pathway (GO-BP) pathways significantly different between the two groups. B, A representative heat map shows the expression profile of the genes listed in GO: Collagen (GO: 0005581). C, The expression of collagen family members (Col3, Col6, Col1) in the quadriceps muscles isolated from WT and KO mice after 3 weeks of a NFD or a 45% HFD (n= 8 each, mean ± SEM). D, Picosirius Red staining of quadriceps femoris observed under polarized microscopy. WT mice fed a normal diet (WT NFD), fed a 45% HFD (WT HFD), and Thbs1-KO mice fed HFD (KO HFD). Thinner (green) and thick collagen fibers (yellow-orange) are shown. E, The quantification of the percentage area positive for Picosirius Red staining. Two sections from each mice were analyzed, four mice for each group (mean ± SEM). *, P < .05; **, P < .005; ***, P < .0005.
Based on these results, we examined the effect of HFD on the gene expression of collagen family members. A short-term (3 wk) HFD significantly increased the expression of Col3a1 and Col6a1 in skeletal muscles (Figure 7C, P < .0005 and P < 0.005, respectively). Col1a1 expression also displayed an upward trend, but the increase was not statistically significant in our experiments (Figure 7C). The expression of Col4a1 encoding a basement membrane collagen did not change upon HFD challenge (data not shown). The Col5a3 expression in skeletal muscles was too low to be reliably compared. These HFD-induced increases of Col3a1 and Col6a1 expression in skeletal muscles were significantly suppressed in Thbs1-null mice (Figure 7C). Then we assessed the fibrillar collagen content in the skeletal muscles with Picosirius Red staining observed under a polarized microscope. The quadriceps femoris isolated from mice fed a control diet displayed loose, mostly thin collagen fibers (Figure 7D, WT CD, green). Under a HFD, wild-type mice showed newly formed thicker collagen fibers (yellow to orange) within the endomysium and perimysium (Figure 7D). In the absence of Thbs1, the diet-induced neoformation of thick collagen fibers was suppressed, and only the preexisting collagen fibers were found in perivascular areas (Figure 7D and E; **, P < .005 and *, P < .05).
Discussion
High-caloric Western diets induce obesity and insulin resistance. The molecular mechanism underlying the link between diet-induced obesity and insulin resistance, however, has not been fully understood. THBS1 is a large glycoprotein highly expressed in hypertrophic visceral adipose tissues of humans (8). THBS1 acts as a multifaceted ECM protein that interacts with other ECM proteins, such as collagens (30, 31), fibronectin (32), and decorin (33). THBS1 is also found in the circulation at elevated levels in obese and insulin-resistant individuals (8). Herein we demonstrate the following: 1) a short-term HFD induces the robust expression of Thbs1 in visceral adipose tissues and elevates the level of circulating THBS1 protein; 2) the Thbs1-null state protects mice from HFD-induced muscle fibrosis and insulin resistance; and 3) Thbs1-null sc adipose tissues develop UCP1-positive brown fat-like structures upon HFD challenge. Our findings propose a metabolic disease model wherein THBS1 released from hypertrophic visceral adipose tissues acts as a metabolic switch to induce muscle fibrosis and insulin resistance as well as adipose tissue dysfunction (Figure 8).
Figure 8.

THBS1 mediates the link between visceral adiposity and muscle fibrosis/insulin resistance. A HFD rapidly induces expansion of visceral adipose tissue ECM and increases THBS1 levels in circulation. THBS1 may induce muscle fibrosis and insulin resistance as well as debrowning of sc adipose tissues in the early stage of the HFD and adipose tissue inflammation in late stages.
In the past, animal studies demonstrated conflicting data with regard to the role of THBS1 in diet-induced obesity and diabetes. In a brief report, Voros and Lijnen (10) reported no weight differences between wild-type and Thbs1 (TSP-1)-KO mice after 15 weeks of a 42% HFD; however, Thbs1-null mice displayed smaller adipose tissue size with a large variability. Li et al (11) demonstrated that Thbs1-null mice were protected from visceral adipose tissue inflammation and insulin resistance induced by 16 weeks of a 60% HFD. In this paper, the authors attributed the phenotypic differences to the THBS1 (TSP-1)-induced macrophage adhesion and migration (11).
While our paper was under review, a new study on TSP-1 suggested that Thbs1-null mice were indeed protected from obesity and insulin resistance after 20 weeks of a 60% HFD; however, the Thbs1-null mice displayed elevated free fatty acid and triglyceride levels (34). The authors attributed these in vivo findings to Thbs1-dependent regulation of inflammation and lipid uptake (34). Prolonged HFD, particularly with the use of supraphysiologically high percentage of fat (60% HFD), however, might have masked the early biological events that should be critical for the development of adipose tissue dysfunction and metabolic deterioration. After a long-term HFD of supraphysiological fat content (60%), the macrophage infiltration in visceral adipose tissue becomes the dominant player in defining metabolic phenotypes (14). Unlike Kong et al (34), we were not able to detect a difference in serum triglyceride levels between wild-type and Thbs1-KO mice. The difference might be due to differences in the duration of HFD feeding and the fat content. Adipose tissue ECM remodeling and gene expression undergo rapid change within 1–2 weeks of a 45% HFD challenge (3). Moreover, a short-term HFD challenge is sufficient to induce insulin resistance in rats (35). In humans, 3-day consumption of a 37% fat diet is enough to induce lipogenic and steroidogenic gene expression in the intestine (36). Unlike others, our study demonstrates the role of THBS1 in the pathogenesis of muscle fibrosis and insulin resistance as well as adipose tissue dysfunction using a short-term (3 wk) HFD challenge, which avoids the potential consequences of macrophage infiltration incurred by a long-term HFD or supraphysiological fat content.
Hyperinsulinemic, euglycemic clamp studies have demonstrated improved insulin-dependent glucose flux in Thbs1-null mice compared with wild-type controls within 3 weeks of a 45% HFD. The improved glucose flux was mediated by skeletal muscles. Notably, Thbs1 is expressed at considerably lower levels in skeletal muscles compared to eWAT. Unlike Thbs1 in eWAT, no HFD-dependent gene induction was noted in skeletal muscles. On the other hand, the robust increase of Thbs1 expression in eWAT coincided with the increase in circulating THBS1 protein during HFD challenge. Within 3 weeks of HFD feeding, however, we were able to see the robust induction of collagen genes, particularly Col3a1 and Col6a1, in skeletal muscles. Increased expression of Col3a1 and Col6a1 in skeletal muscles was observed in conjunction with the formation of thick collagen fibers in the endomysium and perimysium of skeletal muscles. The loss of Thbs1 negates the HFD-induced collagen gene expression and the neoformation of thick collagen fibers.
Tissue fibrosis is the consequence of complex biological processes comprising collagen synthesis, fibril formation, and proteolytic degradation (37). TGF-β1 is known to play the major role in the development of tissue fibrosis (38), and THBS1 (TSP-1) is considered to play a critical role in wound repair and tissue remodeling, partly by activating latent TGF-β (39). Nonetheless, the role of THBS1 in tissue fibrosis and disease process has not been fully defined in vivo due to its pleiotropic effects mediated through multiple downstream targets (39). The difference in collagen gene expression in skeletal muscles was observed only under a HFD challenge, whereas circulating levels of THBS1 at basal state might not be sufficient to cause a significant difference between wild-type and Thbs1-null mice. The role of THBS1 in diet-dependent activation of TGF-βs in skeletal muscles may warrant further investigations. It is also possible that an unidentified endocrine or paracrine factor may act in the downstream of Thbs1 to promote tissue fibrotic changes of skeletal muscle. Notably, tissue THBS1 may act at a higher concentration, in a paracrine manner, at the sites of tissue injury to promote wound healing (40). During obesity progression, the visceral adipose tissues express the highest levels of Thbs1 as shown in this paper and by others (8, 34). Nutritional stress may simulate the biological processes of wound healing in visceral adipose tissues to promote tissue remodeling, whereas increased THBS1 in circulation may induce the muscle fibrosis and insulin resistance at a distance.
Increased collagen content has been observed in insulin-resistant skeletal muscles of humans (41). In mice, a collagen receptor, α2-integrin, was shown to mediate HFD-induced muscle insulin resistance (42). The heterodimer formation of α-integrins with β1-integrin regulates the interactions between cells and ECM proteins including collagens (43). Thbs1-null mice displayed the reduced expression of Col3a1 and Col6a1 in skeletal muscles in association with improved glucose flux. It is plausible that adipose tissue-derived THBS1 may induce insulin resistance of skeletal muscles by activating a set of integrin family members and interacting with newly synthesized collagen fibers. Nonetheless, the exact molecular mechanism by which THBS1 induces muscle fibrosis and insulin resistance remains to be defined. In addition to integrins, CD36 and CD47 are candidate downstream targets of THBS1 (44); either of them or both together may mediate the fibrotic and antiinsulin effects of THBS1 in skeletal muscles. In particular, given the metabolically beneficial phenotype reported on Cd36-null mice (45), the significance of the THBS1-CD36 pathway in the pathogenesis of muscle fibrosis and insulin resistance should be further explored.
We observed a browning phenotype in Thbs1-null inguinal adipose tissues. The eosinophilic, multilocular adipocytes emerge in the sc adipose tissues of Thbs1-null mice with a short-term 45% HFD challenge. During this period, the energy expenditure of Thbs1-null mice displayed higher amplitude and their VCO2 to VO2 ratio is higher than that of wild-type mice. In a short-term HFD, the skeletal muscles of Thbs1-null mice display the protection from HFD-induced muscle fibrosis and insulin resistance. The functional quality of skeletal muscle closely correlates with the metabolic function of adipose tissues (46). Newly discovered peroxisomal proliferator-activated receptor-γ coactivator 1α-dependent myokine, Irisin, mediates the browning phenotype of subcutaneous adipose tissues (47), whereas a cohort of myokines may cross talk with adipose tissues (48). The molecular mechanism through which Thbs1-null mice develop brown fat-like adipocytes upon HFD is beyond the scope of this study; however, we postulate that the protection of skeletal muscles from HFD-induced fibrosis may play a role in the metabolic cross talk between skeletal muscles and adipose tissues.
UCP1 expression is considered as the hallmark of brown fat-like adipocytes. Although we detected the aggregation of UCP1 proteins in the newly developed brown fat-like structures, Western blots that aimed to assess the average UCP1 levels in tissues were not sensitive enough to detect reproducible differences between wild-type and Thbs1-null mice, which were noted with histology. We posit that unlike histological assessment, examining the average UCP1 expression in whole-tissue lysates may not be sensitive enough to detect the presence of the spatially scattered brown fat-like structures in Thbs1-null iWAT; however, despite variability, we detected UCP1 protein expression with Western blot only in iWATs isolated from Thbs1-null mice. A recent paper suggests that brown fat-like adipocytes within inguinal adipose tissues might have been newly produced through de novo adipogenesis (49). We indeed observed increased Pparg2 expression in Thbs1-null iWAT in the early stages of HFD (M.I., unpublished data), which may suggest the presence of increased de novo adipogenesis in Thbs1-null mice. This assumption is consistent with our finding of the increased adipocyte number in Thbs1-null iWAT challenged by a HFD.
In summary, our study sheds new light on the role of THBS1 in HFD-induced muscle fibrosis and insulin resistance. THBS1, a multifaceted ECM protein highly expressed in hypertrophic visceral adipose tissue, may circulate and induce muscle fibrosis and insulin resistance as well as adipose tissue dysfunction. Further investigation of this THBS1-dependent pathway in the pathogenesis of muscle fibrosis and insulin resistance and adipose tissue dysfunction should help us understand the disease processes of obesity and insulin resistance.
Acknowledgments
We thank Jennifer L. Delproposto and Diem Huynh (University of Michigan) for their technical supports. We also thank Drs Li Kang, Owen McGuinnes, and David Wasserman (Vanderbilt University) for the hyperinsulinemic, euglycemic clamp study and tissue glucose uptake assay. We also thank Dr Morton Brown (University of Michigan) for advice on statistical and harmonic analysis. We also thank Drs Robert Lyons (University of Michigan DNA Sequencing Core) and Richard C. McEachin (Department of Computational Medicine and Bioinformatics) for their advice on RNA sequencing.
This work was supported by National Institutes of Health Grants NIH HL106332, DK095137 (to T.-H.C.), AHA Scientist Development Grant 0730028N (to M.I.), 0730028N (M.I.), T32GM007315 (G.M-S.), DK090262 (to C.N.L.), DK084079 (to D.A.B.), DK020572 (to the Michigan Diabetes Training Center), DK089503 (to the Michigan Nutrition Obesity Research Center), and DK059637 (to the Mouse Metabolic Phenotyping Center, Vanderbilt University). The hyperinsulinemic euglycemic study was performed at the Mouse Metabolic Phenotyping Center, Vanderbilt University, supported by National Institutes of Health Grant DK059637.
Disclosure Summary: The authors have nothing to declare in relation to this manuscript.
Footnotes
- ATM
- adipose tissue macrophage
- [14C]2DG
- [14C]-2-deoxyglucose
- ECM
- extracellular matrix
- eWAT
- epididymal white adipose tissue
- GO
- gene ontology
- H&E
- hematoxylin and eosin
- HFD
- high-fat diet
- iWAT
- inguinal white adipose tissue
- KO
- knockout
- NFD
- normal-fat diet
- THBS1
- thrombospondin 1 (TSP-1 or Thbs1)
- UCP1
- uncoupling protein 1
- VCO2
- CO2 production
- VO2
- O2 consumption
- WAT
- white adipose tissue
- WT
- wild type.
References
- 1. Chun TH. Peri-adipocyte ECM remodeling in obesity and adipose tissue fibrosis. Adipocyte. 2012;1:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang G, Greenspan DS. ECM roles in the function of metabolic tissues. Trends Endocrinol Metab. 2012;23:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chun T-H, Inoue M, Morisaki H, et al. Genetic link between obesity and MMP14-dependent adipogenic collagen turnover. Diabetes. 2010;59:2484–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125:577–591 [DOI] [PubMed] [Google Scholar]
- 5. Khan T, Muise ES, Iyengar P, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Divoux A, Tordjman J, Lacasa D, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59:2817–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biology. 2011;3:a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Varma V, Yao-Borengasser A, Bodles AM, et al. Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes. 2008;57:432–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hida K, Wada J, Zhang H, et al. Identification of genes specifically expressed in the accumulated visceral adipose tissue of OLETF rats. J Lipid Res. 2000;41:1615–1622 [PubMed] [Google Scholar]
- 10. Voros G, Lijnen HR. Deficiency of thrombospondin-1 in mice does not affect adipose tissue development. J Thromb Haemost. 2006;4:277–278 [DOI] [PubMed] [Google Scholar]
- 11. Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PLoS One. 2011;6:e26656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lawler J, Sunday M, Thibert V, et al. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101:982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57:3239–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee Y-H, Petkova Anelia P, Mottillo Emilio P, Granneman James G. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012;15:480–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kiernan JA. Collagen type I staining. Biotech Histochem. 2002;77:231. [PubMed] [Google Scholar]
- 17. Hirsch J, Gallian E. Methods for the determination of adipose cell size in man and animals. J Lipid Res. 1968;9:110–119 [PubMed] [Google Scholar]
- 18. Inoue M, Chang L, Hwang J, Chiang S-H, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–633 [DOI] [PubMed] [Google Scholar]
- 19. Morris DL, Oatmen KE, Wang T, Delproposto JL, Lumeng CN. CX(3)CR1 deficiency does not influence trafficking of adipose tissue macrophages in mice with diet-induced obesity. Obesity (Silver Spring). 2012;20:1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schatz MC, Langmead B, Salzberg SL. Cloud computing and the DNA data race. Nat Biotech. 2010;28:691–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech. 2010;28:511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B (Methodological). 1995;57:289–300 [Google Scholar]
- 23. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008;4:44–57 [DOI] [PubMed] [Google Scholar]
- 24. Berglund ED, Li CY, Poffenberger G, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes. 2008;57:1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397 [DOI] [PubMed] [Google Scholar]
- 26. Finegood DT, Bergman RN, Vranic M. Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes. 1987;36:914–924 [DOI] [PubMed] [Google Scholar]
- 27. Steele R, Wall JS, De Bodo RC, Altszuler N. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol. 1956;187:15–24 [DOI] [PubMed] [Google Scholar]
- 28. Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol Endocrinol Metab. 1985;248:E353–EE362 [DOI] [PubMed] [Google Scholar]
- 29. Stupfel M, Perramon A, Gourlet V, Thierry H, Ali M, Lemercerre C. Harmonic analysis of ultradian respiratory rhythms in four small laboratory vertebrates lit in LD12:12. Comp Biochem Physiol A Comp Physiol. 1983;75:293–297 [DOI] [PubMed] [Google Scholar]
- 30. Mumby SM, Raugi GJ, Bornstein P. Interactions of thrombospondin with extracellular matrix proteins: selective binding to type V collagen. J Cell Biol. 1984;98:646–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aho S, Uitto J. Two-hybrid analysis reveals multiple direct interactions for thrombospondin 1. Matrix Biol. 1998;17:401–412 [DOI] [PubMed] [Google Scholar]
- 32. Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winnemöller M. Interactions between thrombospondin and the small proteoglycan decorin: interference with cell attachment. Eur J Cell Biol. 1992;59:47–55 [PubMed] [Google Scholar]
- 34. Kong P, Gonzalez-Quesada C, Li N, Cavalera M, Lee D-W, Frangogiannis NG. Thrombospondin-1 regulates adiposity and metabolic dysfunction in diet-induced obesity enhancing adipose inflammation and stimulating adipocyte proliferation. Am J Physiol Endocrinol Metab. 2013;305:E439–E450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW. Fat feeding causes widespread in vivo insulin resistance, decreased energy expenditure, and obesity in rats. Am J Physiol. 1986;251:E576–E583 [DOI] [PubMed] [Google Scholar]
- 36. Tremblay AJ, Lamarche B, Guay V, Charest A, Lemelin V, Couture P. Short-term, high-fat diet increases the expression of key intestinal genes involved in lipoprotein metabolism in healthy men. Am J Clin Nutr. 2013;98:32–41 [DOI] [PubMed] [Google Scholar]
- 37. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. N Engl J Med. 1994;331:1286–1292 [DOI] [PubMed] [Google Scholar]
- 39. Sweetwyne MT, Murphy-Ullrich JE. Thrombospondin1 in tissue repair and fibrosis: TGF-β-dependent and independent mechanisms. Matrix Biol. 2012;31:178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berria R, Wang L, Richardson DK, et al. Increased collagen content in insulin-resistant skeletal muscle. Am J Physiol Endocrinol Metab. 2006;290:E560–E565 [DOI] [PubMed] [Google Scholar]
- 42. Kang L, Ayala JE, Lee-Young RS, et al. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin α2β1 in mice. Diabetes. 2011;60:416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687 [DOI] [PubMed] [Google Scholar]
- 44. Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2:a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. 2002;109:1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rodnick KJ, Haskell WL, Swislocki AL, Foley JE, Reaven GM. Improved insulin action in muscle, liver, and adipose tissue in physically trained human subjects. Am J Physiol. 1987;253:E489–E495 [DOI] [PubMed] [Google Scholar]
- 47. Bostrom P, Wu J, Jedrychowski MP, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–465 [DOI] [PubMed] [Google Scholar]
- 49. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19:1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]