

Abstract
In addition to the well-characterized role of the sex steroid receptors in regulating fertility and reproduction, reproductive events are also mediated by the hypothalamic-pituitary-adrenal axis in response to an individual's environment. Glucocorticoid secretion in response to stress contributes to the well-characterized suppression of the hypothalamic-pituitary-gonadal axis through central actions in the hypothalamus and pituitary. However, both animal and in vitro studies indicate that other components of the reproductive system are also regulated by glucocorticoids. Furthermore, in the absence of stress, it appears that homeostatic glucocorticoid signaling plays a significant role in reproduction and fertility in all tissues comprising the hypothalamic-pituitary-gonadal axis. Indeed, as central regulators of the immune response, glucocorticoids are uniquely poised to integrate an individual's infectious, inflammatory, stress, nutritional, and metabolic status through glucocorticoid receptor signaling in target tissues. Endocrine signaling between tissues regulating the immune and stress response and those determining reproductive status provides an evolutionary advantage, facilitating the trade-off between reproductive investment and offspring fitness. This review focuses on the actions of glucocorticoids in tissues important for fertility and reproduction, highlighting recent studies that show glucocorticoid signaling plays a significant role throughout the hypothalamic-pituitary-gonadal axis and characterizing these effects as permissive or inhibitory in terms of facilitating reproductive success.
Life history theory asserts that the allocation of energetic resources is a trade-off between survival and reproduction (1, 2). The intrinsic and extrinsic environment will drive the age of the first reproductive event, number and size of offspring, and reproductive lifespan. At the physiological level, the division of resources is likely mediated, in part, through regulation of the stress response by the hypothalamic-pituitary-adrenal (HPA) axis. Activation of the HPA axis results in elevated levels of glucocorticoids, which favors energy mobilization, cardiac output. and sharpened cognition over growth, cellular immunity, and reproduction (3–9). Therefore, when circulating levels of glucocorticoids surpass levels shown to promote fertility, survival occurs at the expense of reproduction. At homeostatic levels, glucocorticoids regulate the timing of puberty onset, mediate the release of sex steroids, and integrate immune regulation of conception and pregnancy progression, suggesting that both stress-induced and physiological levels of glucocorticoids are necessary for fertility (10–12). Here we provide a brief review of data implicating glucocorticoids in fertility and reproduction and highlighting recent developments of the biological role of glucocorticoid signaling in the reproductive tract. Finally, we discuss new avenues of research that connect glucocorticoids with the establishment and maintenance of pregnancy.
The Glucocorticoid Receptor (GR): Mechanisms Generating Signaling Diversity
Initially named for their role in glucose metabolism, glucocorticoids are now recognized to play a pivotal role in a spectrum of biological processes, including immune and cardiovascular function, growth and development, cognition and behavior, and cell proliferation and survival (13–18). The cellular response to glucocorticoids exhibits profound diversity and specificity of action (19–22). For example, glucocorticoids are believed to regulate both pro- and antiinflammatory actions of the innate and adaptive immune systems during the inflammatory response (23–25). The molecular actions of glucocorticoids are mediated by their circulating levels, local metabolism, and intracellular signaling through the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors (26–28). Consistent with the broad effects of glucocorticoids, GR is expressed in nearly all tissues and cell types and is necessary for life after birth (29). The human GR gene consists of 9 exons located on chromosome 5 (Figure 1A). Alternative splicing in exon 9 creates 2 transcriptional isoforms of GR, hGRα and hGRβ, which differ in their carboxy termini (30). hGRα represents the classic GR, functioning as a ligand-dependent transcription factor, and resides primarily in the cytoplasm until bound by ligand. Conversely, hGRβ does not bind glucocorticoids but localizes to the nucleus where it functions as a dominant-negative inhibitor of the transcriptional activity of hGRα (31, 32). In the presence of GR antagonists, hGRβ exerts its own transcriptional regulatory program (33). Depending on their relative expression, ligand availability, and activity, the presence of hGRα and/or hGRβ contributes to the tissue-specific effects of glucocorticoids.
Figure 1.

Organization of and modifications to the human GR gene contribute to signaling diversity A, The human GR gene (NR3C1) is contained in one locus on chromosome 5 (5q31). Nine exons comprise the human GR primary transcript. Exon 1 forms the 5′-untranslated region, whereas exons 2–9 form the protein-coding region. Exon 2 encodes most of the NTD (N-terminal domain), exons 3 and 4 encode the DBD (DNA-binding domain), and exons 5–9 encode the hinge (H) region and LBD (ligand-binding domain). Alternative splicing of the primary transcript results in the α- and β-transcriptional human GR isoforms. B, Translational initiation at 8 different AUG start codons in a single human GR mRNA produces 8 receptor isoforms with progressively shorter NTDs (isoforms A–D3). Alternative posttranslational modifications also contribute to the diversity of human GR. Suggested and validated sites of phosphorylation (P), sumoylation (S), and acetylation (A) are indicated. C, The GR signals as a ligand-dependent transcription factor, where unliganded GR resides primarily in the cytoplasm of cells as part of a multiprotein complex. Cortisol, the most abundant natural glucocorticoid in humans, is converted from inactive cortisone by 11β-HSD I. As a mechanism for local regulation of glucocorticoid action, the biologically active form cortisol can be converted to the inactive cortisone by 11β-HSD II. Upon binding glucocorticoids, GR undergoes a conformational change, dissociates from the heterocomplex, and translocates into the nucleus. Ligand binding is also responsible for a hyperphosphorylated state of the GR protein. Once within the nucleus, GR regulates transcription of target genes by direct binding to GREs (5′-AGAACAnnnTGTTCT-3′, where “n” is any nucleotide) or nGREs (5′-CTCCnGGAGA-3′ nGRE 1, nGRE identified with variable number of spacer nucleotides 0–2), in a composite manner bound to a GRE and interacting with adjacent transcription factor binding sites (STAT5), or tethered to other transcription factors without direct DNA binding (NFκB). 11β-HSB, 11-β hydroxysteroid dehydrogenase; HSP: heat shock protein; NPC: nuclear pore complex; nGRE: negative GRE; p65: nuclear factor NF-κ-B p65 subunit; p50: nuclear factor NF-κ-B p50 subunit.
Multiple translational initiation sites also provide signaling diversity to the hGR (Figure 1B) (34). hGRα and hGRβ share a common mRNA domain with at least 8 alternative translational initiation sites, indicating each splice variant can then generate multiple GR isoforms. The expression of these GR isoforms varies among tissues, and the ratios of various GR isoforms in a given cell can alter their transcriptional potential (34). The profound complexity in the transcription and translation of the hGR gene allows up to 256 different combinations of homo- and heterodimers that exhibit varying expression levels and signaling activities (35). Furthermore, each GR isoform is subject to several posttranslational modifications, including phosphorylation, acetylation, ubiquitination, and sumoylation, which also impact receptor activity (Figure 1B) (20, 36).
Translated hGR is a modular protein, composed of an N-terminal domain that directs transactivation of target genes through binding coregulators and components of the basal transcription machinery; a central DNA-binding domain which interacts with target DNA sequences termed “glucocorticoid response elements” (GREs); and a C-terminal ligand-binding domain that contains steroid and heat shock protein-binding sites (37). Unliganded hGRα resides primarily in the cytoplasm as part of a large multiprotein complex that includes chaperone proteins, namely heat shock proteins 90, 70, and 56 (Figure 1C) (38–40). Following ligand binding, the receptor undergoes a conformational change that results in the dissociation from the multiprotein chaperone complex and translocation into the nucleus (39, 41). Within the nucleus, GR binds as a homodimer directly to either stimulatory or inhibitory GREs to stimulate or repress the expression of target genes (42–44). The interaction of hGRα with GREs is dynamic, with the physical interaction occurring on the order of seconds and the transcriptional result determined by both the GRE sequence and promoter context (45, 46). Alternatively, ligand-activated hGRα can regulate gene expression by physically associating with other transcription factors. These protein-protein interactions of hGRα occur on promoters that do not contain GREs but seem to require tethering to DNA-bound proteins or in a composite manner in which promoters contain both a GRE and the responsive element for GR-interacting transcription factors (47–49). GR-mediated transcriptional repression can occur through direct DNA binding at negative GREs or through protein-protein interactions with DNA-bound transcription factors. An example by which GR tethers is nuclear factor κ-light-chain-enhancer of activated B cells (nuclear factor-κB) or activator protein-1, which results in the transrepression of a wide variety of proinflammatory genes (44, 50). It is highly likely that there are many other GR transcription factor-interacting partners that have not yet been discovered. Cellular processing of GR and the diverse mechanisms by which GR regulates gene transcription contribute to the enormous potential for tissue- and cell-specific effects of glucocorticoids. An overview of the transcriptional and translational regulation of GR and the aspects of GR signaling leading to diversity in signaling are summarized in Figure 1.
Stress and Homeostatic Regulation of Fertility
As a primary mediator of basal and stress-related homeostasis, it is not surprising that stress-induced levels of glucocorticoids have been linked to reduced fecundity. Offspring of female rats subjected to restraint or environment stress during pregnancy experienced fewer and longer pregnancies, with less viable young, as adults (51, 52). Male rats from stressed dams also demonstrate reduced sexual behavior and fertility (53). Stress-induced tradeoffs to reproductive output are evident across species. Other mammals, birds, reptiles, and plants all respond to physical stress by decreasing male and female reproductive function (54–58). Case reports and retrospective studies indicate the response to stress in humans is detrimental to reproductive function throughout human life. High perceived stress during pregnancy is a risk factor for preterm labor and poor outcomes in offspring (59, 60). Psychological or physical stress experienced by adolescents can alter the onset of puberty, shifting the clock on fertility (61, 62).
Influence on Pubertal Timing
Early evidence suggesting the involvement of adrenally produced glucocorticoids in sexual maturation and fertility stems from clinical reports of patients with Addison's disease and Cushing's syndrome (63–65). Both in the case of hypocortisolism (Addison's disease) and hypercorticism (Cushing's syndrome) the onset of puberty is altered. Studies tracking daily cortisol secretion levels in healthy prepubertal girls found a positive association between glucocorticoid secretion and the age of puberty onset (10). Animal models have provided further evidence of a direct contribution by glucocorticoids to the regulation of pubertal timing and offered some insight in the molecular mechanisms by which this occurs (66–68). Both fetal and prepubertal exposure to glucocorticoids appears to be an important determinant of postnatal reproductive development. Offspring in mice of mothers subjected to stress, treated with ACTH, or treated with synthetic glucocorticoids during pregnancy demonstrate a subsequent delay in the onset of puberty (67, 69–71). A model of perinatal exposure in rats resulted in a delay in the timing of vaginal opening, an indicator of puberty in female rats, and disturbances to reproduction in males and females (72). Rats administered the synthetic glucocorticoid dexamethasone at a point later in development but prior to puberty also demonstrate delayed onset of vaginal opening (68). Furthermore, these studies showed that the delayed onset of puberty caused by exposure to glucocorticoids occurs independently of GnRH and LH, suggesting that glucocorticoids target tissues outside of the hypothalamus or pituitary.
The Male Reproductive Tract
The male gonads are also direct targets of glucocorticoid action, mediating stress-induced inhibition of testicular steroidogenesis (see Figure 3) (73, 74). The ability of glucocorticoids to control testosterone biosynthesis in Leydig cells through binding intracellular GR was initially characterized in 1976, and since that time, studies in Leydig cell culture have provided substantial insight into the molecular mechanisms involved (75). Expression of GR has been described in multiple cell types of the testis and expression is conserved across species (76–78). The direct effects of glucocorticoids, mediated by GR, include inhibition of testosterone biosynthetic enzymes and testicular LH receptor number (79–83). Glucocorticoids also inhibit the synthesis of the cholesterol side-chain cleavage enzyme (P450scc), 3β-hydroxysteroid dehydrogenase (3βHSD), and the steroidogenic acute regulatory protein, key regulators of steroidogenesis, in cultured Leydig cells. Downstream mediators of glucocorticoid signaling, namely Annexin 1 and the glucocorticoid-induced leucine zipper (GILZ), may also play a role in homeostatic preservation of fertility. Annexin 1 administered to mice recapitulates the inhibitory effects of glucocorticoids on testosterone release (84). The germline deletion of GILZ in mice results in a significant decrease in circulating corticosterone, a significant increase in circulating testosterone, and male sterility (85). Starting at postnatal day 10, the absence of GILZ results in progressively worse testicular pathology, where germ cells and mature spermatozoa are absent by 2 months of age. The metabolic phenotype in these mice may preclude establishing whether absence of gonadally expressed GILZ is directly responsible for the male phenotype, which may be clarified through cell type-specific deletion with the floxed allele. Interestingly, expression of GR in the testis of GILZ-knockout mice is also significantly decreased, suggesting that both excessive and insufficient local glucocorticoid signaling may be detrimental to the maintenance of male fertility.
Figure 3.
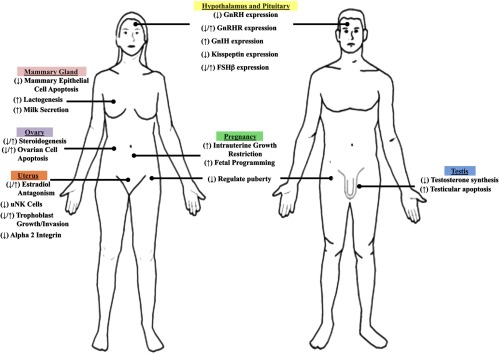
The role of glucocorticoids in tissues of the reproductive tract. The schematic represents several highlighted roles of glucocorticoids (stress-induced or exogenous exposure) in major organs of the reproductive tract. Common central functions in the hypothalamus and pituitary are indicated for the male and female. Sex-specific organs are also indicated. Glucocorticoids induce both repressive and enhancing effects, indicated by right arrow and left arrow, respectively.
The ability of glucocorticoids to act in a tissue- and cell type-specific manner is regulated not only by the relative levels of GR but also by the oxidative or reductive activity of 11βHSD enzymes, which catalyze the conversion of active glucocorticoids to inactive forms and vice versa (86, 87). In vivo, 11βHSD type I exhibits both oxidative and reductive activities, whereas 11βHSD type II is exclusively oxidative (88, 89). Expression of 11βHSD II is tissue specific, with high expression in the kidney, colon, pancreas, and placenta (90–92). In contrast, 11βHSD I is widely expressed, with elevated activity in Leydig cells (93, 94). Considerable debate has centered on the role of 11βHSD I in the testis as a “gate-keeper” of glucocorticoid action. Interestingly, incubating primary rat Leydig cells with corticosterone initially shifts the activity of transfected 11βHSD I to an oxidase, inactivating glucocorticoids (95). The oxidase activity gradually declines over time, but this finding suggests that 11βHSD I may represent a rapid mechanism for modulating glucocorticoid control of testosterone suppression. It is also possible that IIβHSD II plays a role in mediating intracellular glucocorticoid levels. Although expressed at a lower concentration than 11βHSD I, when the activity of IIβHSD II was inhibited in primary rat Leydig cell culture, the ability of corticosterone to suppress testosterone production increased (96). It appears that the hypothalamus and pituitary may possess mechanisms to exert endocrine control over glucocorticoid activity in the testis. LH increases the expression and activity of 11βHSD II, thereby enhancing the capacity for glucocorticoid inactivation, in rat Leydig cells in vitro, indicating both global and local mechanisms exist to regulate glucocorticoid actions in the testis (97).
Coinciding with the glucocorticoid-mediated reduction of testosterone synthesis in Leydig cells, exogenous and stress-induced glucocorticoids cause Leydig cell death, thereby providing 2 contributing mechanisms to stress-derived androgen dysfunction in males (98, 99). Glucocorticoid-induced apoptosis resulting from exogenous exposure has also been described for other cell types in the testis. Spermatogonia, spermatocytes, and spermatids are susceptible to apoptosis by exogenous glucocorticoids in murine models (100, 101). Furthermore, following 7 days of dexamethasone, the seminiferous tubules display widespread alterations to morphology, including vacuolization, disorganization of the germ cell layer, loss of elongated spermatids, and atrophy. The changes to testicular morphology indicate exogenous glucocorticoids have both cytotoxic and apoptotic effects in the testis. Apoptosis in Leydig and germ cells is thought to be mediated by GR, although the exact mechanisms involved in cytotoxicity and apoptosis remain unclear. Deprivation of gonadotropins and testosterone results in stage-specific germ cell apoptosis (102). Yet, glucocorticoid-mediated apoptosis occurs in androgen-dependent and -independent stages, suggesting increased apoptosis induced by exogenous glucocorticoids may not be entirely related to changes in testosterone biosynthesis. Glucocorticoids up-regulate the expression of Bax and Fas ligand (FasL), 2 important proapoptotic proteins, in testicular germ cells in vivo (103, 104). However, it is possible that up-regulated Bax and FasL are secondary to androgen withdrawal induced by glucocorticoids, and further studies are needed to clarify the direct vs indirect contributions of glucocorticoids to germ cell apoptosis, as well as the ability of endogenous cortisol or corticosterone to elicit a similar response (105).
The Female Reproductive Tract
Studies pertaining to the role of glucocorticoids in the female reproductive tract have largely centered on their function in the hypothalamus and pituitary, where it is well established that the stress-activated HPA axis suppresses hypothalamic-pituitary-gonadal (HPG) function. Excellent reviews have summarized the known mechanisms involved, and therefore, only historic highlights and new investigations will be discussed here (106–109). One of the most important functions of the hypothalamus in the HPG axis is to integrate endocrine signals originating from throughout the body to direct a vast number of reproductive functions (Figure 2). The reproductive functions regulated by the hypothalamus are largely mediated by the synthesis and release of GnRH from the preoptic area (110, 111). GnRH is primarily responsible for secretion of the gonadotropins, LH and FSH, from the pituitary. A link between stress and suppression of GnRH release, and therefore LH and FSH secretion, was found almost 30 years ago (112, 113). Since that time, chronic glucocorticoid exposure, simulating the stress response, was found to suppress gonadotropin release by interfering with hypothalamic GnRH and disrupting the menstrual cycles of monkeys (114, 115). The expression of GR in the hypothalamus suggests the effects of glucocorticoids may be direct (116). In fact, dexamethasone-induced GR signaling in GnRH-secreting hypothalamic cell lines represses GnRH promoter activity and subsequent GnRH mRNA levels through its association with a multiprotein complex at negative GRE sites (117). The inhibitory effects of glucocorticoids found in these immortalized cell lines in vitro are further supported by studies evaluating hypothalamic GnRH mRNA expression in adult male rats treated chronically with corticosterone (118). Although chronic stress and elevated levels of glucocorticoids predominantly suppress gonadotropin secretion, the response to transient or acute stress can be variable, either inhibiting or stimulating GnRH receptors (GnRHRs) and gonadotropin subunit expression (119). GnRH drives the synthesis and secretion of LH and FSH through binding of the GnRHR in pituitary gonadotropes (120, 121). Dexamethasone treatment in the mouse pituitary gonadotrope cell line LβT2 indicates glucocorticoids may directly induce GnRHR gene expression. Functional studies in the rat pituitary tumor cell line GH3 transfected with the mouse GnRHR gene identified a promoter region containing an activator protein-1 site important for mediating GnRHR induction by dexamethasone (122). However, the contribution of endogenous glucocorticoids and their intracellular metabolism has not been evaluated with regard to regulation of GnRHR expression. Furthermore, insight into the regulation of GnRHR by glucocorticoids is currently based on discoveries in cell lines and may not represent regulation occurring by endogenous hormone in vivo. Translating these findings into animal models may provide a mechanism by which to discriminate the inhibitory and stimulatory effects of glucocorticoids in the pituitary. Gonadotropin subunit gene expression also demonstrates negative and positive regulation by glucocorticoids. An acute dexamethasone treatment in rats decreased FSHβ mRNA levels in the pituitary by 14-fold (118). However, in primary cultured anterior pituitary cells, glucocorticoids stimulated FSHβ mRNA, which may indicate that glucocorticoid regulation of gonadotropin subunit expression is contextually dependent (123).
Figure 2.
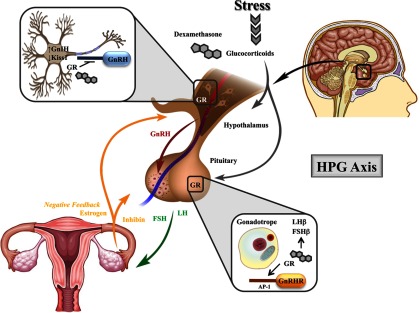
Neuroendocrine regulation of the HPG axis by glugocorticoids. As part of the HPG axis, GnRH is a central driver of the reproductive axis, responsible for the synthesis and secretion of the gonadotropins LH and FSH from the pituitary. In the female reproductive tract, LH and FSH stimulate the ovaries to produce estrogen and are required for ovulation. As part of a classical feedback loop, hormones produced by the ovaries (estrogen and inhibin) inhibit the production of GnRH and the gonadotropins. Stress-induced increases in circulating glucocorticoids or exogenous administration of the synthetic glucocorticoid dexamethasone are largely inhibitory to the reproductive functions of the hypothalamus and pituitary. GR signaling in the hypothalamus is believed to be directly involved in repressing transcription of GnRH. Furthermore, glucocorticoids are also believed to regulate the number of GnIH and kisspeptin (Kiss1) expressing neurons. In the pituitary, both the natural ligand and dexamethasone regulate gonadotropin subunit expression, although both inhibitory and stimulatory effects have been described. In vitro studies suggest dexamethasone directly induces GnRHR expression. These findings indicate glucocorticoids may influence the neuroendocrine functions of the HPG axis both positively and negatively, which provides a mechanism by which glucocorticoids can respond to periods of extreme stress, as well as homeostatic conditions. AP-1, activator protein 1.
In addition to directly regulating GnRH, GnRHR, and pituitary gonadotropes, glucocorticoids may regulate their suppressive effects through novel mediators of the HPG axis (Figure 2). The discovery of gonadotropin-inhibitory hormone (GnIH) and kisspeptin has increased our understanding of GnRH secretion and the way in which glucocorticoids target this process. Avian GnIH and the mammalian orthologs, known as RFamide-related peptides, exert their inhibitory effects through direct contact with GnRH neurons and regulation of the LHβ and FSHβ subunits in the pituitary, although there is some disparity across species (124–129). GR is expressed in GnIH neurons, and stress triggers the induction of GnIH expression, which can be blocked by adrenalectomy (130). Furthermore, neonatal exposure to dexamethasone results in increased number of GnIH-expressing neurons, up-regulation of the GnIH receptor in GnRH neurons, and lower GnRH mRNA expression in adult mice (131). These mice also demonstrate a delayed pubertal onset, which suggests that glucocorticoid regulation of GnIH may be part of the mechanism by which stress controls reproductive maturation and homeostasis.
Initially identified by loss-of-function mutations leading to hypogonadotropic hypogonadism, kisspeptin, encoded by the Kiss1 gene, is a powerful stimulator of GnRH secretion and gonadotropin release (132, 133). GR colocalizes to Kiss1 expressing neurons in mice and rats, and Kiss1 expression is repressed in response to stress or exogenous glucocorticoids (134–136). The tissue-specific deletion of GR in kisspeptin-expressing neurons eliminates stress-induced suppression of Kiss1 (137). Although the absence of GR in kisspeptin-containing neurons did not prevent the acute suppression of the HPG axis following stress, GR-Kiss1-deficient mice show accelerated recovery of reproductive function following stress. Thus, GR signaling in kisspeptin neurons is not the sole mediator of the glucocorticoid response to stress but contributes to the many mechanisms along the HPG axis that integrate the reproductive system and stress response. Due to the multifaceted nature by which the body integrates the HPA and HPG axis, delineating a function for GR signaling in each contributing tissue is difficult. The use of tissue- and cell-specific deletion models will certainly contribute to a greater understanding of the local vs systemic actions of glucocorticoids. Interestingly, the reproductive consequences of pituitary GR deletion have not been reported, although the negative feedback regulation of corticosterone in response to stress is impaired in these mice and likely results in suppression of reproductive activities of the pituitary (138). Certainly, evaluating both basal and stress-induced secretion of gonadotropes is of interest in this mouse model and would contribute to the understanding of primary vs secondary effects of glucocorticoid signaling in the pituitary.
In addition to the effects of glucocorticoids and GR in tissues of the central nervous system, GR expression has also been demonstrated in the ovary, where glucocorticoids directly regulate steroid biosynthesis (Figure 3) (139–142). In rat and porcine granulosa cells maintained in culture, glucocorticoids enhance gonadotropin-stimulated production of progesterone (141, 143). However, in cultured human granulosa cells, cortisol and dexamethasone inhibit LH-stimulated steroidogenesis, indicating that in vitro glucocorticoids exhibit both stimulatory and inhibitory effects in the ovary (144). Similarly, glucocorticoids have been ascribed both pro- and antiinflammatory functions in the ovary. Glucocorticoid exposure in the developing human ovary results in decreased germ cell density as a result of increased apoptosis of oogonia (145). However, glucocorticoids also act as potent cytoprotective agents in the ovary, protecting against apoptosis induced by cAMP, p53, and TNF-α stimulation in immortalized rat granulosa cells (146). In primary bovine luteal cells, the endogenous glucocorticoid cortisol suppresses TNF and interferon γ-induced apoptosis by restricting expression of caspase 8 and 3, thereby preserving corpus luteum function (147). The antiapoptotic effects of glucocorticoids may also play a role in minimizing damage related to follicle rupture. Ligand-activated GR signaling restricts the expression and activity of extracellular matrix metalloproteinase 9 in human ovarian surface epithelial cells, suppressing the inflammation-associated proteolytic injury to the ovarian surface during ovulation (148). Unfortunately, the protective effects of glucocorticoids during the ovulatory process also convey protection against apoptosis in ovarian epithelial tumors, which emphasizes the need for ligands capable of selectively activating GR functions (149).
The activity of glucocorticoids is also regulated locally within the ovary by 11βHSD I and II, which likely contribute to the ability of glucocorticoids to act in an explicit manner during ovarian steroidogenesis and apoptosis. Both isoforms have been identified in various ovarian cell types across species, and the expression of 11βHSD I and II is developmentally restricted during preovulatory follicular development (150–154). In response to gonadotropin-induced luteinization, granulosa cells express increased levels of 11βHSD I mRNA but reduced levels of 11βHSD II mRNA, as compared with granulosa cells before the LH surge (155). The expression and activity of 11βHSD II is elevated in the corpus luteum late in rat pregnancy (150). During this same period, mRNA expression of 11βHSD I is not detectable, coinciding with the inactivation of glucocorticoids that can block luteal regression. Several studies have reported a link between glucocorticoid metabolism and pregnancy outcome (12). Furthermore, reported activity of ovarian 11βHSD dehydrogenase has been correlated with the clinical outcomes of in vitro fertilization and embryo transfer, where activity was found to be inversely proportional to outcome, although activity varied in subsequent treatment cycles (156, 157). Findings in women indicate that regulation of local glucocorticoid activity may determine reproductive success, in which both low levels of 11βHSD I and glucocorticoids in the ovarian follicular fluid are associated with in vitro fertilization success (158).
Mammary glands are also a target of glucocorticoid action (Figure 3). This tissue expresses GR and is responsive to glucocorticoids in a developmental stage-specific manner (159, 160). Early during mammary gland development, undifferentiated cells respond to glucocorticoid treatment by inducing expression of cell cycle inhibitors and demonstrating diminished proliferation rates (160). However, unlike most organs, a majority of the growth and development of the mammary gland occurs during the postnatal period, with the greatest changes appearing after the onset of pregnancy and during early lactation. As with the progressive increase in mammary growth and development, circulating glucocorticoid levels and mammary GR expression also increase during pregnancy and remain high through parturition (161–164). Following parturition, glucocorticoids play an important role in lactogenesis and milk secretion. The first experiments that linked glucocorticoids to milk secretion involved hormone ablation experiments, in which both lactose synthesis and secretory activation are diminished in rats following adrenalectomy (165, 166). Unfortunately, the ability to link the effects of glucocorticoids specifically to the mammary gland following adrenalectomy is somewhat overshadowed by changes to the hormonal milieu following endocrine ablation. Studies in mouse mammary explants, whole-organ cultures, and conditional knockout mice have further characterized the role of glucocorticoids in milk protein synthesis and secretion (reviewed in Ref. 167). Interestingly, transplantation of embryonic mammary buds from GR-deficient mice into wild-type recipients resulted in abnormal ductal morphogenesis but no overt phenotypic changes to the mammary gland during pregnancy (168). Targeted deletion of GR from mammary epithelial cells during lobuloalveolar development led to incomplete epithelial penetration of the mammary fat pad that persists through pregnancy (169). Although epithelial GR-deficient mice are capable of milk production, their pups are 20% smaller, indicating intact GR signaling in the mammary gland is important but not essential for both maternal reproductive success and outcomes in offspring.
Unlike the heterogeneous response to apoptotic stimuli in the ovary, glucocorticoids primarily induce an antiapoptotic effect in cells of the mammary gland. During postlactational involution, circulating glucocorticoid levels drop and mammary epithelial cells die by programmed cell death. Glucocorticoid treatment at the time of weaning prevents involution and apoptosis, suggesting glucocorticoids act as survival factors in mammary epithelial cells (170). The molecular mechanism controlling the switch between lactation and involution in mammary epithelial cells includes activation of the signal transducer and activator of transcription (STAT)-3 and inhibition of STAT5 expression and activation (171, 172). Glucocorticoids are able to modulate early involution by regulating STAT3 activation and STAT5 inactivation and degradation, preventing apoptosis (173). Furthermore, expression profiling by microarray analysis identified several hundred genes differentially regulated by dexamethasone treatment in postlactating epithelial cells, including several cell-cycle regulators, indicating that several cell maintenance pathways may also be regulated by glucocorticoids (160). The protective effects provided by glucocorticoids appear to extend to certain breast cancer cell lines as well, in which pretreatment with dexamethasone prevents chemotherapy-induced apoptosis in the estrogen receptor (ER) α-negative human mammary epithelial cell line MCF10A-Myc (174, 175). Gene expression profiling in these cells indicates GR activation predominately regulates expression of signal transduction, metabolism, and transcription factor genes, including the essential up-regulation of serum and glucocorticoid-inducible protein kinase 1 (SGK1) and MAPK phosphatase-1 (MKP1). Interestingly, in the ERα-positive cell lines MCF-7, ZR-75-1, and Con-8, glucocorticoids inhibit cell growth through arrest of the cell cycle at the G0/G1 phase (176, 177). However, in the ERα-positive cell line BT-474, dexamethasone hinders cell-growth inhibition induced by the cytotoxic drug trastuzumab, which suggests that the variable effects of glucocorticoids in vitro may represent cell type-specific effects or different mechanisms of regulating cell growth and cell death (178). In mammary explant studies, pretreatment with dexamethasone significantly attenuated paclitaxel-induced apoptosis, decreasing tumor responsiveness to chemotherapy (179). The prosurvival or antiapoptotic signaling induced by dexamethasone in cells of the mammary gland is concerning because glucocorticoids are routinely used in breast cancer treatment as antiemetics and antihypersensitivity agents. However, by understanding the gene expression and signaling pathway targets of glucocorticoids in mammary cells, combinatorial use of kinases and/or kinase inhibitors with selective GR modulators may enhance the efficacy of treatments and alleviate drug resistance.
The Uterus and the Establishment of Pregnancy
Compared with the well-established role of the ER and progesterone receptor in the uterus, direct evidence implicating GR signaling in uterine biology has only recently been described (180, 181). Mifepristone, which has long been acknowledged for its antiprogestin activity, is routinely utilized as a potent high-affinity GR antagonist (182). It is surprising that the GR binding activity of Mifepristone has largely been ignored by those studying its actions in the uterus. We now know that glucocorticoids regulate many key aspects of early pregnancy, including the effects of the maternal immune system, embryo attachment and invasion, along with the growth and development of the fetus (Figure 3) (12). These actions are likely the reason that considerable interest has been given to the administration of glucocorticoids to improve pregnancy rates and outcomes (183, 184). Animal studies support the pregnancy-promoting potential of glucocorticoids (185). It is well established that uterine receptivity and embryo implantation are determined by uterine natural killer (NK) cells, cytokines, dendritic cells, and macrophages, cells regulated by the immune system (186). The immune modulatory effects of glucocorticoids provide a mechanistic link between stress suppression, the immune system, and reproductive function (Figure 4). However, unlike the tissues central to the HPG axis, the uterus has remained largely understudied in regard to direct glucocorticoid action.
Figure 4.
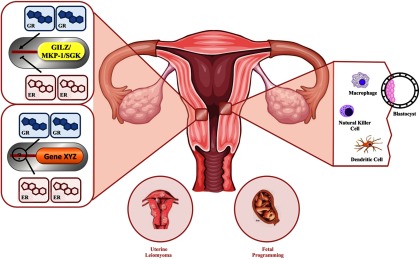
Impact of glucocorticoid signaling in the uterus. Glucocorticoids regulate many of the signal transduction and biological processes that are important for uterine function and successful reproduction. In a human uterine leiomyoma cell line and human uterine endometrial cell line, glucocorticoids regulate many unique gene targets and also target many estradiol-regulated genes. In endometrial cells, estradiol antagonizes the transcription of common immune-modulatory GR-target genes, GILZ, MAPK phosphatase 1 (MKP-1), and SGK. Altered GR and ERα recruitment to the promoter of GILZ is likely part of the mechanism by which GILZ expression is antagonized. It is not yet known what mechanisms are involved in the coregulation of common glucocorticoid and estradiol target genes. As potent regulators of the immune system, glucocorticoids inhibit aspects of the inflammatory-like response to estradiol. The immunosuppressant actions of glucocorticoids may play a role in preventing immune rejection of the implanting blastocyst. Clinical studies have indicated that glucocorticoids can influence uterine NK cells, enhancing pregnancy outcomes in some women. Macrophages and dendritic cells play a role in the immune control of endometrial receptivity to embryo implantation and are immune cell types regulated by glucocorticoids. Glucocorticoids can act in both early and late pregnancy to affect the growth and development of the fetus. Both prenatal stress and maternal exposure to exogenous glucocorticoids can lead to fetal programming of the HPA axis. MKP-1: MAPK phosphatase 1; SGK: serum- and glucocorticoid-induced protein kinase.
GR was originally identified in the uterus more than 30 years ago, and since then, glucocorticoid signaling has been found to regulate uterine estrogen and ER action in animal models of hormone supplementation/ablation (187–189). Dexamethasone blocks estradiol-induced edema of the stroma and myometrium, vasodilation and vascular permeability, uterine eosinophil chemotactic factor activity, and infiltration of eosinophils (Figure 4) (190–192). Furthermore, both glucocorticoids and estradiol are able to coregulate expression of GR and ER gene targets in the rat uterus and human uterine cell lines (Figure 4) (191). Glucocorticoid and estradiol antagonism appears to be pervasive, reported in cell types originating from tissues throughout the body (193–196). However, the mechanism by which antagonism occurs is contextual, targeting diverse genes and promoter elements, and reports of glucocorticoid and estradiol antagonism in the rodent uterus are unable to discriminate between primary effects and effects stemming from the hypothalamus or pituitary. In order to discern the primary effects and the involvement of GR in the mechanisms regulating glucocorticoid and estradiol antagonism in the uterus, cell lines were evaluated for whole-genome gene expression regulated by dexamethasone and estradiol. In the human uterine leiomyoma cell line UtLM, dexamethasone and estradiol regulate almost 10 000 gene probes, including several annotated in pathways required for cellular homeostasis (197). Ingenuity Pathway Analysis identified genes involved in cell growth, development, and differentiation as significantly regulated by glucocorticoids in human uterine leiomyoma cells, and the biological function associated with these changes was confirmed through cell cycle assays. The ability of glucocorticoids to antagonize the proliferative effects of estradiol in leiomyoma cells is in agreement with the biological antagonism of the uterotrophic effects of estradiol (198). A mechanism of glucocorticoid and estradiol transcriptional coregulation was determined in human uterine endometrial cells for the classic glucocorticoid-responsive gene GILZ (199). Both GR and ER are able to bind the promoter of GILZ, and the relative levels of receptor recruitment correspond to changes in expression, which suggests glucocorticoids and estradiol can directly regulate uterine gene expression. This notion is further supported by evidence from whole-genome microarray analysis in endometrial cells, which identified several common targets of glucocoritcoids and estradiol, many of which require GR and ER for coregulation (200). Interestingly, numerous gene targets were regulated only in the presence of glucocorticoids and estradiol but not by either ligand alone. This subset of genes represents unique targets of glucocorticoids in the presence of estradiol that have been previously unrecognized in terms of uterine physiology. The identification of novel glucocorticoid-regulated genes in single-cell populations of the uterus may indicate a mechanism by which glucocorticoids exert their estrogen-antagonistic effects throughout the uterus, and future studies should examine regulation of these targets in vivo.
In addition to direct transcriptional regulation, antagonism of glucocorticoid signaling by estradiol in the uterus may occur through regulation of chaperone proteins. Expression of heat shock proteins 90 and 70 is increased in the myometrium and endometrium in response to estradiol and during pregnancy leading up to parturition, which may act to inhibit GR activity in these uterine cell types (201). Enzymatic regulation of glucocorticoid signaling in the uterus is also likely, as both type I and II 11βHSD have been localized to the uterus, demonstrating cell-type and cyclic variation (202–206). In addition to the modulatory role in the uterus, the 11βHSD isoenzymes mediate glucocorticoid actions in the decidua, for the establishment of pregnancy, and in the placenta throughout pregnancy. In the decidua, 11βHSD I is predominantly expressed, although very is known regarding the role of 11βHSD I here (207). Changes in the expression level of 11βHSD I parallels increased decidual cell apoptosis, indicating a mechanism for programmed cell death during pregnancy (208). 11βHSD I in utero also plays an important biological role during early pregnancy, where mated rams treated with an 11βHSD I inhibitor showed impaired embryo development (209). Regulation of local glucocorticoid levels by 11βHSD II may also be important for early pregnancy. Expression of both 11βHSD isoforms in human endometrial stromal cells is enhanced during decidualization and may play a role in embryo attachment and implantation (210, 211). Placental 11βHSD II plays a significant role in guarding the fetus from high levels of maternal glucocorticoids, where overexposure of the developing fetus to excessive glucocorticoid levels is linked to lower birth weight. Human and animal models of intrauterine growth restriction report reduced expression/activity of 11βHSD II in the placenta (212–214). These observations indicate that regulation of the activity of glucocorticoids is essential for the initiation and throughout pregnancy.
Glucocorticoids regulate many processes that are thought to both negatively and positively impact key aspects of early pregnancy. Resident inflammatory cells of the uterus, and those recruited at the onset of pregnancy, are uniquely positioned to sense antigens and signals from both the maternal environment and embryo to mediate early pregnancy events (Figure 4). For example, NK cells in the uterine mucosa contribute to the cytokine response at the maternal-fetal interface, and imbalance in the temporal and spatial distribution of NK cells in the uterine mucosa corresponds to a high rate of miscarriage (215). Daily glucocorticoids suppressed levels of uterine NK cells in women with high numbers of uterine NK cells and a history of recurrent miscarriages (216). Furthermore, in 2 case reports of individuals with a total of 29 recurrent miscarriages, both patients had successful pregnancies and deliveries following glucocorticoid treatment (184, 217). These data suggest that glucocorticoids may play a role in improving the intrauterine environment. Synthetic glucocorticoids in vitro have been reported to increase proliferation in a GR-positive choriocarcinoma cell line (a model for first-trimester human trophoblasts), as well as enhance Matrigel invasion and up-regulate expression of the promatrix metalloproteinase-2 (218). However, these studies are in opposition to those that find dexamethasone suppressed expression of matrix metalloproteinase-9, necessary for cytotrophoblast invasion in vitro, and migration, indicating that glucocorticoids can both positively and negatively impact trophoblast invasion (219). Glucocorticoids have been shown to negatively impact trophoblast growth, reduce proliferation, and induce apoptosis (218, 220–222). Prior to invasion, embryo implantation is facilitated by cell-surface adhesion molecules on the trophoblast and endometrium. Dexamethasone treatment of cytotrophoblasts significantly reduced cell surface expression of the α 2 integrin subunit, indicating that glucocorticoids may regulate embryo attachment, as well as invasion (223). The balance between pregnancy-promoting and -inhibiting effects suggests that the concentration and activity of glucocorticoids are tightly linked to early pregnancy success.
In Utero Development and Fetal Programming
Unfortunately, a higher frequency of growth-restricted fetuses is a well-documented side effect of glucocorticoid treatment during pregnancy (224–227). The consequences of intrauterine growth restriction extend beyond the immediate perinatal period. Data has linked fetal exposure to excessive glucocorticoids triggered by an adverse maternal environment or exogenous exposure and programming of the HPA axis (Figure 3) (228–232). Fetal programming during development by glucocorticoid exposure has been widely reported in the literature and thus will only be briefly reviewed here (see recent reviews in Refs. 233–236). Epidemiologic studies suggest that elevations in glucocorticoid exposure during fetal life, in addition to intrauterine growth restriction, may result in fetal programming of cardiovascular, metabolic, and neuroendocrine disorders in adult life. In the placenta, conversion of maternal cortisol or corticosterone to their inactive forms by 11βHSD II provides a protective barrier against fetal exposure to high levels of glucocorticoids (237). However, following exogenous administration of synthetic glucocorticoids or in times of glucocorticoid excess, this sentry of the placenta may be ineffective in protecting the fetus from overexposure. In the rat, maternal exposure to dexamethasone during gestation results in reduced birth weight and hypertensive adult offspring (238, 239). A singleton-bearing, nonhuman primate model of dexamethasone treatment from midterm revealed a dose-dependent reduction in postnatal growth, impaired glucose tolerance, and hyperinsulemia in offspring (240). Although the impact of prenatal synthetic glucocorticoid exposure on HPA function in humans is not well studied compared with other mammalian species, a follow-up report 30 years following offspring of individuals who received antenatal betamethasone revealed signs of insulin resistance (241). Furthermore, fetal exposure to synthetic glucocorticoids is associated with persistent neurologic consequences in humans, including cortical thinning (242). Overexposure to endogenous glucocorticoids, through reduced or absent activity of placental 11βHSD II, has also been linked to reduced birth weight and programming effects very similar to those caused by antenatal exposure to exogenous glucocorticoids (243–245). Alterations to the HPA axis may involve persistently altered expression of the hippocampal GR, an important component of the negative feedback loop that terminates stress-induced HPA axis activation (246). Such effects appear to be transgenerational, demonstrating permanent changes in the molecular regulation of the HPA axis following prenatal stress or glucocorticoid exposure (247). Persistent changes in gene expression that remain after the inciting event are thought to be mediated by epigenetic mechanisms that affect both mRNA and mRNA variant expression (248). Intrauterine growth restriction in the rat causes changes in histone association with GR, variant and total mRNA levels, and protein expression in the hippocampus (249). Furthermore, fetal glucocorticoid exposure caused significant variations in genome-wide promoter methylation, ultimately leading to profound changes to the epigenetic landscape. These differences may underlie the long-term changes in HPA axis function following antenatal glucocorticoid treatment, although it is unclear what mechanisms prevail. Despite these detrimental effects, the antenatal use of glucocorticoids has been employed for more than 50 years to accelerate fetal lung maturation, reduce respiratory distress syndrome, and reduce mortality in preterm infants, which represent 10% of all pregnancies (250). Until a selective GR agonist is demonstrated with discriminating effects in the lung, glucocorticoid treatment in preterm infants represents their primary mechanism for survival.
Summary and Future Perspectives
Through the use of glucocorticoid ablation/supplementation experiments, in vitro tissue explants and cell lines, and limited conditional knockout models, we now possess a greater understanding of how glucocorticoids mediate fertility (Figure 3). However, the role and mechanism of GR signaling in tissues of the reproductive tract are far from completely understood and will benefit from studies that support previous tissue explant and cell line studies. Glucocorticoids exert a range of positive and negative effects throughout the HPG axis. Furthermore, the differential expression of enzymes that regulate the activation and inactivation of glucocorticoids contributes to the complex signaling pathways involved in stress-mediated reproductive consequences and allow for the local regulation of reproductive functions through intracellular metabolism of glucocorticoids (251). The lack of tissue-specific GR ablation studies in organs of the reproductive tract has so far precluded the discrimination between primary effects in the hypothalamus and pituitary and secondary effects in the mammary glands and gonads. Furthermore, it is important to consider the effects of glucocorticoids demonstrated experimentally as they relate to homeostatic or stress-induced levels of glucocorticoids, where endocrine supplementation may represent acute or chronic stress levels but not homeostatic levels. Physiologic glucocorticoid levels and regulated local activity most likely act as rheostats, governing the permissive or inhibitory actions as they pertain to the reproductive system, which may be contrary to those actions occurring in times of extreme stress or exposure to exogenous forms of glucocorticoids. Acknowledging a conventional function for glucocorticoids in the establishment and maintenance of fertility, as well as mediating stress-induced reproductive changes, may alter the commonly held view that glucocorticoids are hormones primarily of the stress response system.
Acknowledgments
We thank Dr. Robert Oakley and Dr. Carl Bortner for critical reading of the manuscript and National Institute of Environmental Health Sciences Arts and Photography for contributions to figure preparation.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ER
- estrogen receptor
- GILZ
- glucocorticoid-induced leucine zipper
- GnIH
- gonadotropin-inhibitory hormone
- GnRHR
- GnRH receptor
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HPA
- hypothalamic-pituitary-adrenal
- HPG
- hypothalamic-pituitary-gonadal
- HSD
- hydroxysteroid dehydrogenase
- NK
- natural killer
- STAT
- signal transducer and activator of transcription.
References
- 1. Stearns SC. Life-history tactics: a review of the ideas. Q Rev Biol. 1976;51(1):3–47 [DOI] [PubMed] [Google Scholar]
- 2. Wingfield JC, Sapolsky RM. Reproduction and resistance to stress: when and how. J Neuroendocrinol. 2003;15(8):711–724 [DOI] [PubMed] [Google Scholar]
- 3. Sayers G. The adrenal cortex and homoestasis. Physiol Rev. 1950;30(3):241–320 [DOI] [PubMed] [Google Scholar]
- 4. Welt ID, Stetten D, Jr, Ingle DJ, Morley EH. Effect of cortisone upon rates of glucose production and oxidation in the rat. J Biol Chem. 1952;197(1):57–66 [PubMed] [Google Scholar]
- 5. Galosy RA, Clarke LK, Vasko MR, Crawford IL. Neurophysiology and neuropharmacology of cardiovascular regulation and stress. Neurosci Biobehav Rev. 1981;5(1): 137–175 [DOI] [PubMed] [Google Scholar]
- 6. Stolz G, Aschoff JC, Born J, Aschoff J. VEP, physiological and psychological circadian variations in humans. J Neurol. 1988;235(5):308–313 [DOI] [PubMed] [Google Scholar]
- 7. Elenkov IJ, Chrousos GP. Stress hormones, Th1/Th2 patterns, pro/anti-inflammatory cytokines and susceptibility to disease. Trends Endocrinol Metab. 1999;10(9):359–368 [DOI] [PubMed] [Google Scholar]
- 8. Schwartz NB, McCormack CE. Reproduction: gonadal function and its regulation. Annu Rev Physiol, 1972;34:425–472 [DOI] [PubMed] [Google Scholar]
- 9. Stier KS, Almasi B, Gasparini J, Piault R, Roulin A, Jenni L. Effects of corticosterone on innate and humoral immune functions and oxidative stress in barn owl nestlings. J Exp Biol. 2009;212(Pt 13):2085–209191 [DOI] [PubMed] [Google Scholar]
- 10. Shi L, Wudy SA, Buyken AE., Maser-Gluth C, Hartmann MF, Remer T. Prepubertal glucocorticoid status and pubertal timing. J Clin Endocrinol Metab. 2011;96(6):E891–E898 [DOI] [PubMed] [Google Scholar]
- 11. Dubey AK, Plant TM. A suppression of gonadotropin secretion by cortisol in castrated male rhesus monkeys (Macaca mulatta) mediated by the interruption of hypothalamic gonadotropin-releasing hormone release. Biol Reprod,. 1985;33(2):423–431 [DOI] [PubMed] [Google Scholar]
- 12. Michael AE., Papageorghiou AT. Potential significance of physiological and pharmacological glucocorticoids in early pregnancy. Hum Reprod Update. 2008;14(5):497–517 [DOI] [PubMed] [Google Scholar]
- 13. Olefsky JM, Kimmerling G. Effects of glucocorticoids on carbohydrate metabolism. Am J Med Sci. 1976;271(2):202–210 [DOI] [PubMed] [Google Scholar]
- 14. Munck A, Koritz SB. Studies on the mode of action of glucocorticoids in rats. I. Early effects of cortisol on blood glucose and on glucose entry into muscle, liver and adipose tissue. Biochim Biophys Acta. 1962; 57:310–317 [DOI] [PubMed] [Google Scholar]
- 15. Swingle WW, Davanzo JP, Glenister D, Wagle G, Osborne M, Rowen R. Effect of mineralo- and glucoccoticoids on fasted adrenalectomized dogs subjected to electroshock. Proc Soc Exp Biol Med. 1960;104:184–188 [DOI] [PubMed] [Google Scholar]
- 16. Ballard PL, Ballard RA. Glucocorticoid receptors and the role of glucocorticoids in fetal lung development. Proc Natl Acad Sci USA. 1972;69(9):2668–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmid PG, Eckstein JW, Abboud FM. Comparison of effects of deoxycorticosterone and dexamethasone on cardiovascular responses to norepinephrine. J Clin Invest. 1967;46(4):590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chrousos GP, Charmandari E, Kino T. Glucocorticoid action networks–an introduction to systems biology. J Clin Endocrinol Metab. 2004;89(2):563–564 [DOI] [PubMed] [Google Scholar]
- 19. Lamberts SW, Huizenga AT, de Lange P, de Jong FH, Koper JW. Clinical aspects of glucocorticoid sensitivity. Steroids. 1996;61(4):157–160 [DOI] [PubMed] [Google Scholar]
- 20. Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem. 2011;286(5):3177–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burd CJ, Archer TK. Chromatin architecture defines the glucocorticoid response. Mol Cell Endocrinol. 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Viegas LR, Hoijman E, Beato M, Pecci A. Mechanisms involved in tissue-specific apopotosis regulated by glucocorticoids. J Steroid Biochem Mol Biol. 2008;109(3–5):273–278 [DOI] [PubMed] [Google Scholar]
- 24. Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab. 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013;154(3):993–1007 [DOI] [PubMed] [Google Scholar]
- 26. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amelung D, Hubener HJ, Roka L, Meyerheim G. Conversion of cortisone to compound F. J Clin Endocrinol Metab. 1953;13(9):1125–1126 [DOI] [PubMed] [Google Scholar]
- 28. Ramamoorthy S, Cidlowski JA. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev. 2013; 24:41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cole TJ, Blendy JA, Monaghan AP, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9(13):1608–1621 [DOI] [PubMed] [Google Scholar]
- 30. Hollenberg SM, Weinberger C, Ong ES, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318(6047):635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oakley RH, Jewell CM, Yudt MR, Bofediato DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor β isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274(39):27857–27866 [DOI] [PubMed] [Google Scholar]
- 32. Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor β, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95(6):2435–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor β binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27(6):2266–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18(3):331–342 [DOI] [PubMed] [Google Scholar]
- 35. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anbalagan M, Huderson B, Murphy L, Rowan BG. Post-translational modifications of nuclear receptors and human disease. Nucl Recept Signal. 2012;10:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giguère V, Hollenberg SM, Rosenfeld MG, Evans RM. Functional domains of the human glucocorticoid receptor. Cell. 1986;46(5):645–652 [DOI] [PubMed] [Google Scholar]
- 38. Denis M, Gustafsson JA, Wikström AC. Interaction of the Mr = 90,000 heat shock protein with the steroid-binding domain of the glucocorticoid receptor. J Biol Chem. 1988;263(34):18520–18523 [PubMed] [Google Scholar]
- 39. Pratt WB, Hutchison KA, Scherrer LC. Steroid receptor folding by heat-shock proteins and composition of the receptor heterocomplex. Trends Endocrinol Metab. 1992;3(9):326–333 [DOI] [PubMed] [Google Scholar]
- 40. Sanchez ER. Chaperoning steroidal physiology: lessons from mouse genetic models of Hsp90 and its cochaperones. Biochim Biophys Acta. 2012;1823(3):722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Terry LJ, Shows EB, Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science. 2007;318(5855):1412–1416 [DOI] [PubMed] [Google Scholar]
- 42. NIH Workshop on Mechanism of Action of Thyroid Hormone Protein-DNA interactions at steroid hormone regulated genes. Endocr Res. 1989;15(4):417–440 [DOI] [PubMed] [Google Scholar]
- 43. Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17(3):245–261 [DOI] [PubMed] [Google Scholar]
- 44. Surjit M, Ganti KP, Mukherji A, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145(2):224–241 [DOI] [PubMed] [Google Scholar]
- 45. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324(5925):407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287(5456):1262–1265 [DOI] [PubMed] [Google Scholar]
- 47. Miner JN, Yamamoto KR. Regulatory crosstalk at composite response elements. Trends Biochem Sci. 1991;16(11):423–426 [DOI] [PubMed] [Google Scholar]
- 48. Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-κ B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15(2):943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jonat C, Rahmsdorf HJ, Park KK, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62(6):1189–1204 [DOI] [PubMed] [Google Scholar]
- 50. Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol. 2007;72(4):799–809 [DOI] [PubMed] [Google Scholar]
- 51. Herrenkohl LR. Prenatal stress reduces fertility and fecundity in female offspring. Science. 1979;206(4422):1097–1099 [DOI] [PubMed] [Google Scholar]
- 52. Carney EW, Crissman JW, Liberacki AB, Clements CM, Breslin WJ. Assessment of adult and neonatal reproductive parameters in Sprague-Dawley rats exposed to propylene glycol monomethyl ether vapors for two generations. Toxicol Sci. 1999;50(2):249–258 [DOI] [PubMed] [Google Scholar]
- 53. Crump CJ, Chevins PF. Prenatal stress reduces fertility of male offspring in mice, without affecting their adult testosterone levels. Horm Behav. 1989;23(3):333–343 [DOI] [PubMed] [Google Scholar]
- 54. De Rensis F, Scaramuzzi RJ. Heat stress and seasonal effects on reproduction in the dairy cow–a review. Theriogenology. 2003;60(6):1139–1151 [DOI] [PubMed] [Google Scholar]
- 55. Schmid B, Tam-Dafond L, Jenni-Eiermann S, Arlettaz R, Schaub M, Jenni L. Modulation of the adrenocortical response to acute stress with respect to brood value, reproductive success and survival in the Eurasian hoopoe. Oecologia. 2013;173(1):33–44 [DOI] [PubMed] [Google Scholar]
- 56. Lucas LD, French SS. Stress-induced tradeoffs in a free-living lizard across a variable landscape: consequences for individuals and populations. PLoS One. 2012;7(11):e49895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Akers SW, Mitchell CA. Seismic stress effects on reproductive structures of tomato, potato, and marigold. HortScience. 1985;20(4):684–686 [PubMed] [Google Scholar]
- 58. Jin Y, Yang H, Wei Z, Ma H, Ge X. Rice male development under drought stress: phenotypic changes and stage-dependent transcriptomic reprogramming. Mol Plant. 2013;6(5):1630–1645 [DOI] [PubMed] [Google Scholar]
- 59. Mancuso RA, Schetter CD, Rini CM, Roesch SC, Hobel CJ. Maternal prenatal anxiety and corticotropin-releasing hormone associated with timing of delivery. Psychosom Med. 2004;66(5):762–769 [DOI] [PubMed] [Google Scholar]
- 60. Rice F, Jones I, Thapar A. The impact of gestational stress and prenatal growth on emotional problems in offspring: a review. Acta Psychiatr Scand. 2007;115(3):171–183 [DOI] [PubMed] [Google Scholar]
- 61. Magner JA, Rogol AD, Gorden P. Reversible growth hormone deficiency and delayed puberty triggered by a stressful experience in a young adult. Am J Med. 1984;76(4):737–742 [DOI] [PubMed] [Google Scholar]
- 62. Genazzani AD, Ricchieri F, Lanzoni C, Strucchi C, Jasonni VM. Diagnostic and therapeutic approach to hypothalamic amenorrhea. Ann N Y Acad Sci. 2006;1092:103–113 [DOI] [PubMed] [Google Scholar]
- 63. Marilus R, Dickerman Z, Kaufman H, Varsano I, Laron Z. Addison's disease associated with precocious sexual development in a boy. Acta Paediatr Scand. 1981;70(4):587–589 [DOI] [PubMed] [Google Scholar]
- 64. Zadik Z, Cooper M, Chen M, Stern N. Cushing's disease presenting as pubertal arrest. J Pediatr Endocrinol. 1993;6(2):201–204 [PubMed] [Google Scholar]
- 65. Dupuis CC, Storr HL, Perry LA, et al. Abnormal puberty in paediatric Cushing's disease: relationship with adrenal androgen, sex hormone binding globulin and gonadotrophin concentrations. Clin Endocrinol (Oxf). 2007;66(6):838–843 [DOI] [PubMed] [Google Scholar]
- 66. Killian DB, Kiesling DO, Wulff FP, Stewart AN. Effects of adrenalectomy and glucocorticoids on puberty in gilts reared in confinement. J Anim Sci. 1987;64(1):231–236 [DOI] [PubMed] [Google Scholar]
- 67. Smith JT, Waddell BJ. Increased fetal glucocorticoid exposure delays puberty onset in postnatal life. Endocrinology. 2000;141(7):2422–2428 [DOI] [PubMed] [Google Scholar]
- 68. Kinouchi R, Matsuzaki T, Iwasa T, et al. Prepubertal exposure to glucocorticoid delays puberty independent of the hypothalamic Kiss1-GnRH system in female rats. Int J Dev Neurosci. 2012;30(7):596–601 [DOI] [PubMed] [Google Scholar]
- 69. Politch JA, Herrenkohl LR. Prenatal ACTH and corticosterone: effects on reproduction in male mice. Physiol Behav. 1984;32(1):135–137 [DOI] [PubMed] [Google Scholar]
- 70. Harvey PW, Chevins PF. Deleterious effects of adrenocorticotrophic hormone administration during late pregnancy upon offspring somatic, neurological, and sexual development in mice. Teratology. 1987;35(2):229–238 [DOI] [PubMed] [Google Scholar]
- 71. Politch JA, Herrenkohl LR. Effects of prenatal stress on reproduction in male and female mice. Physiol Behav. 1984;32(1):95–99 [DOI] [PubMed] [Google Scholar]
- 72. Benesova O, Pavlik A. Perinatal treatment with glucocorticoids and the risk of maldevelopment of the brain. Neuropharmacology. 1989;28(1): 89–97 [DOI] [PubMed] [Google Scholar]
- 73. Orr TE, Mann DR. Role of glucocorticoids in the stress-induced suppression of testicular steroidogenesis in adult male rats. Horm Behav,. 1992;26(3):350–363 [DOI] [PubMed] [Google Scholar]
- 74. Hardy MP, Gao HB, Dong Q, et al. Stress hormone and male reproductive function. Cell Tissue Res. 2005;322(1):147–153 [DOI] [PubMed] [Google Scholar]
- 75. Evain D, Morera AM, Saez JM. [Glucocorticoids receptors in rat testis: their role in Leydig cells specific function and DNA synthesis (author's transl)]. Ann Endocrinol (Paris). 1976;37(2):101–102 [PubMed] [Google Scholar]
- 76. Schultz R., Isola J, Parvinen M, et al. Localization of the glucocorticoid receptor in testis and accessory sexual organs of male rat. Mol Cell Endocrinol. 1993; 95(1–2):115–120 [DOI] [PubMed] [Google Scholar]
- 77. Herrera-Luna CV, Budik S, Aurich C. Gene expression of ACTH, glucocorticoid receptors, 11βHSD enzymes, LH-, FSH-, GH receptors and aromatase in equine epididymal and testicular tissue. Reprod Domest Anim. 2012;47(6):928–935 [DOI] [PubMed] [Google Scholar]
- 78. Haeussler S, Claus R. Expression of the glucocorticoid receptor and 11β-hydroxysteroid dehydrogenase 2 in pig testes cells along fetal development. Reprod Fertil Dev. 2007;19(5):664–669 [DOI] [PubMed] [Google Scholar]
- 79. Bambino TH, Hsueh AJ. Direct inhibitory effect of glucocorticoids upon testicular luteinizing hormone receptor and steroidogenesis in vivo and in vitro. Endocrinology. 1981;108(6):2142–2148 [DOI] [PubMed] [Google Scholar]
- 80. Hales DB, Payne AH. Glucocorticoid-mediated repression of P450scc mRNA and de novo synthesis in cultured Leydig cells. Endocrinology. 1989;124(5):2099–2104 [DOI] [PubMed] [Google Scholar]
- 81. Wang X, Walsh LP, Reinhart AJ, Stocco DM. The role of arachidonic acid in steroidogenesis and steroidogenic acute regulatory (StAR) gene and protein expression. J Biol Chem. 2000;275(26):20204–20209 [DOI] [PubMed] [Google Scholar]
- 82. Payne AH, Sha LL. Multiple mechanisms for regulation of 3 β-hydroxysteroid dehydrogenase/delta 5—Δ4-isomerase, 17α-hydroxylase/C17-20 lyase cytochrome P450, and cholesterol side-chain cleavage cytochrome P450 messenger ribonucleic acid levels in primary cultures of mouse Leydig cells. Endocrinology. 1991;129(3):1429–1435 [DOI] [PubMed] [Google Scholar]
- 83. Xiao YC, Huang YD, Hardy DO, Li XK, Ge RS. Glucocorticoid suppresses steroidogenesis in rat progenitor Leydig cells. J Androl. 2010;31(4):365–371 [DOI] [PubMed] [Google Scholar]
- 84. Cover PO, Baanah-Jones F, John CD, Buckingham JC. Annexin 1 (lipocortin 1) mimics inhibitory effects of glucocorticoids on testosterone secretion and enhances effects of interleukin-1β. Endocrine. 2002;18(1):33–39 [DOI] [PubMed] [Google Scholar]
- 85. Suarez PE, Rodriquez EG, Soundarjan R, et al. The glucocorticoid-induced leucine zipper (gilz/Tsc22d3-2) gene locus plays a crucial role in male fertility. Mol Endocrinol. 2012;26(6):1000–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Monder C, White PC. 11 β-Hydroxysteroid dehydrogenase. Vitam Horm. 1993;47:187–271 [PubMed] [Google Scholar]
- 87. Stewart PM, Krozowski ZS. 11 β-Hydroxysteroid dehydrogenase. Vitam Horm. 1999;57:249–324 [PubMed] [Google Scholar]
- 88. Agarwal AK, Monder C, Eckstein B, White PC. Cloning and expression of rat cDNA encoding corticosteroid 11β-dehydrogenase. J Biol Chem. 1989;264(32):18939–18943 [PubMed] [Google Scholar]
- 89. Naray-Fejes-Toth A, Rusvai E, denault DL, St Germain DL, Fejes-Tóth G. Expression and characterization of a new species of 11 β-hydroxysteroid dehydrogenase in Xenopus oocytes. Am J Physiol. 1993;265(6 Pt 2):F896–F900 [DOI] [PubMed] [Google Scholar]
- 90. Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 11β-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol. 1994;105(2):R11–R17 [DOI] [PubMed] [Google Scholar]
- 91. Brown RW, Chapman KE, Kotelevtsev Y, et al. Cloning and production of antisera to human placental 11 β-hydroxysteroid dehydrogenase type 2. Biochem J. 1996;313(Pt 3):1007–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Seckl JR, Walker BR. Minireview: 11β-hydroxysteroid dehydrogenase type 1—a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142(4):1371–1376 [DOI] [PubMed] [Google Scholar]
- 93. Tannin GM, Agarwal AK, Monder C, New MI, White PC. The human gene for 11β-hydroxysteroid dehydrogenase. Structure, tissue distribution, and chromosomal localization. J Biol Chem. 1991;266(25):16653–16658 [PubMed] [Google Scholar]
- 94. Leckie CM, Welberg LA, Seckl JR. 11β-Hydroxysteroid dehydrogenase is a predominant reductase in intact rat Leydig cells. J Endocrinol. 1998;159(2):233–238 [DOI] [PubMed] [Google Scholar]
- 95. Ge RS, Hardy MP. Initial predominance of the oxidative activity of type I 11β-hydroxysteroid dehydrogenase in primary rat Leydig cells and transfected cell lines. J Androl. 2000;21(2):303–310 [PubMed] [Google Scholar]
- 96. Ge RS, Dong Q, Niu EM, et al. 11β-Hydroxysteroid dehydrogenase 2 in rat leydig cells: its role in blunting glucocorticoid action at physiological levels of substrate. Endocrinology. 2005;146(6):2657–2664 [DOI] [PubMed] [Google Scholar]
- 97. Wang Q, Zhang P, Gao HB. Luteinizing hormone induces expression of 11β-hydroxysteroid dehydrogenase type 2 in rat Leydig cells. Reprod Biol Endocrinol. 2009;7:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gao HB, Tong MH, Hu YQ, Guo QS, Ge R, Hardy MP. Glucocorticoid induces apoptosis in rat leydig cells. Endocrinology. 2002;143(1):130–138 [DOI] [PubMed] [Google Scholar]
- 99. Chen Y, Wang Q, Wang FF, Gao HB, Zhang P, et al. Stress induces glucocorticoid-mediated apoptosis of rat Leydig cells in vivo. Stress. 2012;15(1):74–84 [DOI] [PubMed] [Google Scholar]
- 100. Yazawa H, Sasagawa I, Nakada T. Apoptosis of testicular germ cells induced by exogenous glucocorticoid in rats. Hum Reprod. 2000;15(9):1917–1920 [DOI] [PubMed] [Google Scholar]
- 101. Orazizadeh M, Khorsandi LS, Hashemitabar M. Toxic effects of dexamethasone on mouse testicular germ cells. Andrologia. 2010;42(4):247–253 [DOI] [PubMed] [Google Scholar]
- 102. Sinha Hikim AP, Swerdloff RS. Hormonal and genetic control of germ cell apoptosis in the testis. Rev Reprod. 1999;4(1):38–47 [DOI] [PubMed] [Google Scholar]
- 103. Mahmoud H, Mahmoud O, Layasadat K, Naeim A, et al. Dexamethasone effects on Bax expression in the mouse testicular germ cells. Folia Histochem Cytobiol. 2009;47(2):237–241 [DOI] [PubMed] [Google Scholar]
- 104. Khorsandi LS, Hashemitabar M, Orazizadeh M, Albughobeish N, et al. Dexamethasone effects on fas ligand expression in mouse testicular germ cells. Pak J Biol Sci. 2008;11:2231–2236 [DOI] [PubMed] [Google Scholar]
- 105. Kondo T, Goto S, Ihara Y, et al. Diethylstilbestrol attenuates antioxidant activities in testis from male mice. Free Radic Res. 2002;36(9):957–966 [DOI] [PubMed] [Google Scholar]
- 106. Li XF, Knox AM, O'Byrne KT. Corticotrophin-releasing factor and stress-induced inhibition of the gonadotrophin-releasing hormone pulse generator in the female. Brain Res. 2010;1364:153–163 [DOI] [PubMed] [Google Scholar]
- 107. Whirledge S, Cidlowski JA. Glucocorticoids, stress, and fertility. Minerva Endocrinol. 2010;35(2):109–125 [PMC free article] [PubMed] [Google Scholar]
- 108. Nakamura K, Sheps S, Arck PC. Stress and reproductive failure: past notions, present insights and future directions. J Assist Reprod Genet. 2008;25(2–3):47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Viau V. Functional cross-talk between the hypothalamic-pituitary-gonadal and -adrenal axes. J Neuroendocrinol. 2002;14(6):506–513 [DOI] [PubMed] [Google Scholar]
- 110. Kobayashi T, Kigawa T, Mizuno M, Amenomori Y, Watanabe T. Gonadotropin-releasing activity in rat hypothalamic extract. Endocrinol Jpn. 1966;13(4):430–437 [DOI] [PubMed] [Google Scholar]
- 111. Christensen A, Bentley GE, Cabrera R, et al. Hormonal regulation of female reproduction. Horm Metab Res,. 2012;44(8):587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Matteri RL, Watson JG, Moberg GP. Stress or acute adrenocorticotrophin treatment suppresses LHRH-induced LH release in the ram. J Reprod Fertil,. 1984;72(2):385–393 [DOI] [PubMed] [Google Scholar]
- 113. Gilbeau PM, Smith CG. Naloxone reversal of stress-induced reproductive effects in the male rhesus monkey. Neuropeptides. 1985;5(4–6):335–338 [DOI] [PubMed] [Google Scholar]
- 114. Plant TM. A study of the role of the postnatal testes in determining the ontogeny of gonadotropin secretion in the male rhesus monkey (Macaca mulatta). Endocrinology. 1985;116(4):1341–1350 [DOI] [PubMed] [Google Scholar]
- 115. Hayashi KT, Moberg GP. Influence of the hypothalamic-pituitary-adrenal axis on the menstrual cycle and the pituitary responsiveness to estradiol in the female rhesus monkey (Macaca mulatta). Biol Reprod. 1990;42(2):260–265 [DOI] [PubMed] [Google Scholar]
- 116. Poisson M, Pertuiset BF, Moguilewsky M, Magdelenat H, Martin PM. [Steroid receptors in the central nervous system. Implications in neurology]. Rev Neurol (Paris). 1984;140(4):233–248 [PubMed] [Google Scholar]
- 117. Chandran UR, Attardi B, Friedman R, Dong KW, Roberts JL, DeFranco DB. Glucocorticoid receptor-mediated repression of gonadotropin-releasing hormone promoter activity in GT1 hypothalamic cell lines. Endocrinology. 1994;134(3):1467–1474 [DOI] [PubMed] [Google Scholar]
- 118. Gore AC, Attardi B, DeFranco DB. Glucocorticoid repression of the reproductive axis: effects on GnRH and gonadotropin subunit mRNA levels. Mol Cell Endocrinol. 2006; 256(1–2):40–48 [DOI] [PubMed] [Google Scholar]
- 119. Kotitschke A, Sadie-Van Gijsen H, Avenant C, Fernandes S, Hapgood JP. Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Mol Endocrinol. 2009;23(11):1726–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hyde CL, Childs G, Wahl LM, Naor Z, Catt KJ. Preparation of gonadotroph-enriched cell populations from adult rat anterior pituitary cells by centrifugal elutriation. Endocrinology. 1982;111(4):1421–1423 [DOI] [PubMed] [Google Scholar]
- 121. Cheng KW, Leung PC. The expression, regulation and signal transduction pathways of the mammalian gonadotropin-releasing hormone receptor. Can J Physiol Pharmacol. 2000;78(12):1029–1052 [PubMed] [Google Scholar]
- 122. Maya-Nunez G, Conn PM. Transcriptional regulation of the GnRH receptor gene by glucocorticoids. Mol Cell Endocrinol. 2003;200(1–2):89–98 [DOI] [PubMed] [Google Scholar]
- 123. Kilen SM, Szabo M, Strasser GA, McAndrews JM, Ringstrom SJ, Schwartz NB. Corticosterone selectively increases follicle-stimulating hormone β-subunit messenger ribonucleic acid in primary anterior pituitary cell culture without affecting its half-life. Endocrinology. 1996;137(9):3802–3807 [DOI] [PubMed] [Google Scholar]
- 124. Tsutsui K, Saigoh E, Ukena K, et al. A novel avian hypothalamic peptide inhibiting gonadotropin release. Biochem Biophys Res Commun. 2000;275(2):661–667 [DOI] [PubMed] [Google Scholar]
- 125. Kriegsfeld LJ, Mei DF, Bentley GE, et al. Identification and characterization of a gonadotropin-inhibitory system in the brains of mammals. Proc Natl Acad Sci USA. 2006;103(7):2410–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ciccone NA, Dunn IC, Boswell T, et al. Gonadotrophin inhibitory hormone depresses gonadotrophin α and follicle-stimulating hormone β subunit expression in the pituitary of the domestic chicken. J Neuroendocrinol. 2004;16(12):999–1006 [DOI] [PubMed] [Google Scholar]
- 127. Ubuka T, Ukena K, Sharp PJ, Bentley GE, Tsutsui K. Gonadotropin-inhibitory hormone inhibits gonadal development and maintenance by decreasing gonadotropin synthesis and release in male quail. Endocrinology. 2006;147(3):1187–1194 [DOI] [PubMed] [Google Scholar]
- 128. Bentley GE, Perfito N, Ukena K, Tsutsui K, Wingfield JC. Gonadotropin-inhibitory peptide in song sparrows (Melospiza melodia) in different reproductive conditions, and in house sparrows (Passer domesticus) relative to chicken-gonadotropin-releasing hormone. J Neuroendocrinol. 2003;15(8):794–802 [DOI] [PubMed] [Google Scholar]
- 129. Sari IP, Rao A, Smith JT, Tilbrook AJ, Clarke IJ. Effect of RF-amide-related peptide-3 on luteinizing hormone and follicle-stimulating hormone synthesis and secretion in ovine pituitary gonadotropes. Endocrinology. 2009;150(12):5549–5556 [DOI] [PubMed] [Google Scholar]
- 130. Kirby ED, Geraghty AC, Ubuka T, Bentley GE, Kaufer D. Stress increases putative gonadotropin inhibitory hormone and decreases luteinizing hormone in male rats. Proc Natl Acad Sci USA. 2009;106(27):11324–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Soga T, Dalpatadu SL, Wong DW, Parhar IS. Neonatal dexamethasone exposure down-regulates GnRH expression through the GnIH pathway in female mice. Neuroscience. 2012;218:56–64 [DOI] [PubMed] [Google Scholar]
- 132. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100(19):10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349(17):1614–1627 [DOI] [PubMed] [Google Scholar]
- 134. Takumi K, Iijima N, Higo S, Ozawa H. Immunohistochemical analysis of the colocalization of corticotropin-releasing hormone receptor and glucocorticoid receptor in kisspeptin neurons in the hypothalamus of female rats. Neurosci Lett. 2012;531(1):40–45 [DOI] [PubMed] [Google Scholar]
- 135. Kinsey-Jones JS, Li XF, Knox AM, et al. Down-regulation of hypothalamic kisspeptin and its receptor, Kiss1r, mRNA expression is associated with stress-induced suppression of luteinising hormone secretion in the female rat. J Neuroendocrinol. 2009;21(1):20–29 [DOI] [PubMed] [Google Scholar]
- 136. Grachev P, Li XF, O'Byrne K. Stress regulation of kisspeptin in the modulation of reproductive function. Adv Exp Med Biol. 2013;784:431–454 [DOI] [PubMed] [Google Scholar]
- 137. Wang O, Lanjuin A, Ho C, Basko J, Dulac C, Majzoub J. Disruption of Glucocorticoid Receptor Signaling in Kisspeptin Neurons Accelerates the Recovery of Reproductive Function in the Post-Traumatic Stress Period. Endocr Rev. 2012;33(Meeting Abstracts):p. OR12-2 [Google Scholar]
- 138. Schmidt MV, Sterlemann V, Wagner K, et al. Postnatal glucocorticoid excess due to pituitary glucocorticoid receptor deficiency: differential short- and long-term consequences. Endocrinology. 2009;150(6):2709–2716 [DOI] [PubMed] [Google Scholar]
- 139. Schreiber JR, Nakamura K, Erickson GF. Rat ovary glucocorticoid receptor: identification and characterization. Steroids. 1982;39(5):569–584 [DOI] [PubMed] [Google Scholar]
- 140. Hirst MA, Northrop JP, Danielsen M, Ringold GM. High level expression of wild type and variant mouse glucocorticoid receptors in Chinese hamster ovary cells. Mol Endocrinol. 1990;4(1):162–170 [DOI] [PubMed] [Google Scholar]
- 141. Adashi EY, Jones PB, Hsueh AJ. Synergistic effect of glucocorticoids on the stimulation of progesterone production by follicle-stimulating hormone in cultured rat granulosa cells. Endocrinology. 1981;109(6):1888–1894 [DOI] [PubMed] [Google Scholar]
- 142. Andersen CY. Possible new mechanism of cortisol action in female reproductive organs: physiological implications of the free hormone hypothesis. J Endocrinol. 2002;173(2):211–217 [DOI] [PubMed] [Google Scholar]
- 143. Channing CP, Tsai V, Sachs D. Role of insulin, thyroxin and cortisol in luteinization of porcine granulosa cells grown in chemically defined media. Biol Reprod. 1976;15(2):235–247 [DOI] [PubMed] [Google Scholar]
- 144. Michael AE, Pester LA, Curtis P, Shaw RW, Edwards CR, Cooke BA. Direct inhibition of ovarian steroidogenesis by cortisol and the modulatory role of 11 β-hydroxysteroid dehydrogenase. Clin Endocrinol (Oxf). 1993;38(6):641–644 [DOI] [PubMed] [Google Scholar]
- 145. Poulain M, Frydman N, Duquenne C, et al. Dexamethasone induces germ cell apoptosis in the human fetal ovary. J Clin Endocrinol Metab. 2012;97(10):E1890–E1897 [DOI] [PubMed] [Google Scholar]
- 146. Sasson R, Amsterdam A. Pleiotropic anti-apoptotic activity of glucocorticoids in ovarian follicular cells. Biochem Pharmacol. 2003;66(8):1393–1401 [DOI] [PubMed] [Google Scholar]
- 147. Komiyama J, Nishimura R, Lee HY, et al. Cortisol is a suppressor of apoptosis in bovine corpus luteum. Biol Reprod. 2008;78(5):888–895 [DOI] [PubMed] [Google Scholar]
- 148. Rae MT, Price D, Harlow CP, Critchley HO, Hillier SG. Glucocorticoid receptor-mediated regulation of MMP9 gene expression in human ovarian surface epithelial cells. Fertil Steril. 2009;92(2):703–708 [DOI] [PubMed] [Google Scholar]
- 149. Runnebaum IB, Brüning A. Glucocorticoids inhibit cell death in ovarian cancer and up-regulate caspase inhibitor cIAP2. Clin Cancer Res. 2005;11(17):6325–6332 [DOI] [PubMed] [Google Scholar]
- 150. Waddell BJ, Benediktsson R, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 2 in the rat corpus luteum: induction of messenger ribonucleic acid expression and bioactivity coincident with luteal regression. Endocrinology. 1996;137(12):5386–5391 [DOI] [PubMed] [Google Scholar]
- 151. Condon J, Ricketts ML, Whorwood CB, Stewart PM. Ontogeny and sexual dimorphic expression of mouse type 2 11β-hydroxysteroid dehydrogenase. Mol Cell Endocrinol. 1997;127(2):121–128 [DOI] [PubMed] [Google Scholar]
- 152. Evagelatou M, Peterson SL, Cooke BA. Leukocytes modulate 11β-hydroxysteroid dehydrogenase (11β-HSD) activity in human granulosa-lutein cell cultures. Mol Cell Endocrinol. 1997;133(2):81–88 [DOI] [PubMed] [Google Scholar]
- 153. Tetsuka M, Thomas FJ, Thomas MJ, Anderson RA, Mason JI, Hillier SG. Differential expression of messenger ribonucleic acids encoding 11β-hydroxysteroid dehydrogenase types 1 and 2 in human granulosa cells. J Clin Endocrinol Metab. 1997;82(6):2006–2009 [PubMed] [Google Scholar]
- 154. Tetsuka M, Yamamoto S, Hayashida N, et al. Expression of 11β-hydroxysteroid dehydrogenases in bovine follicle and corpus luteum. J Endocrinol. 2003;177(3):445–452 [DOI] [PubMed] [Google Scholar]
- 155. Tetsuka M, Milne M, Simpson GE, Hillier SG, et al. Expression of 11β-hydroxysteroid dehydrogenase, glucocorticoid receptor, and mineralocorticoid receptor genes in rat ovary. Biol Reprod. 1999;60(2):330–335 [DOI] [PubMed] [Google Scholar]
- 156. Michael AE, Gregory L, Walker SM, et al. Ovarian 11β-hydroxysteroid dehydrogenase: potential predictor of conception by in-vitro fertilisation and embryo transfer. Lancet. 1993;342(8873):711–712 [DOI] [PubMed] [Google Scholar]
- 157. Michael AE, Gregory L, Piercy EC, Walker SM, Shaw RW, Cooke BA. Ovarian 11 β-hydroxysteroid dehydrogenase activity is inversely related to the outcome of in vitro fertilization-embryo transfer treatment cycles. Fertil Steril. 1995;64(3):590–598 [PubMed] [Google Scholar]
- 158. Michael AE, Collras TD, Norgate DP, Gregory L, Wood PJ, Cooke BA. Relationship between ovarian cortisol:cortisone ratios and the clinical outcome of in vitro fertilization and embryo transfer (IVF-ET). Clin Endocrinol (Oxf). 1999;51(5):535–540 [DOI] [PubMed] [Google Scholar]
- 159. Shyamala G. Specific cytoplasmic glucocorticoid hormone receptors in lactating mammary glands. Biochemistry. 1973;12(16):3085–3090 [DOI] [PubMed] [Google Scholar]
- 160. Hoijman E, Rocha-Viegas L, Kalko SG, et al. Glucocorticoid alternative effects on proliferating and differentiated mammary epithelium are associated to opposite regulation of cell-cycle inhibitor expression. J Cell Physiol. 2012;227(4):1721–1730 [DOI] [PubMed] [Google Scholar]
- 161. Burke CW, Roulet F. Increased exposure of tissues to cortisol in late pregnancy. Br Med J. 1970;1(5697):657–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Genazzani AR, Fraioli F, Hurlimann J, Fioretti P, Felber JP. Immunoreactive ACTH and cortisol plasma levels during pregnancy. Detection and partial purification of corticotrophin-like placental hormone: the human chorionic corticotrophin (HCC). Clin Endocrinol (Oxf). 1975;4(1):1–14 [DOI] [PubMed] [Google Scholar]
- 163. Alexandrova M. Glucocorticoid receptor of rat mammary gland during pregnancy and lactation. Endocrinol Exp. 1986;20(2–3):293–300 [PubMed] [Google Scholar]
- 164. Lindsay JR, Nieman LK. The hypothalamic-pituitary-adrenal axis in pregnancy: challenges in disease detection and treatment. Endocr Rev. 2005;26(6):775–799 [DOI] [PubMed] [Google Scholar]
- 165. Folley SJ, Greenbaum AL. Effects of adrenalectomy and of treatment with adrenal cortex hormones on the arginase and phosphatase levels of lactating rats. Biochem J. 1946;40(1):46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Chatterton RT, Jr, Harris JA, King WJ, Wynn RM. Ultrastructural alterations in mammary glands of pregnant rats after ovariectomy and hysterectomy: effect of adrenal steroids and prolactin. Am J Obstet Gynecol. 1979;133(6):694–702 [DOI] [PubMed] [Google Scholar]
- 167. Casey TM, Plaut K. The role of glucocorticoids in secretory activation and milk secretion, a historical perspective. J Mammary Gland Biol Neoplasia. 2007;12(4):293–304 [DOI] [PubMed] [Google Scholar]
- 168. Kingsley-Kallesen M, Mukhopadhyay SS., Wyszomierski SL, Schanler S, Schütz G. The mineralocorticoid receptor may compensate for the loss of the glucocorticoid receptor at specific stages of mammary gland development. Mol Endocrinol. 2002;16(9):2008–2018 [DOI] [PubMed] [Google Scholar]
- 169. Wintermantel TM, Bock D, Fleig V, Greiner EF, Schütz G. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol Endocrinol. 2005;19(2):340–349 [DOI] [PubMed] [Google Scholar]
- 170. Feng Z, Marti A, Jehn B, Altermatt HJ, Chicaiza G, Jaggi R. Glucocorticoid and progesterone inhibit involution and programmed cell death in the mouse mammary gland. J Cell Biol. 1995;131(4):1095–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kritikou EA, Sharkey A, Abell K, et al. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130(15):3459–3468 [DOI] [PubMed] [Google Scholar]
- 172. Humphreys RC, Hennighausen L. Signal transducer and activator of transcription 5a influences mammary epithelial cell survival and tumorigenesis. Cell Growth Differ. 1999;10(10):685–694 [PubMed] [Google Scholar]
- 173. Bertucci PY, Quaglino A, Pozzi AG, Kordon EC, Pecci A. Glucocorticoid-induced impairment of mammary gland involution is associated with STAT5 and STAT3 signaling modulation. Endocrinology. 2010;151(12):5730–5740 [DOI] [PubMed] [Google Scholar]
- 174. Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64(5):1757–1764 [DOI] [PubMed] [Google Scholar]
- 175. Aziz MH, Shen H, Maki CG. Glucocorticoid receptor activation inhibits p53-induced apoptosis of MCF10Amyc cells via induction of protein kinase Cϵ. J Biol Chem. 2012;287(35):29825–29836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Lippman M, Bolan G, Huff K. The effects of glucocorticoids and progesterone on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res. 1976;36(12):4602–4609 [PubMed] [Google Scholar]
- 177. Goya L, Maiyar AC, Ge Y, Firestone GL. Glucocorticoids induce a G1/G0 cell cycle arrest of Con8 rat mammary tumor cells that is synchronously reversed by steroid withdrawal or addition of transforming growth factor-α. Mol Endocrinol. 1993;7(9):1121–1132 [DOI] [PubMed] [Google Scholar]
- 178. Sumikawa T, Shigeoka Y, Igishi T, et al. Dexamethasone interferes with trastuzumab-induced cell growth inhibition through restoration of AKT activity in BT-474 breast cancer cells. Int J Oncol. 2008;32(3):683–688 [PubMed] [Google Scholar]
- 179. Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD. Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol Ther. 2006;5(8):933–940 [DOI] [PubMed] [Google Scholar]
- 180. Wetendorf M, DeMayo FJ. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol Cell Endocrinol. 2012;357(1–2):108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Carpenter KD, Korach KS. Potential biological functions emerging from the different estrogen receptors. Ann NY Acad Sci. 2006;1092:361–373 [DOI] [PubMed] [Google Scholar]
- 182. Couzinet B, Le Strat N, Ulmann A, Baulieu EE, Schaison G. Termination of early pregnancy by the progesterone antagonist RU 486 (Mifepristone). N Engl J Med. 1986;315(25):1565–1570 [DOI] [PubMed] [Google Scholar]
- 183. Boomsma CM, Keay SD, Macklon NS. Peri-implantation glucocorticoid administration for assisted reproductive technology cycles. Cochrane Database Syst Rev, 2007(1): p. CD005996. [DOI] [PubMed] [Google Scholar]
- 184. Quenby S, Farquharson R, Young M, Vince G. Successful pregnancy outcome following 19 consecutive miscarriages: case report. Hum Reprod. 2003;18(12):2562–2564 [DOI] [PubMed] [Google Scholar]
- 185. Duong HT, Piotrowska-Tomala KK, Acosta TJ, et al. Effects of cortisol on pregnancy rate and corpus luteum function in heifers: an in vivo study. J Reprod Dev. 2012;58(2):223–230 [DOI] [PubMed] [Google Scholar]
- 186. Robertson SA. Immune regulation of conception and embryo implantation-all about quality control? J Reprod Immunol. 2010;85(1):51–57 [DOI] [PubMed] [Google Scholar]
- 187. Prodi G, De Giovanni C, Galli MC, et al. 17 β-Estradiol, 5α-dihydrotestosterone, progesterone and cortisol receptors in normal and neoplastic human endometrium. Tumori. 1979;65(2):241–253 [DOI] [PubMed] [Google Scholar]
- 188. Atkinson S, Adams NR. Adrenal glands alter the concentration of oestradiol-17β and its receptor in the uterus of ovariectomized ewes. J Endocrinol. 1988;118(3):375–380 [DOI] [PubMed] [Google Scholar]
- 189. Rabin DS, Johnson EO, Brandon DD, Liapi C, Chrousos GP. Glucocorticoids inhibit estradiol-mediated uterine growth: possible role of the uterine estradiol receptor. Biol Reprod. 1990;42(1):74–80 [DOI] [PubMed] [Google Scholar]
- 190. Howe RS, Lee YH, Fischkoff SA, Teuscher C, Lyttle CR. Glucocorticoid and progestin regulation of eosinophil chemotactic factor and complement C3 in the estrogen-treated rat uterus. Endocrinology. 1990;126(6):3193–3199 [DOI] [PubMed] [Google Scholar]
- 191. Rhen T, Grissom S, Afshari C, Cidlowski JA. Dexamethasone blocks the rapid biological effects of 17β-estradiol in the rat uterus without antagonizing its global genomic actions. FASEB J. 2003;17(13):1849–1870 [DOI] [PubMed] [Google Scholar]
- 192. Rhen T, Cidlowski JA. Estrogens and glucocorticoids have opposing effects on the amount and latent activity of complement proteins in the rat uterus. Biol Reprod. 2006;74(2):265–274 [DOI] [PubMed] [Google Scholar]
- 193. Cvoro A, Yuan C, Paruthiyil S, Miller OH, Yamamoto KR, Leitman DC. Cross talk between glucocorticoid and estrogen receptors occurs at a subset of proinflammatory genes. J Immunol. 2011;186(7):4354–4360 [DOI] [PubMed] [Google Scholar]
- 194. Gong H, Jarzynka MJ, Cole TJ, et al. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68(18):7386–7393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Zhang Y, Leung DY, Nordeen SK, Goleva E. Estrogen inhibits glucocorticoid action via protein phosphatase 5 (PP5)-mediated glucocorticoid receptor dephosphorylation. J Biol Chem. 2009;284(36):24542–24552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Tynan SH, Lundeen SG, Allan GF. Cell type-specific bidirectional regulation of the glucocorticoid-induced leucine zipper (GILZ) gene by estrogen. J Steroid Biochem Mol Biol. 2004;91(4–5):225–239 [DOI] [PubMed] [Google Scholar]
- 197. Whirledge S, Dixon D, Cidlowski JA. Glucocorticoids regulate gene expression and repress cellular proliferation in human uterine leiomyoma cells. Horm Cancer. 2012;3(3):79–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Gunin AG, Emelianov V, Tolmachev AS. Effect of adrenocorticotrophic hormone on the development of oestrogen-induced changes and hyperplasia formation in the mouse uterus. Reproduction. 2002;123(4):601–611 [DOI] [PubMed] [Google Scholar]
- 199. Whirledge S, Cidlowski JA. Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology. 2013;154(1):499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Whirledge S, Xiaojiang X, Cidlowski JA. Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biol Reprod. 2013;89(3):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Wu WX, Derks JB, Zhang Q, Nathanielsz PW. Changes in heat shock protein-90 and -70 messenger ribonucleic acid in uterine tissues of the ewe in relation to parturition and regulation by estradiol and progesterone. Endocrinology. 1996;137(12):5685–5693 [DOI] [PubMed] [Google Scholar]
- 202. Albiston AL, Smith RE, Krozowski ZS. Changes in the levels of 11β-hydroxysteroid dehydrogenase mRNA over the oestrous cycle in the rat. J Steroid Biochem Mol Biol. 1995;52(1):45–48 [DOI] [PubMed] [Google Scholar]
- 203. Arcuri F, Monder C, Lockwood CJ, Schatz F. Expression of 11 β-hydroxysteroid dehydrogenase during decidualization of human endometrial stromal cells. Endocrinology. 1996;137(2):595–600 [DOI] [PubMed] [Google Scholar]
- 204. Roland BL, Funder JW. Localization of 11β-hydroxysteroid dehydrogenase type 2 in rat tissues: in situ studies. Endocrinology. 1996;137(3): 1123–1128 [DOI] [PubMed] [Google Scholar]
- 205. Burton PJ, Krozowski ZS, Waddell BJ. Immunolocalization of 11β-hydroxysteroid dehydrogenase types 1 and 2 in rat uterus: variation across the estrous cycle and regulation by estrogen and progesterone. Endocrinology. 1998;139(1):376–382 [DOI] [PubMed] [Google Scholar]
- 206. Thompson A, Han VK, Yang K. Spatial and temporal patterns of expression of 11β-hydroxysteroid dehydrogenase types 1 and 2 messenger RNA and glucocorticoid receptor protein in the murine placenta and uterus during late pregnancy. Biol Reprod. 2002;67(6):1708–1718 [DOI] [PubMed] [Google Scholar]
- 207. McDonald SE, Henderson TA, Gomez-Sanchez CE, Critchley HO, Mason JI. 11β-hydroxysteroid dehydrogenases in human endometrium. Mol Cell Endocrinol. 2006;248(1–2):72–78 [DOI] [PubMed] [Google Scholar]
- 208. Chan J, Rabbitt EH, Innes BA, et al. Glucocorticoid-induced apoptosis in human decidua: a novel role for 11β-hydroxysteroid dehydrogenase in late gestation. J Endocrinol. 2007;195(1):7–15 [DOI] [PubMed] [Google Scholar]
- 209. Dorniak P, Welsh TH, Jr, Bazer FW, Spencer TE. Cortisol and interferon tau regulation of endometrial function and conceptus development in female sheep. Endocrinology. 2013;154(2):931–941 [DOI] [PubMed] [Google Scholar]
- 210. Arcuri F, Battistini S, Hausknecht V, Cintorino M, Lockwood CJ, Schatz F. Human endometrial decidual cell-associated 11β-hydroxysteroid dehydrogenase expression: its potential role in implantation. Early Pregnancy. 1997;3(4):259–264 [PubMed] [Google Scholar]
- 211. Kuroda K, Venkatakrishnan R, Salker MS, et al. Induction of 11β-HSD 1 and activation of distinct mineralocorticoid receptor- and glucocorticoid receptor-dependent gene networks in decidualizing human endometrial stromal cells. Mol Endocrinol. 2013;27(2):192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wächter R, Masarik L, Bürzle M, Mallik A, von Mandrach U. Differential expression and activity of 11β-hydroxysteroid dehydrogenase in human placenta and fetal membranes from pregnancies with intrauterine growth restriction. Fetal Diagn Ther. 2009;25(3):328–335 [DOI] [PubMed] [Google Scholar]
- 213. Wyrwoll CS, Seckl JR, Holmes MC. Altered placental function of 11β-hydroxysteroid dehydrogenase 2 knockout mice. Endocrinology. 2009;150(3):1287–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Shams M, Kilby MD, Somerset DA, et al. 11β-Hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Hum Reprod. 1998;13(4):799–804 [DOI] [PubMed] [Google Scholar]
- 215. Clifford K, Flanagan AM, Regan L. Endometrial CD56+ natural killer cells in women with recurrent miscarriage: a histomorphometric study. Hum Reprod. 1999;14(11):2727–2730 [DOI] [PubMed] [Google Scholar]
- 216. Quenby S, Feigenbaum SL, Bass KE, et al. Prednisolone reduces preconceptual endometrial natural killer cells in women with recurrent miscarriage. Fertil Steril. 2005;84(4):980–984 [DOI] [PubMed] [Google Scholar]
- 217. Ogasawara M, Aoki K. Successful uterine steroid therapy in a case with a history of ten miscarriages. Am J Reprod Immunol. 2000;44(4):253–255 [DOI] [PubMed] [Google Scholar]
- 218. Mandl M, Ghaffari-Tabrizi N, Haas J, Nähammer G, Desoye G. Differential glucocorticoid effects on proliferation and invasion of human trophoblast cell lines. Reproduction. 2006;132(1):159–167 [DOI] [PubMed] [Google Scholar]
- 219. Librach CL, Barratt S, Kaur M, Baker PN. Interleukin-1 β regulates human cytotrophoblast metalloproteinase activity and invasion in vitro. J Biol Chem. 1994;269(25):17125–17131 [PubMed] [Google Scholar]
- 220. Ma Y, Ryu JS, Dulay A, Segal M, Guller S. Regulation of plasminogen activator inhibitor (PAI)-1 expression in a human trophoblast cell line by glucocorticoid (GC) and transforming growth factor (TGF)-β. Placenta. 2002;23(10):727–734 [DOI] [PubMed] [Google Scholar]
- 221. Crocker IP, Barratt S, Kaur M, Baker PN. The in-vitro characterization of induced apoptosis in placental cytotrophoblasts and syncytiotrophoblasts. Placenta. 2001;22(10):822–830 [DOI] [PubMed] [Google Scholar]
- 222. Audette MC, Greenwood SL, Sibley CP, et al. Dexamethasone stimulates placental system A transport and trophoblast differentiation in term villous explants. Placenta. 2010;31(2):97–105 [DOI] [PubMed] [Google Scholar]
- 223. Ryu JS, Majeska RJ, Ma Y, LaChapelle L, Guller S. Steroid regulation of human placental integrins: suppression of α2 integrin expression in cytotrophoblasts by glucocorticoids. Endocrinology. 1999;140(9):3904–3908 [DOI] [PubMed] [Google Scholar]
- 224. Reinisch JM, Simon NG, Karow WG, Gandelman R. Prenatal exposure to prednisone in humans and animals retards intrauterine growth. Science. 1978;202(4366):436–438 [DOI] [PubMed] [Google Scholar]
- 225. Katz VL., Thorp JM, Jr, Bowes WA., Jr Severe symmetric intrauterine growth retardation associated with the topical use of triamcinolone. Am J Obstet Gynecol. 1990;162(2):396–397 [DOI] [PubMed] [Google Scholar]
- 226. Seckl JR. Glucocorticoids and small babies. Q J Med. 1994;87(5):259–262 [PubMed] [Google Scholar]
- 227. McDonald TJ, Franko KL, Brown JM, Jenkins SL, Nathanielsz PW, Nijland MJ. Betamethasone in the last week of pregnancy causes fetal growth retardation but not adult hypertension in rats. J Soc Gynecol Investig. 2003;10(8):469–473 [DOI] [PubMed] [Google Scholar]
- 228. Maccari S, Darnaudery M, Morley-Fletcher S, Zuena AR. Prenatal stress and long-term consequences: implications of glucocorticoid hormones. Neurosci Biobehav Rev. 2003; 27(1–2):119–127 [DOI] [PubMed] [Google Scholar]
- 229. Zucchi FC, et al. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PLoS One. 2013; 8(2): e56967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13(7):269–277 [DOI] [PubMed] [Google Scholar]
- 231. Entringer S. Impact of stress and stress physiology during pregnancy on child metabolic function and obesity risk. Curr Opin Clin Nutr Metab Care. 2013;16(3):320–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Crudo A, Suderman M, Moisiadis VG, et al. Glucocorticoid programming of the fetal male hippocampal epigenome. Endocrinology. 2013;154(3):1168–1180 [DOI] [PubMed] [Google Scholar]
- 233. Reynolds RM. Glucocorticoid excess and the developmental origins of disease: two decades of testing the hypothesis–2012 Curt Richter Award Winner. Psychoneuroendocrinology. 2013;38(1):1–11 [DOI] [PubMed] [Google Scholar]
- 234. Khulan B, Drake AJ. Glucocorticoids as mediators of developmental programming effects. Best Pract Res Clin Endocrinol Metab. 2012;26(5):689–700 [DOI] [PubMed] [Google Scholar]
- 235. Hogg K, Price EM, Hanna CW, Robinson WP. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther. 2012;92(6):716–726 [DOI] [PubMed] [Google Scholar]
- 236. Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 2011;59(3):279–289 [DOI] [PubMed] [Google Scholar]
- 237. Brown RW, Chapman KE, Murad P, Edwards CR, Seckl JR. Purification of 11 β-hydroxysteroid dehydrogenase type 2 from human placenta utilizing a novel affinity labelling technique. Biochem J. 1996;313(Pt 3):997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Levitt NS, Lindsay RS, Holmes MC, Seckl JR. Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology. 1996;64(6):412–418 [DOI] [PubMed] [Google Scholar]
- 239. Ortiz LA, Quan A, Zarzar F, Weinberg A, Baum M. Prenatal dexamethasone programs hypertension and renal injury in the rat. Hypertension. 2003;41(2):328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. de Vries A, Holmes MC, Heijnis A, et al. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest. 2007;117(4):1058–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Dalziel SR, Walker NK, Parag V, et al. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-year follow-up of a randomised controlled trial. Lancet. 2005;365(9474):1856–1862 [DOI] [PubMed] [Google Scholar]
- 242. Davis EP, Sandman CA, Buss C, Wing DA, Head K. Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol Psychiatry. 2013;piiS0006-3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Lindsay RS, Lindsay RM, Edwards CR, Seckl JR. Inhibition of 11-β-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension. 1996;27(6):1200–1204 [DOI] [PubMed] [Google Scholar]
- 244. Welberg LA, Seckl JR, Holmes MC. Inhibition of 11β-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur J Neurosci. 2000;12(3):1047–1054 [DOI] [PubMed] [Google Scholar]
- 245. Holmes MC, Abrahamson CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11β-Hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci. 2006;26(14):3840–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269–301 [DOI] [PubMed] [Google Scholar]
- 247. Iqbal M, Moisiadis VG, Kostaki A, Matthews SG. Transgenerational effects of prenatal synthetic glucocorticoids on hypothalamic-pituitary-adrenal function. Endocrinology. 2012;153(7):3295–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res. 2009;88(5):400–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Ke X, Schober ME, McKnight RA, et al. Intrauterine growth retardation affects expression and epigenetic characteristics of the rat hippocampal glucocorticoid receptor gene. Physiol Genomics. 2010;42(2):177–189 [DOI] [PubMed] [Google Scholar]
- 250. Crudo A, Petropoulos S, Moisiadis VG, et al. Prenatal synthetic glucocorticoid treatment changes DNA methylation states in male organ systems: multigenerational effects. Endocrinology. 2012;153(7):3269–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Chapman K, Holmes M, Seckl J. 11β-Hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93(3):1139–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]