

Abstract
Palmoplantar keratodermas (PPKs) are a group of disorders that are diagnostically and therapeutically problematic in dermatogenetics1-3. Punctate PPKs are characterized by circumscribed hyperkeratotic lesions on palms and soles with considerable heterogeneity. In 18 families with autosomal dominant punctate PPK (OMIM #148600), we report heterozygous loss-of-function mutations in AAGAB, encoding alpha- and gamma-adaptin binding protein p34, at a previously linked locus on 15q22. p34, a cytosolic protein with a Rab-like GTPase domain, was shown to bind both clathrin adaptor protein complexes, indicative of a role in membrane traffic. Ultrastucturally, lesional epidermis showed abnormalities in intracellular vesicle biology. Immunohistochemistry showed hyperproliferation within the punctate lesions. Knockdown of p34 in keratinocytes led to increased cell division, which was linked to greatly increased epidermal growth factor receptor (EGFR) protein expression and tyrosine phosphorylation. We hypothesize that p34 deficiency may impair endocytic recycling of growth factor receptors such as EGFR, leading to increased signaling and proliferation.
Palmoplantar keratoderma (PPK) describes a group of hereditary skin disorders characterized by thickening (hyperkeratosis) of the palm and sole epidermis1. Historically, PPKs have been classified according to the pattern of lesions (diffuse, focal, punctate, striate), as well as histopathologic changes (epidermolytic, non-epidermolytic, porokeratotic). Molecular genetics now allows for improved classification, diagnosis and personalized medicine approaches to treatment. Three distinct types of inherited punctate PPK are recognized by OMIM2. These are: PPKP1 (also known as the Buschke-Fischer-Brauer type, OMIM #148600), mapped to 15q22 Ref.4-6; PPKP2 (porokeratotic type, OMIM #175860), not yet mapped; and PPKP3 (also known as acrokeratoelastoidosis, OMIM #101850), where preliminary linkage suggests a possible locus on 2p25-p12 Ref.7. An additional locus was identified at 8q24.13-8q24.21 in two Chinese families whose phenotype resembles PPKP1 clinically and histologically8. Here we studied a collection of 18 kindreds with PPKP1 from Scotland, Ireland, Japan and Tunisia, 11 of which had a family history consistent with autosomal dominant inheritance (Supplementary Fig. 1). In our PPKP1 case collection, there was considerable phenotypic variation (Figure 1). In all families, onset was typically during the first to second decades of life, with the appearance of small circumscribed lesions on palms and soles. These consistently increase in number with advancing age and later coalesce to form larger lesions. In some families, lesions remained subtle (Figure 1a&b), whereas in other kindreds, the phenotype, resembling HPV-induced papilloma-like lesions, was much more severe, painful and debilitating (Figure 1c&d). Histology of lesional palmar epidermis from 3 unrelated kindreds – Families 1, 11 and 15 – all showed very similar findings of a well-defined central epidermal depression associated with hypergranulosis and a prominent layer of overlying orthokeratosis (Figure 1e). Immunohistochemical staining for cell proliferation marker Ki67 showed continuous staining of the basal cell (proliferative) compartment of the epidermis beneath the hyperkeratotic lesions (Figure 1f), indicative of a hyperproliferative form of hyperkeratosis rather than a retention hyperkeratosis due to defective desquamation.
Figure 1. Clinical and histological features of punctate palmoplantar keratoderma.

(a) The palms of the proband in PPKP1 Family 1 show numerous small, hard, slightly indented hyperkeratotic lesions (arrow; see inset), which typically appear around age 10 and increase in number throughout life.
(b) On the soles, lesions tend to coalesce at pressure points and so the presentation can resemble a focal form of keratoderma.
(c&d) In some cases, the phenotype is much more severe, as seen here in the proband from Family 15.
(e) This H&E stained section shows a punch sample of skin incorporating a well defined central epidermal depression associated with hypergranulosis and a prominent layer of overlying orthokeratosis. Scale bar = 500 μm.
(f) The proliferation marker Ki67 shows an increase in the numbers of keratinocytes in cell cycle within the floor of the epidermal depression (arrowheads) compared to perilesional epidermis (arrow). Scale bar = 100 μm.
Microsatellite linkage to the previously reported 15q22 locus was observed in a large Tunisian kindred originating from Saudi Arabia (Supplementary Fig. 2), with a maximum 2-point lod score of 8.18 at θ=0 obtained with D15S983 (Supplementary Table 1). The family was reported previously9. A 6.24 Mb critical interval was defined by visible recombination with markers D15S993 (proximal) and D15S977 (distal) containing 80 RefSeq genes. This locus overlaps the previously reported intervals for PPKP1 (Supplementary Table 2). In parallel, the proband in Family 1 (of Scottish ancestry) was subjected to whole exome sequencing. Of 28,513 SNPs identified, we elected to concentrate on loss-of-function variants located within the critical interval defined by our linkage data and that previously reported. Of 106 unique “stop-gained” SNPs in the exome dataset, only 3 SNPs, all heterozygous in the proband, were located on chromosome 15 (Supplementary Table 3). Two were located well outside the interval: a known SNP rs57809907 predicting p.E417X in DYX1C1; and a second previously unreported variant of unknown consequence predicting p.L439X in GABRG3. The remaining variant, c.481C>T, predicting p.R161X in AAGAB fell within our linkage interval and that initially reported5, but was just outside the interval reported in a Chinese kindred6. This variant was absent from current dbSNP and 1000Genomes datasets. The mutation was confirmed by conventional sequencing and co-segregated with the PPKP1 phenotype in Family 1 (Supplementary Fig. 3a&b). By conventional sequencing, a second loss-of-function mutation, c.348_349del, was identified and found to fully co-segregate in the linked kindred and two additional Tunisian families (Supplementary Fig. 3c&d). Sequencing of the remaining 16 families revealed a total of 8 mutations in AAGAB (2 nonsense; 5 frameshift and 1 splice site mutation), as detailed in Supplementary Table 4. AAGAB, consisting of 10 exons spanning 53,708 bp of genomic DNA, encodes alpha- and gamma-adaptin binding protein p34, identified from a yeast 2-hybrid screen using a subunit of the AP-1 adaptor protein complex as bait10. The positions of the mutations in relation to the p34 protein organization are shown in Figure 2.
Figure 2. Identification of mutations in AAGAB in PPKP1 families.
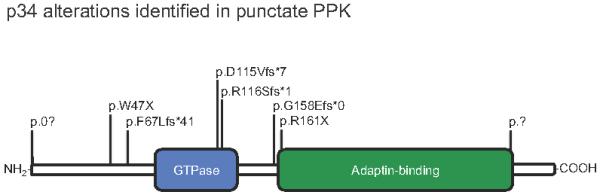
Protein domain organization of the p34 protein encoded by AAGAB, showing positions of all 8 alterations identified. Further details of the mutations are listed in Table 4. Examples of sequence traces are shown in Supplementary Fig. 3.
By quantitative RT-PCR, we confirmed that p34 is expressed at broadly comparable levels in skin, HeLa cells, primary epidermal keratinocytes and the commonly used keratinocyte cell line HaCaT11 (Figure 3a). By QRT-PCR, we also showed that p34 message is very widely expressed across tissues, including skin (Supplementary Fig. 4). Two independent siRNAs were developed that showed near-complete knockdown of p34 protein in HaCaT cells (Figure 3b). Treatment of HaCaT cells with either of these potent siRNAs resulted in an approximately 2-fold increase in cell counts over time (Figure 3c), thus mirroring the increased epidermal proliferation observed in lesional epidermis (Figure 1f).
Figure 3. AAGAB is expressed in skin and keratinocytes and its depletion leads to increased cell numbers over time.
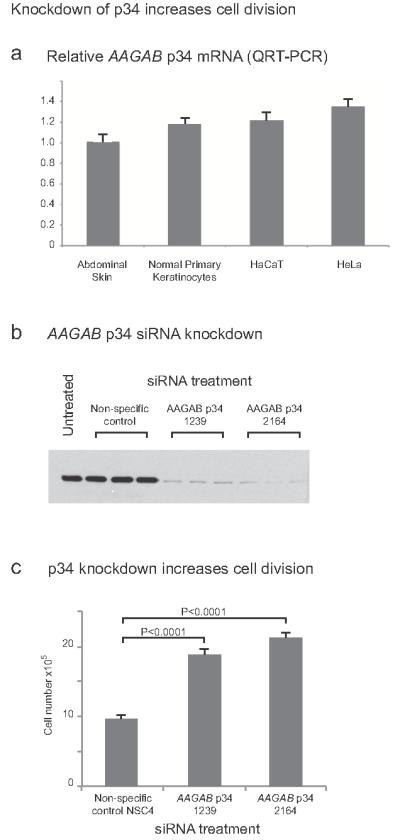
(a) QRT-PCR showed that AAGAB mRNA is expressed in skin and is present at broadly comparable levels in HeLa cells, primary epidermal keratinocytes and the epidermal keratinocyte cell line HaCaT. Conventional RT-PCR showed expression in palmoplantar epidermis comparable to abdominal skin (data not shown).
(b) Western blotting shows that two independent siRNAs (designated 1239 and 2164) potently knocked down p34 expression in HaCaT cells. Non-specific control siRNA = NSC4 (inverted lacZ sequence).
(c) AAGAB knockdown by either siRNA leads to an approximate 2-fold increase in HaCaT cell division at 96 hours post-transfection. Similar data were seen at early time-points (not shown). Error bars show standard deviation. Non-specific control siRNA = NSC4 (inverted lacZ sequence).
The coating of clathrin-coated vesicles consists of clathrin and adaptor complexes, both of which have to be recruited to the appropriate membrane from the cytoplasm12,13. The two most abundant types of adaptor protein complexes are AP-1, which is responsible for sorting proteins between the trans-Golgi network (TGN) and endosomes, and AP-2, which is responsible for sorting proteins at the plasma membrane. Both are heterotypic complexes with AP-1 containing a γ-adaptin subunit and AP-2 containing an α-adaptin subunit. Although cDNAs encoding p34 were the predominant species of clone from the original yeast 2-hybrid screen10, difficulties in obtaining specific antibodies meant that further biochemical confirmation of these protein-protein interactions were not presented. Here, using two independent antibodies to p34 made in-house, we confirm by immunoprecipitation followed by western blotting that this protein indeed interacts with both AP-1 and AP-2 complexes in the cytosol (Figure 4a). Cytosolic localization was confirmed by immunocytochemistry and using both N- and C-terminal GFP-tagged p34 constructs (Figure 4b), although C-terminal tagging also led to some nuclear accumulation of the fusion protein. Cell fractionation studies showed that p34 is found in the cytosol but not in membrane or clathrin-coated vesicle fractions in HeLa cells (Figure 4c). Near-complete siRNA knockdown of p34 (Figure 4d) did not lead to an overt change in the plasma membrane or TGN localization of AP-2 or AP-1, respectively (Figure 4e). Neither AP-1 nor AP-2 co-localized with N- or C-terminal GFP fusions of p34 (Supplementary Fig. 5). Essentially identical diffuse cytoplasmic localization data were obtained in the keratinocyte cell line HaCaT (not shown). Overall, these data confirm that p34 is a cytosolic protein that binds to AP-1 and AP-2, however, p34 does not follow these protein complexes into their membrane-associated vesicle populations on intracellular membranes or at the plasma membrane, respectively. This is consistent with a possible chaperone role, as previously suggested10. For example, p34 might either prevent soluble clathrin from assembling with soluble adaptor complexes in the cytosol; p34 might be involved in vesicle uncoating; or this protein might aid recruitment of soluble adaptors to membranes10. Bioinformatics analysis revealed the presence of a GTPase domain, which is most closely related to the Rab superfamily of vesicular trafficking proteins (Figure 2a; Supplementary Fig. 6). It is not known if this GTPase domain is functional. If so, this may be indicative of a role in active transport of cytosolic adaptor complexes rather than a more passive-acting chaperone function14.
Figure 4. p34 associates with AP-1 and AP-2 in the cytosol.
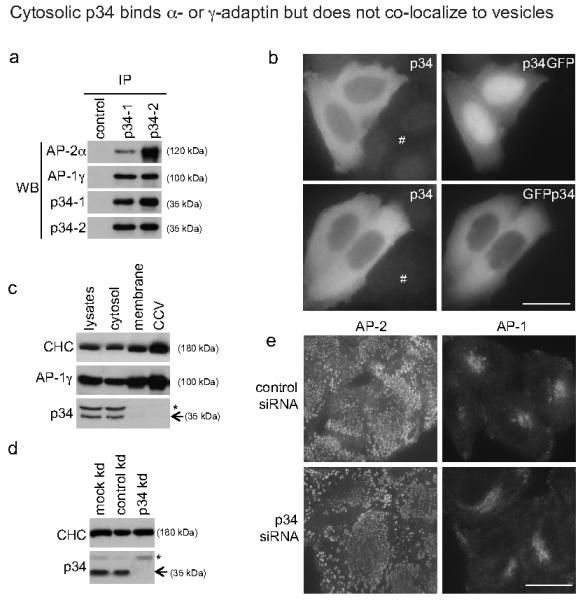
(a) Native immunoprecipitation (IP) of HeLa cytosolic extracts was performed with antibodies against p34 (p34-1 and p34-2), followed by Western blotting (WB) with antibodies against p34, AP-1 or AP-2. Note the association of both AP-1 and AP-2 adaptor complexes with p34.
(b) HeLa cells were transiently transfected with either C-terminally (p34GFP) or N-terminally (GFPp34) GFP-tagged p34 constructs. Both constructs show a mainly cytosolic localization and this is consistent with the staining pattern (albeit faint) observed using an antibody against p34 in either transfected or untransfected cells (#).
(c) HeLa lysates were subjected to fractionation into cytosol, membrane, and clathrin-coated vesicle (CCV) fractions, and then Western blotted for clathrin heavy chain (CHC), AP-1 and p34. Note that p34 is found principally in the cytosolic fraction and is virtually undetectable in membrane or CCV fractions in which AP-1 and CHC are enriched. (* = cross reacting band)
(d) HeLa cells were treated with siRNA to knock down expression of p34 (kd), and western blotted for clathrin heavy chain (CHC) and p34 (* = cross reacting band). Note the efficient knockdown of p34 (>95%).
(e) Localization of AP-1 and AP-2 visualized by immunofluorescence following knock down of p34 (kd). Note that the AP-2-containing membrane-associated vesicles or AP-1-trans-Golgi network appear unperturbed. Bar = 20 μm.
Ultrastructural analysis of lesional plantar skin revealed mild acanthosis, a reduction in the granular cell layer and compact orthokeratosis (Supplementary Fig. 7). In basal keratinocytes (Figure 5), there was a large increase in the number of small vesicles close to the cell membrane and prominent dilatation of the Golgi apparatus (Supplementary Fig. 7). These ultrastructural features are consistent with a vesicle transport defect.
Figure 5. Transmission electron microscopy of lesional plantar skin reveals vesicle abnormalities within basal keratinocytes.

Transmission electron microscopy of a basal epidermal keratinocyte (E) within PPKP1 lesional epidermis reveals an abnormal abundance of membrane-bound vesicles abutting the plasma membrane (arrows), close to the cutaneous basement membrane (*). D = dermis. Further electron micrographs are shown in Supplementary Fig. 7.
We hypothesized that a possible mechanism whereby perturbation of vesicle trafficking could lead to increased epidermal proliferation might involve recycling of the epidermal growth factor receptor (EGFR). EGFR is known to undergo a process of rapid turnover – a process that requires a number of sorting steps including clathrin- and AP-2-dependent endocytosis15,16. Internalized ligand-bound EGFR is either shuttled to late endosomes/lysosomes for degradation or into recycling endosomes, where, following removal of the bound ligand and dephosphorylation, the receptor is returned to the plasma membrane via vesicle fusion. Knockdown of p34 in HaCaT cells led to a modest ~2.5-fold increase in EGFR mRNA levels as determined by QRT-PCR (data not shown), however, a >10-fold increase in EGFR total protein expression was seen by western blot (Figure 6a), consistent with decreased EGFR turnover. Importantly, phosphorylation of EGFR at tyrosine-992, indicating active EGFR signaling17, was increased >20-fold (Figure 6b). The regulation of EGFR signaling via clathrin-mediated endocytosis is a complex process involving Rab proteins15, dynamin18, AP-2, the Grb2 adaptor protein, and multiple mechanisms relating to post-translational modification of the receptor itself19. Disruption of EGFR endocytosis by knockdown of the motor protein dynamin has been shown to produce decreased protein turnover with increased EGFR signaling18, very similar to what we observed here by knockdown of p34.
Figure 6. Knockdown of p34 greatly increases EGFR protein expression.
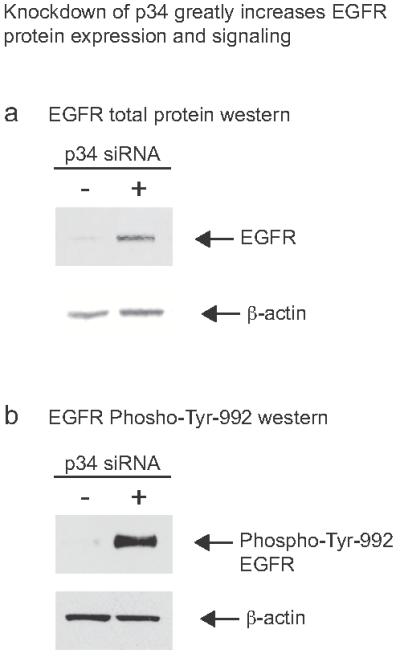
(a) Knockdown of p34 with siRNA 1239 leads to greatly increased EGFR protein compared to non-specific control siRNA NSC4. Part of the blot was stained for β-actin as a loading control and to normalize EGFR quantification. Using the LI-COR Odyssey system, the observed upregulation was found to be >10-fold (data not shown). +, p34 siRNA; −, NSC4 siRNA control.
(b) Knockdown of p34 with siRNA 1239 also leads to greatly increased phosphorylation of tyrosine-992 on the EGFR protein compared to non-specific control siRNA NSC4. Using the LI-COR Odyssey system, the observed increase in phosphorylation of this residue was found to be >20-fold (data not shown).
Here, using a combination of genetic linkage and whole exome sequencing, we have identified a gene for the autosomal dominant form of punctate palmoplantar keratoderma previously linked to 15q22 Ref.5,6,9, known as PPKP1 (OMIM #148600). The causative gene, AAGAB, encodes alpha- and gamma-adaptin binding protein p34 Ref.10. AP-2 is known to be involved in EGFR endocytosis19 and we confirm here that p34 binds cytosolic AP-2 (Figure 4) and show that p34 deficiency increases epidermal cell proliferation (Figure 1f & Figure 3). Overall, p34 appears to play a hitherto unrecognized role in the control of cell division, possibly by contributing to normal endocytotic recycling of receptor tyrosine kinases such as EGFR15,16,18,19. Other genes involved in intracellular vesicle trafficking have been linked to inherited skin disease where hyperkeratosis and/or keratoderma form part of the phenotypic constellation. This includes SNAP29 in cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma (CEDNIK syndrome)20 and VPS33B in arthrogryposis, renal dysfunction, and cholestasis (ARC syndrome)21, both of which encode proteins required for vesicle membrane fusion in the epidermis and other tissues. Moreover, loss-of-function mutations in AP1S1, which encodes the small σ1A subunit of the AP-1 complex, have been shown to cause the recessive neurocutaneous disorder MEDNIK (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratodermia)22. Similarly, defects in multiple proteins involved in melanosome transport and fusion lead to the pigmentation disorder Griscelli syndrome23 and mutations in the AP-3 subunit protein β3A have been linked to Hermansky-Pudlak syndrome type 2, which consists of oculocutaneous albinism and other features24. Thus, a growing number of genes involved in vesicle transport have been linked to genetic skin disease, plus or minus extra-cutaneous phenotypes.
Disruption of EGFR signaling is a feature of neoplasia. There are anecdotal reports of cancer occurring in association with punctate PPK families25, however, without genetic testing we cannot say that the p34 gene was involved in those cases. Although a few cases of cancer were reported in the larger families studied here, we did not observe co-segregation with the p34 mutations and so any causal link remains unproven.
It remains unclear why haploinsufficiency for this widely expressed protein leads to a phenotype limited to palm and sole epidermis, although this is not unusual in hereditary skin disease, e.g. haploinsufficiency for the desmosomal proteins desmoplakin and desmoglein-1 Ref.26,27, or keratin 1 Ref.28, all of which are structural proteins expressed throughout the epidermis, lead to striate palmoplantar keratoderma29. Another unanswered question relates to why the hyperkeratotic lesions are focal. These lesions are late-onset, appearing in the first or second decades of life, and increase in number with increasing age. This might be explained by a “second hit” mutation in AAGAB and so we examined this by sequencing the gene using genomic DNA derived from microdissected lesional tissue from more than one patient. This revealed neither loss-of-heterozygosity nor a second compound heterozygous p34 mutation (data not shown). Immunohistochemistry was also attempted, however, neither the in-house nor commercial p34 antibodies available gave a signal on fixed tissue. A further possibility is the occurrence of a second somatic mutation in a gene other than AAGAB.
In conclusion, this study expands the molecular diagnostic repertoire for keratodermas and reveals that the AP-1 and AP-2 associated protein p34 is involved in control of epidermal cell division.
Online Methods
Affected individuals and phenotypes
Individuals from 18 apparently unrelated families presented with PPK at dermatology clinics in Scotland, Ireland, Japan and Tunisia. Clinical examination and histological analysis of affected persons was consistent with a diagnosis of the Buschke-Fischer-Brauer type of punctate PPK (PPKP1; OMIM reference number 148600)2. In severely affected cases, where keratoderma resembled plantar warts, presence of human papilloma virus was excluded in the clinical work-up. EDTA blood samples were obtained from the patients with written informed consent and Institutional Ethics Committee approval that complied with the Declaration of Helsinki Principles. DNA was extracted following standard procedures.
Linkage analysis
Microsatellite genotypes were generated for members of Family 15 using standard protocols. Lod scores were calculated using FASTLINK, assuming a mutant allele frequency of 1 in 50,000 30,31. The lod scores were re-calculated using a 10% chance of non-penetrance because of difficulties obtaining a definitive clinical ascertainment in some individuals in Family 15 (the family reside in a remote mountainous area of Tunisia).
Exome sequencing
Exome enrichment was performed using the Agilent SureSelect 50 Mb exome enrichment system (Agilent UK, Wokingham, UK), using the protocol for Applied Biosystems SOLiD (Protocol version 1.0, May 2010), followed by sequencing on the ABI SOLiD platform (Applied Biosystems, Foster City CA), giving an average sequencing depth of >30x across the exome. Next generation sequencing and SNP calling was performed by The East Anglian Sequencing and Informatics Hub (EASIH; http://www.easih.ac.uk).
Histology
Routine hematoxylin and eosin staining of paraffin-embedded tissue was performed using standard protocols. Immunohistochemical staining Ki67 for the cell proliferation marker Ki67 was performed using polyclonal antibody HPA000451 (Sigma-Aldrich, Poole, UK).
Mutation analysis of AAGAB gene
The individual exons of AAGAB were amplified by PCR using the genomic primers listed in Supplementary Table 5 and the following cycling conditions: One cycle of 94°C for 5 min; 35 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 1 min; and 1 final extension at 72°C for 5 min.
Cell culture
HaCaT keratinocytes were maintained in a monolayer in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Paisley, UK) containing 10% fetal calf serum (FCS). Normal human keratinocytes were obtained from CELLnTECH and cultured in the defined growth medium CnT57 (TCS Cellworks Ltd, Buckingham, UK). HeLa cells were cultured in DMEM supplemented with 10% FCS, 2mM L-glutamine, 50 units/ml penicillin and 50μg/ml streptomycin (Sigma-Aldrich).
Reverse transcriptase PCR
Total RNA was extracted from cultured HaCaT cells and primary keratinocytes using an RNeasy Mini kit (Qiagen, Crawley, UK). 1 μg of RNA was reverse transcribed using AMV Reverse Transcriptase (Promega, Southampton, UK) and oligo(dT)15 (Roche Diagnostics, Welwyn Garden City, UK). 1 μl of reaction mix was used in subsequent PCR using Expand High Fidelity PCR buffer containing 1.5mM MgCl2 and 0.5 units of High Fidelity DNA Polymerase (Roche Diagnostics). Primers AAGAB5intFor and AAGAB8intRev (see Supplementary Table 5) were used to amplify a 292 bp fragment using the following program: one cycle of 94°C for 5 min; 35 cycles of 94°C for 30 s, 52°C for 30 s and 72°C for 30 s; 72°C for 5 min.
Isolation of RNA and gene expression analysis
RNA was isolated from abdominal skin, primary keratinocytes, HaCaT and HeLa cells using an RNeasy mini kit (Qiagen) and the RNA was treated with RNase-Free DNase to remove any contaminating genomic DNA. 1μg of each was reversed transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). 1μl of reaction were used in quantitative PCR using Perfecta qPCR ToughMix™, ROX™ (Quanta Biosciences) on a 7900HT Fast Real-Time PCR system (Applied Biosystems). Taqman® Gene Expression Assays (Life Technologies) for AAGAB (part number Hs01027607_m1) and GAPDH (part number 4310884E) were used as per manufacturer’s recommendations.
Multiple tissue expression analysis
A cDNA clone was generated to act as a standard, as follows: cDNA generated following reverse transcription of HaCaT RNA was used as template for amplification of a 940 bp AAGAB cDNA fragment using the primers P34clone1 and P34clone2 (see Supplementary Table 5). The cDNA was cloned as an EcoR I–Hind III fragment into pcDNA™3.1(−)/myc-HisA (Life Technologies). A normalized cDNA panel from 48 different human tissues (Origene Technologies, Inc., Maryland, USA) was quantified using Taqman® Gene Expression Assay Hs01027607_m1 (Life Technologies). The standard used in this assay was plasmid DNA encoding wild type AAGAB. The standard curve was calculated using the formulae described by Applied Biosystems.
siRNA knockdown of AAGAB in HaCaT cells
siRNA oligonucleotides targeting AAGAB and a scrambled negative control were obtained from Eurofins MWG Operon. The sequences of the siRNAs used were AAGAB1239 UGU AAG AGA GUG AGG AAU A and AAGAB2164 GGA AAG UAC UGC AAA UAA A. Non-specific control siRNA (NSC4) sequence (a scrambled bacterial lacZ sequence) was UAG CGA CUA AAC ACA UCA AUU’. 1.2 × 105 HaCaT cells were added to wells of a 6 well dish containing DMEM supplemented with 10% FCS and transfected with siRNAs at a final concentration of 5 nM using Lipofectamine RNAiMAX Reagent (Invitrogen). Cells were incubated for 72 hours or 96 hours before harvesting for RNA extraction and protein isolation or for the cell proliferation analysis.
Cell proliferation analysis
HaCaT cells were treated with siRNAs as above and incubated for 96 hours. Cells were washed with PBS then detached from the dishes using 0.05% Trypsin-EDTA. Counts of viable cells (trypan-blue staining) were obtained using a hemocytometer.
AAGAB p34 antibodies
Two affinity purified rabbit polyclonal antibodies were made in-house (J.H. group), designated p34-1 and p34-2, using GST-fusion protein consisting of residues 142-315 (C-terminus) of rat p34 (85% identical and 91% similar to human p34). These antibodies were used for the HeLa cell experiments (western and immunofluorescence). For the HaCaT cell experiments (W.H.I.M. group), rabbit anti-AAGAB polyclonal antibody was used (Sigma-Aldrich). Immunofluorescence and western blotting of HeLa cells was repeated using the Sigma-Aldrich antibody with very similar results (W.H.I.M. group, data not shown). Similarly, western blotting and immunofluorescence of HaCaT cells was repeated using p34-2 with similar results (W.H.I.M. group, data not shown).
Western blotting of HaCaT lysates and EGFR quantification
Cells were rinsed in ice-cold PBS then lyzed in SDS lysis buffer (1% SDS, 20 mM Tris-HCl pH 8.0, 137 mM NaCl, 10% glycerol, 2 mM EDTA) plus protease inhibitor cocktail (Sigma-Aldrich), phosphatase inhibitor cocktail (Sigma-Aldrich) and 25 U/ml Benzonase (Merck, Darmstadt, Germany), incubating at room temperature for 10 min with agitation. Lysates were cleared by centrifugation at 10,000 g for 10min at 4°C then protein concentrations determined using Bio-Rad Protein Assay (Bio-Rad Laboratories, Hemel Hempsted, UK). For each sample, 10 μg of total protein were separated on NuPAGE 4-12% Bis-Tris polyacrylamide gels (Invitrogen) in MOPS buffer then blotted onto HyBond-C Extra (GE Healthcare, Amersham, UK). Membranes were probed with 1:1000 dilution of the appropriate primary antibody (rabbit anti-AAGAB; Sigma-Aldrich), rabbit anti-EGFR (Cell Signaling Technologies, Beverly MA), or 1:10000 dilution mouse anti-beta-actin (Sigma-Aldrich), then 1:1000 dilution of goat anti-rabbit-HRP or rabbit anti-mouse-HRP (Dako, Glostrup, Denmark) as the secondary antibody. Immobilon Western Chemiluminescent HRP substrate (Millipore, Watford, UK) was used for visualization. For detection of phospho-EGFR, blots were blocked in 3% BSA in PBS containing 0.1% Tween 20 (PBST) and 10mM beta-glycerophosphate for 1h at room temperature then incubated overnight in 1/1000 dilution of phospho-EGF receptor (Tyr-992) antibody (Cell Signalling Technology) in PBST containing 10 mM beta-glycerophosphate at 4°C. Blots were washed in 3 changes of PBST containing 10mM beta-glycerophosphate then incubated in 1/1000 dilution of goat anti-rabbit HRP (DAKO) in PBST plus 10 mM beta-glycerophosphate for 1 h at room temperature. Following washing as before, blots were developed using ECL. For detection of beta-actin, blots were blocked and antibodies were diluted in 5% non-fat milk in PBST under the same conditions as above. A dilution of 1/10,000 was used for the anti-actin antibody (Sigma-Aldrich) and 1/1000 for the secondary rabbit anti-mouse HRP antibody (DAKO). Quantification of protein bands on blots was done using a LI-COR Odyssey infra-red imaging system.
HeLa cell transfection
Transfection of HeLa cells with siRNAs was achieved with Oligofectamine (Invitrogen) following the manufacturer’s instructions, using 25 mM siRNA and incubating for 72 h. Knockdown of p34 was achieved using an siGENOME SMARTpool (#M-0159891) and control knockdowns were performed with a non-targeting siRNA (#D-001810-10), both (Thermo-Fisher, Epsom, UK). Transient transfections with GFP-tagged plasmid DNA was achieved using HeLa Monster transfection reagent (Cambridge Bioscience, Cambridge, UK) following the manufacturer’s instructions and transfecting 48 h prior to imaging. For immunofluorescence, cells were grown on glass bottom coverslips (Mat Tek, Ashland MA), fixed in 3% formaldehyde in PBS at room temperature and permeabilized with 0.1% Triton X-100. The cells were imaged with a Zeiss Axiovert 200 inverted microscope using a Zeiss Plan Achromat 63x oil immersion objective, a Hamamatsu ORCA-ER2 camera and Improvision Openlab software (Perkin-Elmer, Cambridge, UK).
Western blot, immunofluorescence and immunoprecipitation in HeLa cells
These were performed with antibodies raised in-house including p34 (p34-1, p34-2), AP-2α, AP-1γ, clathrin heavy chain 10. Antibody P34-2 was used to detect p34 unless otherwise specified. N- and C- terminally tagged p34 were constructed from rat cDNA 10 (note that human and rat AP-1γ, share 99% amino acid identity) by PCR amplification and cloned in-frame into pEGFP-N3 (p34GFP) or pEGFP-C1 (GFPp34; Takara Bio Europe/Clontech, Saint-Germain-en-Laye, France) using Hind III and Kpn I restriction sites. The sequence of both constructs was verified by Sanger sequence analysis.
Immunoprecipitation and clathrin-coated vesicle isolation in HeLa cell extracts
For immunoprecipitation, cells were lyzed in PBS containing 1% IGEPAL (Sigma-Aldrich), cleared of debris by centrifugation and antibody-protein complexes captured using protein-A Sepharose (Pharmacia, Milton Keynes, UK). The samples were then analyzed by PAGE electrophoresis and Western blotting. Blots were probed with various antibodies followed by ECL Plus Western Blotting Detection System (GE Healthcare). Control CCV-enriched fractions were prepared as described previously 32, except the final pelleting step was performed at 86,900 × g RCFmax (40,000 rpm, TLA-110) for 30 min to improve yield.
Ultrastructural analysis
Skin biopsy specimens were cut into small pieces (of <1 mm3) and fixed in half-strength Karnovsky fixative for 4 hours at room temperature. After washing in 0.1 M phosphate buffer (pH 7.4), the samples were immersed in 1.3% aqueous osmium tetroxide (TAAB Laboratories, Berkshire, UK) for 2 hours, followed by incubation in 2% uranyl acetate (Bio-Rad), and dehydrated in a graded ethanol series, and then embedded in epoxy resin via propylene oxide (TAAB Laboratories). Ultra-thin sections were stained with uranyl acetate and lead citrate and examined in a Philips CM10 transmission electron microscope (Philips, Eindhoven, The Netherlands).
Supplementary Material
ACKNOWLEDGEMENTS
The authors dedicate this paper to their erstwhile colleague, the late dermatologist and cell biologist Susan M. Morley, who treated some of the patients studied here. We thank Margaret “Scottie” Robinson, University of Cambridge and Colin Watts, University of Dundee for insightful discussions, and Tayside Tissue Bank, Dundee for providing skin samples. Specialist Sequencing and Bioinformatics Services were provided by the East Anglian Sequencing and Informatics Hub (EASIH), University of Cambridge, UK which is supported by National Institute for Health Research and Cambridge Biomedical Research Centre (http://www.easih.ac.uk). This work was supported by a Programme Grant (Reference 092530/Z/10/Z) and a Strategic Award (Reference 098439/Z/12/Z) from the Wellcome Trust; a project grant from PC Project (www.pachyonychia.org); and by an A*STAR Research Attachment Program (ARAP) fellowship to O. M.
REFERENCES
- 1.Itin PH, Fistarol SK. Palmoplantar keratodermas. Clin Dermatol. 2005;23:15–22. doi: 10.1016/j.clindermatol.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Stevens HP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. Literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640–651. [PubMed] [Google Scholar]
- 3.Kelsell DP, Stevens HP. The palmoplantar keratodermas: much more than palms and soles. Mol Med Today. 1999;5:107–113. doi: 10.1016/s1357-4310(98)01428-2. [DOI] [PubMed] [Google Scholar]
- 4.Emmert S, et al. 47 patients in 14 families with the rare genodermatosis keratosis punctata palmoplantaris Buschke-Fischer-Brauer. Eur J Dermatol. 2003;13:16–20. [PubMed] [Google Scholar]
- 5.Martinez-Mir A, et al. Identification of a locus for type I punctate palmoplantar keratoderma on chromosome 15q22-q24. J Med Genet. 2003;40:872–878. doi: 10.1136/jmg.40.12.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao M, et al. Refined localization of a punctate palmoplantar keratoderma gene to a 5.06-cM region at 15q22.2-15q22.31. Br J Dermatol. 2005;152:874–878. doi: 10.1111/j.1365-2133.2005.06488.x. [DOI] [PubMed] [Google Scholar]
- 7.Jung EG. Acrokeratoelastoidosis. Humangenetik. 1973;17:357–358. doi: 10.1007/BF00273193. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XJ, et al. Identification of a locus for punctate palmoplantar keratodermas at chromosome 8q24.13-8q24.21. J Invest Dermatol. 2004;122:1121–1125. doi: 10.1111/j.0022-202X.2004.22507.x. [DOI] [PubMed] [Google Scholar]
- 9.El Amri I, et al. Clinical and genetic characteristics of Buschke-Fischer-Brauer’s disease in a Tunisian family. Ann Dermatol Venereol. 2010;137:269–275. doi: 10.1016/j.annder.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Page LJ, Sowerby PJ, Lui WW, Robinson MS. Gamma-synergin: an EH domain-containing protein that interacts with gamma-adaptin. J Cell Biol. 1999;146:993–1004. doi: 10.1083/jcb.146.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boukamp P, et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson MS, Bonifacino JS. Adaptor-related proteins. Curr Opin Cell Biol. 2001;13:444–453. doi: 10.1016/s0955-0674(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 13.Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Horgan CP, McCaffrey MW. Rab GTPases and microtubule motors. Biochem Soc Trans. 2011;39:1202–1206. doi: 10.1042/BST0391202. [DOI] [PubMed] [Google Scholar]
- 15.Ceresa BP. Regulation of EGFR endocytic trafficking by rab proteins. Histol Histopathol. 2006;21:987–993. doi: 10.14670/HH-21.987. [DOI] [PubMed] [Google Scholar]
- 16.Rappoport JZ, Simon SM. Endocytic trafficking of activated EGFR is AP-2 dependent and occurs through preformed clathrin spots. J Cell Sci. 2009;122:1301–1305. doi: 10.1242/jcs.040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Downward J, Parker P, Waterfield MD. Autophosphorylation sites on the epidermal growth factor receptor. Nature. 1984;311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- 18.Sousa LP, et al. Suppression of EGFR endocytosis by dynamin depletion reveals that EGFR signaling occurs primarily at the plasma membrane. Proc Natl Acad Sci U S A. 2012;109:4419–4424. doi: 10.1073/pnas.1200164109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goh LK, Huang F, Kim W, Gygi S, Sorkin A. Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J Cell Biol. 2010;189:871–883. doi: 10.1083/jcb.201001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sprecher E, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet. 2005;77:242–251. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gissen P, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 22.Montpetit A, et al. Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord. PLoS Genet. 2008;4:e1000296. doi: 10.1371/journal.pgen.1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: a model system to study vesicular trafficking. Pigment Cell Melanoma Res. 2009;22:268–282. doi: 10.1111/j.1755-148X.2009.00558.x. [DOI] [PubMed] [Google Scholar]
- 24.Tarpey PS, et al. Mutations in the gene encoding the Sigma 2 subunit of the adaptor protein 1 complex, AP1S2, cause X-linked mental retardation. Am J Hum Genet. 2006;79:1119–1124. doi: 10.1086/510137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennion SD, Patterson JW. Keratosis punctata palmaris et plantaris and adenocarcinoma of the colon. A possible familial association of punctate keratoderma and gastrointestinal malignancy. J Am Acad Dermatol. 1984;10:587–591. doi: 10.1016/s0190-9622(84)80262-5. [DOI] [PubMed] [Google Scholar]
- 26.Armstrong DK, et al. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8:143–148. doi: 10.1093/hmg/8.1.143. [DOI] [PubMed] [Google Scholar]
- 27.Wan H, et al. Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol. 2004;150:878–891. doi: 10.1111/j.1365-2133.2004.05996.x. [DOI] [PubMed] [Google Scholar]
- 28.Whittock NV, et al. Frameshift mutation in the V2 domain of human keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol. 2002;118:838–844. doi: 10.1046/j.1523-1747.2002.01750.x. [DOI] [PubMed] [Google Scholar]
- 29.McLean WH. Genetic disorders of palm skin and nail. J Anat. 2003;202:133–141. doi: 10.1046/j.1469-7580.2003.00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cottingham RW, Jr., Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 31.Schaffer AA, Gupta SK, Shriram K, Cottingham RW., Jr. Avoiding recomputation in linkage analysis. Hum Hered. 1994;44:225–237. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 32.Hirst J, Miller SE, Taylor MJ, von Mollard GF, Robinson MS. EpsinR is an adaptor for the SNARE protein Vti1b. Mol Biol Cell. 2004;15:5593–5602. doi: 10.1091/mbc.E04-06-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.