

Abstract
γδ T cells, a lineage of innate-like lymphocytes, are distinguished from conventional αβ T cells in their antigen-recognition, cell activation requirements and effector functions. γδ T cells have been implicated in the pathology of several human autoimmune and inflammatory diseases and their corresponding mouse models, but their specific roles in these diseases have not been elucidated. We report that γδTCR+ cells including both the CD27−CD44hi and CD27+CD44lo subsets infiltrate islets of pre-diabetic non-obese diabetic (NOD) mice. Moreover, NOD CD27−CD44hi and CD27+CD44lo γδ T cells were pre-programmed to secrete IL-17, or IFN-γ upon activation. Adoptive transfer of T1D to T and B lymphocyte-deficient NOD recipients was greatly potentiated when γδ T cells, and specifically the CD27− γδ T cell subset, were included compared to transfer of αβ T cells alone. Antibody-mediated blockade of IL-17 prevented T1D transfer in this setting. Moreover, introgression of genetic Tcrd deficiency onto the NOD background provided robust T1D protection, supporting a non-redundant, pathogenic role of γδ T cells in this model. The potent contributions of CD27− γδ T cells and IL-17 to islet inflammation and diabetes reported here suggest that these mechanisms may also underlie human T1D.
INTRODUCTION
T cell receptor (TCR) γδ T cells are a highly conserved lineage of lymphocytes that use gene rearrangement to encode their defining antigen receptors (1) and provide non-redundant contributions to host protection against a broad spectrum of infectious diseases. In contrast to αβ T cells, γδ T cell receptors recognize antigen directly, without a requirement for processing and presentation by antigen-presenting cells (APC) (2-4). Moreover, whereas TCR engagement in the thymus is essential for conventional αβ T cell maturation, this event is necessary for development of some, but not all, TCRγδ+ thymocytes (5). These and other signatory properties have led to their classification as innate-like T cells, along with subsets of αβ T cells restricted by either the MHC class I-like molecule CD1d (invariant NKT cells; iNKT) or MHC-related protein 1 (MR1) (mucosal-associated invariant T cells; MAIT) (6-8); (9). Thus, γδ T cells can contribute to host protection by responding either to microbial non-peptidic antigens or to endogenous self ‘stress antigens’ upregulated at sites of inflammation. Most γδ T cells do not circulate according to the patterns of conventional αβ T cells, and they often respond much more rapidly than conventional lymphocytes, invoking the concept of lymphoid stress-surveillance (8). These unconventional activation requirements, their ready capacity to associate with tissues, and broad functional potentials (10-12) suggest that γδ T cells may also act in settings of autoimmune disease (13).
Recent data point to disease-protective roles for CD1d-restricted iNKT and MR-1-restricted MAIT cells and, in contrast, a pathogenic contribution of γδ T cells in several inflammatory and autoimmune diseases, including type 1 diabetes (T1D). For example, earlier studies of both human T1D and the non-obese diabetic (NOD) mouse showed decreased iNKT-cell frequencies and reduced ability to secrete cytokines such as IL-4 compared to healthy controls (14, 15). Introgression of the Cd1d-null mutation onto the NOD background disrupted iNKT cell function and further aggravated diabetes (16). A disease-suppressive role for MAIT cells was suggested in human multiple sclerosis (17) and demonstrated in an experimental autoimmune encephalomyelitis (EAE) mouse model (18). In contrast to these proposed disease-protective functions, γδTCR+ cells composed a high proportion of T cells in the lesions of multiple sclerosis (MS) patients (19), suggesting a pathogenic role for this T cell subset. Indeed, similar to findings in human MS, γδ T cells have been found at high frequency in the brains of mice with EAE (20). Moreover, γδ T cells are reportedly involved in other experimentally induced mouse models of autoimmune disease, including collagen-induced arthritis and colitis (5, 21-23). Here we provide evidence for a potent pathogenic role for γδ T cells in a spontaneous mouse model of T1D.
The NOD mouse has been extensively studied as a spontaneous model of human T1D, with which it shares both genetic risk variants and features of autoimmune pathogenesis (24, 25). However, γδ T cells have been reported to serve both protective and pathogenic roles in the NOD mouse model. γδ T cells purified from among the intraepithelial lymphocyte (IEL) population of NOD mice could prevent T1D when transferred to NOD recipients that had undergone neonatal thymectomy (26). Transfer of T1D protection by T cells pre-conditioned with aerosolized insulin depended on γδ T cells in the inoculum (27). Recently, γδ T cells were reported to protect NOD mice from T1D via a TGF-β-dependent mechanism (28). These studies support a T1D-suppressive function for γδ T cells in the NOD mouse. In contrast, γδ T cell clones isolated from the spleen and pancreatic lymph nodes (PLN) of NOD mice were reactive against insulin (29), predicting a pathogenic role for such cells. In addition, in islet biopsies from human diabetic patients, γδ TCR sequences were predominant among the T cell clonotypes identified (30), suggesting that γδT cells infiltrate the pancreatic islets in human T1D. Collectively these data suggest that γδ T cells may participate in T1D pathogenesis.
Here we used a genetic approach, supported by in vivo immunological analyses, to identify a critical mechanism by which γδ T cells contribute to NOD mouse T1D pathogenesis. We provide evidence that IL-17 is an important mediator facilitating the contributions of CD27− γδ T cell subset to islet inflammation and diabetes. Given the strong similarities between the NOD mouse model and human T1D, these data suggest a potential role of γδ T cells and IL-17 in the human disease.
MATERIALS AND METHODS
Mice
All mice used in this study were maintained in a specific pathogen-free facility at the Hospital for Sick Children. Spontaneous T1D incidence at age 6 months in NOD/Jsd animals is 83% in females and 35% in males. NOD.B6;129P2-Tcrdtm1Mom/J (NOD.TcRδ-KO) mice were a gift from Dr. Robert Tigelaar (Yale University, CT). All procedures performed on these mice were performed according to guidelines and animal use protocols of the Hospital for Sick Children animal care committee.
DNA preparation and microsatellite and SNP genotyping
DNA was prepared from tail biopsies by digestion in Tissue Digest Solution B (AutoGen AG00122) with 2ug/mL Proteinase K (Qiagen #19131). SNP genotyping was performed using Illumina Mouse MD Linkage Panel (Illumina, CA) at The Centre for Applied Genomics at the Hospital for Sick Children. For high-resolution microsatellite genotyping, DNA was diluted 1/20 prior to introduction into PCR reactions. Novel microsatellite markers were generated as previously described (31). DNA was amplified for 35 cycles with the following conditions: 30s at 94°C, 30s at 55°C, 1min at 72°C using Multiplex Master Mix (Qiagen #206143). The PCR product was diluted 1/20, and 1uL was mixed with a 10uL mixture of formamide and a 500LIZ size standard (1mL HI-DI formamide per 7μL size standard). Samples were run on the 3730XL DNA Analyzer (Applied Biosystems). Alleles were sized, in comparison to standards, by viewing their electropherograms in GeneMapper (Applied Biosystems).
Spontaneous diabetes assessment
Blood and/or urine glucose levels were measured in mice biweekly. Animals were classified as diabetic when blood glucose exceeded 16mmol/L or urine glucose exceeded 250mg/dL. Statistical analysis on T1D life table data was performed using the Log-rank (Mantel-Cox) test, Prism software (Version 5.0b, GraphPad Software Inc).
Insulitis assessment
Pancreata were dissected and immediately immersed in OCT media (Tissue-Tek, Torrance, CA), frozen at −78°C in 2-methylbutane over dry ice, and stored at −70°C. Preparation of frozen sections was performed with a Leica CM 3050 Cryostat (Leica Canada). To maximize analysis of independent islet infiltrates, three 5-μm sections were cut at least 400 μm apart. Pancreatic sections were stained with Mayer’s hematoxylin and eosin Y (H+E, Sigma) to visualize leukocyte infiltration. Assessment of insulitis severity in pancreatic sections was performed as previously described (32). Briefly, islets were graded according to the following criteria: 0, no visible infiltrates; 1, peri-insulitis as indicated by peri-vascular and peri-islet infiltrates; 2, 25-50% of the islet interior occluded by leukocytes displaying invasive infiltrates; 3, 50-80% of the islet interior invaded by leukocytes; or 4, invasive insulitis involving >80% of the islet field.
Magnetic bead depletions
For purification of total spleen T cells, cell suspensions were stained with biotinylated rat antibodies specific for CD49b (clone DX5, Becton Dickinson, San Jose, CA), CD105 (clone MJ7/18, Sunnybrook Research Institute (SRI)), and microbeads specific for biotin, CD11c, CD11b, and CD19 (Miltenyi Biotec, Cologne, Germany). For purification of splenic αβT cells, cell suspensions were stained as above, with the addition of a biotinylated hamster antibody against mouse TCRγ (clone GL3, SRI). Labeled cells were depleted using an AutoMACS Pro Separator (Miltenyi Biotec). The purity of eluted cells was assessed by flow cytometry using hamster antibodies against TCRβ (clone H57-597, SWRI) and TCRγ (clone GL3, SRI).
Flow cytometry
Single cell suspensions were prepared from thymus, spleen, pooled peripheral lymph nodes, or the small intestine epithelium. Isolation of lymphocytes from intestinal epithelium was performed as previously described (33). Viable cell counts were determined by trypan blue exclusion, and cells were stained in staining media (1X HBSS (GibcoBRL, Gaithersburg, MD), 10mM Hepes, 2% calf serum (Sigma, St. Louis, MO)) with flourochrome-conjugated antibodies: TCRγ-PE (clone GL3, SRI), TCRβ-FITC (clone H57-597, SRI), CD19-FITC (clone 6D5, SRI), CD44-APC (clone IM781, eBioscience, San Diego, CA), CD27-biotin (clone LG.7F9, eBioscience), and avidin-APCCy7 (Invitrogen, Carlsbad, CA). Flourescence was analyzed using a BD (Becton Dickinson, San Jose, CA) LSR-II flow cytometer. Dead cells were excluded using 1ug/mL propidium iodide. At least 400 γδTCR+ events were acquired for islet flow cytometry, and a minimum of 1,000 γδTCR events were acquired for analysis of other tissues.
Cell stimulation and intracellular cytokine staining
Cell suspensions were prepared as described above, then resuspended at a concentration of 5 x 106/mL in DMEM with 10% FBS, 1% nonessential amino acids, 2 mM l-glutamine, 50 μM 2-ME, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate. Cells were incubated for 5 hours at 37C in the presence of 50ng/mL PMA, 1ug/mL Ionomycin and GolgiPlug protein transport inhibitor (BD Biosciences, 1:1000 dilution). Cells were washed, then resuspended in staining media (1X HBSS (GibcoBRL, Gaithersburg, MD), 10mM Hepes, 2% calf serum (Sigma, St. Louis, MO)) with flourochrome-conjugated antibodies against cell surface antigens: TCRγ-PE (clone GL3, SRI), TcRβ-PECy7 (clone H57-597, eBioscience), CD19-PECy7 (clone 6D5, eBioscience), CD27-biotin (clone LG.7F9, eBioscience), Fixable Blue viability dye (Invitrogen) and avidin-APCCy7 (Invitrogen, Carlsbad, CA). Cells were washed, then fixed and permeabilized according to reagent manufacturer’s instructions (BD Cytofix/Cytoperm Kit, #554714). Cells were then stained with flourochrome-conjugated antibodies against cytokines: IFN-γ-APC (clone XMG1.2, BD Pharmingen), IL-17A-FITC (clone eBio17B7, eBioscience).
Cell sorting
For sorting of CD27−γδT cells, splenic cell suspensions were depleted of non-T cells (as above) then stained with TCRγ-PE (clone GL3, SRI), TCRβ-FITC (clone H57-597, SRI), CD27-biotin (clone LG.7F9, eBioscience), and avidin-APCCy7 (Invitrogen, Carlsbad, CA) and 1μg/mL propidium iodide. Sorting was performed on a MoFlo cell sorter (Dako Cytomation). Cells were gated on PI-, forward vs side scatter, and 2 populations were collected: FITC+ (αβT cells) and PE+APCCy7− (CD27−γδT cells). Purity of each of these cell populations was >96%.
T cell adoptive transfers
Either total NOD splenic T cells or NOD splenic αβT cells were purified using magnetic bead depletion, as described above. 107 T cells were resuspended in 200μL sterile PBS then transferred to 4-5 week old female NOD.SCID recipients by tail vein intravenous (i.v.) injection. For transfer of αβT cells and CD27− γδT cells, these sorted populations were pooled, 107 cells were resuspended in 200μL sterile PBS and transferred to 4-5 week old female NOD.SCID recipients by i.v. tail vein injection. Highly purified, live CD27− γδT cells were present in this innoculum at their orthotopic frequency, and numbered approximately 80,000-100,000 cells among the 107 T cells injected. Recipients were monitored for T1D until 100 days post-transfer. In IL-17 neutralization study, 10μg IL-17 neutralizing antibody (R&D Systems, clone 50104, MAB421) or rat IgG2a isotype control antibody (R&D Systems, clone 54447, MAB006) in 100μL sterile PBS was administered by i.p. injection twice per week, as described previously (34), beginning on the day of T cell transfer. This monoclonal antibody was raised against E. coli-derived recombinant mouse IL 17 residues Thr22-Ala158 (NCBI Accession Number Q62386) and recognizes murine IL-17A. This antibody also shows ~40% reactivity with recombinant mouse (rm) IL-17A/IL-17F heterodimer, but no cross-reactivity with rmIL-17B, rmIL 17C, rmIL-17D, rmIL-17E, or homodimeric rmIL-17F (see R&D Systems, http://www.rndsystems.com/Products/mab421/). The dosage and regimen of anti-IL-17 was based on a previous publication demonstrating this regimen to be effective at controlling antigen-specific Th17 cells in the EAE model (35). Treatment was continued until recipients became diabetic or until 100 days post-adoptive transfer.
Islet preparations
Islets were prepared by published methods (36) with slight modifications. Briefly, a surgical clamp was placed over the ampulla to block the bile pathway to the duodenum. Three mL of ice-cold HBSS (GibcoBRL) with 1mM CaCl2, 10mM HEPES and 2mg/mL Collagenase Type IV (Worthington Biochemical Corp., Lakewood NJ, CLS-4, Lot #40C11795) was slowly perfused into the pancreas through a needle inserted into the common bile duct. The pancreas was dissected and placed in 2mL HBSS with 1mM CaCl2, 10mM HEPES and 2mg/mL Collagenase Type IV and incubated at 37°C for 10min. The suspension was shaken to disaggregate the tissue, then allowed to digest for another 2min at 37°C. The tissue was washed by addition of 50mL ice-cold staining media and centrifuged at 290 x g for 2 min. The supernatant was discarded and the wash was repeated. The islets were resuspended, passed through a 70μM sterile cell strainer (BD Falcon 35-2350) and retained on the strainer. Then, the enriched islet preparation was resuspended and hand picked under a dissecting microscope. The purified islets were incubated at 37°C for 5 minutes in enzyme-free cell dissociation buffer (Invitrogen #13151-014). The resulting single cell suspension was used for antibody staining and flow cytometry analysis.
RESULTS
γδT cells potentiate αβT cell mediated transfer of T1D to NOD.SCID recipients
To elucidate potential pathogenic or protective role(s) of γδ T cells in T1D, we tested the impact of this cell subset on adoptive transfer of T1D to an immuno-deficient, T1D-protected strain. Splenic total T cells, purified αβ T cells alone or purified γδ T cells alone were transferred into T and B cell-deficient NOD.SCID recipients. Consistent with our own and others’ previous data (J. Danska, personal communication; (37)), transfer of 107 total splenic T cells from 12-14 week old female pre-diabetic NOD mice resulted in 100% T1D incidence in NOD.SCID recipients by 100 days post-transfer (Fig 1, solid black trace). In contrast, removal of γδ T cells from the inocula rendered NOD αβ T cells less potent in transferring T1D to only 50% of recipients (Fig 1, dashed trace, p<0.0001 relative to total T cells, n≥9 recipients per group), suggesting that γδ T cells contribute to disease immunopathogenesis. Notably, splenic γδ T cells alone were unable to transfer disease (Fig 1, grey trace, n=9 recipients) underscoring the requirement for cooperation with αβ T cells to potentiate T1D in this adoptive transfer setting.
Fig 1. Both αβT and γδT cells are required for efficient T cell-mediated T1D transfer to NOD.SCID recipients.

Splenocytes were harvested from 12- to 14-week old NOD mice. B cells and APC, and in some cases γδTCR+ cells were depleted by magnetic bead purification. Purification of γδT only was performed by FACS sorting. Either 107 splenic total T cells (containing αβT and γδT), 107 splenic αβT cells alone, or 200,000 γδT alone were transferred to 4-5-week old female NOD.SCID recipients by tail vein i.v. injection. Recipients of NOD total T cells (αβT+γδT, black solid trace), all developed T1D within 100 days. In contrast, recipients of NOD αβT cells alone (αβT, dashed trace) were protected from T1D compared to recipients of both T cell subsets (p<0.0001). γδT cells alone were not able to transfer T1D in this setting (p=0.0141 compared to NOD αβT cells alone; no recipients diabetic). P values are Log-rank Mantel-Cox test comparisons of survival curves, n≥9 recipients per group).
CD27−CD44hi γδT cells infiltrate the islets preceding T1D
A pathogenic role for γδT cells might be reflected by their presence at the site of beta cell destruction. Hence, Islets of Langerhans were purified, enzymatically dissociated and infiltrating leukocytes assessed by flow cytometry (Fig 2A, C). γδ T cells were detected in NOD islets at two time points, 8 weeks and 12 weeks of age, preceding T1D onset (Fig 2A). Interestingly, the percentage of intra-islet γδ T cells within the CD19−αβTCR− population was greater at 12 weeks compared to 8 weeks of age (p<0.05, Fig 2B). Although TCR-mediated thymic selection does not constrain the γδ TCR repertoire, it does pre-program γδ T cell effector function (5, 38). Two subsets of γδ T cells can be distinguished by expression of the tumor necrosis factor receptor (TNFR) family member CD27. CD27− γδ T cell progenitors mature without cognate ligand engagement into CD44hi IL-17-secreting cells, whereas γδ progenitors that bind ligand mature into CD27+ cells with lower CD44 levels and primarily secrete IFN-γ (8, 12), phenotypes that remain stable over time in the periphery (5, 12). To define the frequencies of these functionally distinct subsets in various anatomical compartments, γδ T cells were assessed for CD27 and CD44 expression by flow cytometry in spleen, multiple peripheral lymph nodes (LN), and in highly purified islets from pre-diabetic 8- and 12-week old NOD mice (Figs 2C, D, E). At both pre-diabetic time points assessed, the majority of γδ T cells in all peripheral LN tested were CD27+CD44lo, with a much lower frequency of CD27−CD44hi γδ T cells (Fig 2C, D, E). In 8-week old NOD mice, the splenic γδ T cells pool also had a low frequency of CD27−CD44hi compared to CD27+CD44lo cells. The relative frequencies of CD27−CD44hi and CD27+CD44lo γδ T subsets in 8-week old NOD mice were similar to those observed in age-matched non-autoimmune-prone CD1 strain mice (Fig S2). In contrast, CD27−CD44hi γδ T cells were enriched in the spleens of 12-week old NOD mice, relative to other peripheral LN (p<0.01, Fig 2C, E). This finding was consistent with our observations that splenic T cells from 12-week old donors displayed enhanced diabetogenicity relative to equal numbers of LN T cells when transferred into NOD.SCID recipients (Table S1). In both 8- and 12-week old NOD mice, islet infiltrating γδTCR+ cells included both the CD27−CD44hi and CD27+CD44lo subsets (Fig. 2C) suggesting that the effector functions of either or both cell types might contribute to islet pathology. However, we noticed that within the γδTCR+ fraction, there was a greater frequency of CD27−CD44hi T cells in the islets than in any LN compartments examined (p<0.01; Fig. 2D, E). Moreover, CD27−CD44hi γδ T cells were present at time points corresponding to the early stages of insulitis. These data showed that γδ T cells with potential to produce IL-17 accumulate in the islets prior to T1D onset and thus might contribute to pathogenesis in this model.
Fig 2. CD27− γδTcR+ cells infiltrate NOD islets during T1D pathogenesis.
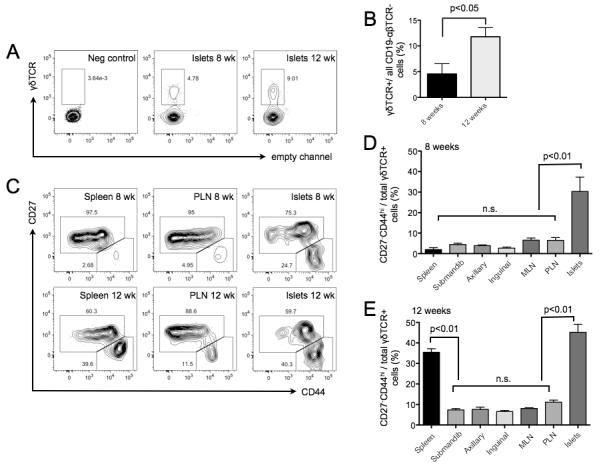
A) Flow cytometry-based enumeration of γδT cells in the pancreatic islets of 8- and 12-week old NOD mice. Pancreata were perfused with collagenase and the islets were prepared (Materials and Methods). Cells of interest were gated based on excluding propidium iodide (live cells), followed by forward and side scatter profile. Stains for both αβTcR and CD19 were included to exclude B cells and αβT cells from subsequent analysis. Within the CD19- αβTCR- population, an intra-islet γδTCR+ population was identified in both 8- and 12-week old NOD mice. Representative plots were chosen from n≥5 biological replicates. B) The frequency of γδTCR+ cells among non-B, non-αβT cells is shown for islet samples from 8- and 12-week old NOD mice. The frequency of intra-islet γδT cells is significantly higher among 12-week-old NOD mice, compared to 8-week old NOD mice (p<0.05, 2-tailed non-parametric t-test, n≥5 biological replicates). C) Within the γδTCR+ population, the expression of CD27 and CD44 was used to discriminate two subsets: the CD27+CD44low (putatively IFN-γ secreting), and the CD27− CD44hi (putatively IL-17 secreting) γδT cells. FACS profiling of these subsets was performed in spleen, various lymph nodes, and islets from 8- and 12-week old NOD mice. Representative plots were chosen from n≥5 biological replicates. D) Statistical of analysis of multiple lymphoid compartments reveals that CD27−CD44hi γδT cells were present at high frequency in the islets of 8-week old NOD mice, compared to all other lymphoid compartments in 8-week old NOD mice (p<0.01, 2-tailed non-parametric t-test, n≥5 biological replicates). E) In samples from 12-week old NOD mice, CD27−CD44hi γδT cells were present at elevated frequencies in the spleen and islets, compared to all lymph nodes tested (p<0.01, 2-tailed non-parametric t-test, n=8 biological replicates).
CD27−CD44hi γδ T cells are pre-programmed to secrete IL-17
A unique feature of γδ T cells is their expression of surface markers that are highly predictive of cytokine profile upon activation. In other strains of mice, CD27− γδ T cells are pre-programmed to produce IL-17 upon activation, whereas CD27+ γδ T cells are skewed toward IFN-γ production (12, 38). To confirm that this correlation between CD27 surface expression and cytokine potential was also observed in the NOD mouse, we assessed IL-17 and IFN-γ production in γδ T cells stimulated ex vivo. Cells were activated by exposure to PMA and ionomycin in the presence of a protein transport inhibitor then assessed for CD27, IL-17A and IFN-γ expression by flow cytometry. Without activation, these cells displayed no evidence of basal IL-17A or IFN-γ production (Fig 3A). In contrast, activated NOD γδ T cells from the spleen, thymus, and pooled peripheral LN all showed appreciable IL-17A+ and IFN-γ+ populations (Fig 3A, upper row). CD27 expression was examined on IL-17+ (blue trace) and IFN-γ+ (red trace) γδ T cells (Fig 3A, lower row). As expected, IL-17+ γδ T cells expressed low levels of CD27, while IFN-γ+ γδ T cells were CD27+ (Fig 3A, lower row). These data confirmed that like other strains, NOD mouse CD27−CD44hi γδ T cells are pre-programmed to secrete IL-17 upon activation, and suggested that the CD27−CD44hi γδ T cells observed in pre-diabetic NOD mouse islets (Fig 2) may contribute pathogenic effects via IL-17.
Fig 3. CD27− γδT cells mediate T1D transfer via an IL-17 dependent mechanism.
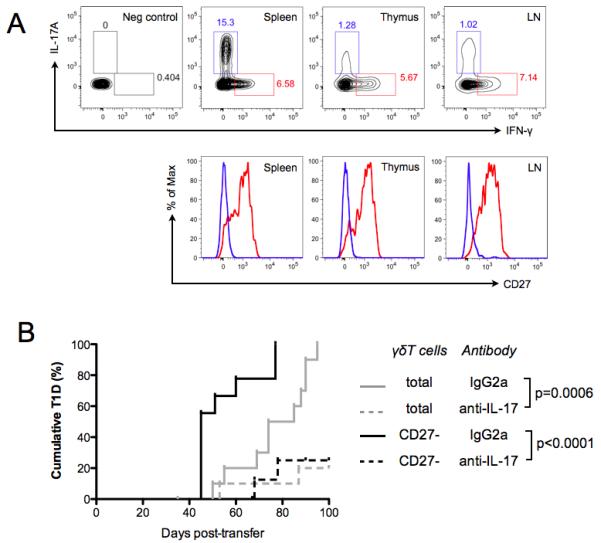
A) Cells from NOD spleen, thymus, or pooled peripheral LN were cultured in the presence of 50ng/mL PMA and 1ug/mL Ionomycin for 5 hours. Cells were stained for surface markers (γδTCR, αβTCR, CD19, CD44, CD27, and a fixable viability dye), then permeabilized and stained with antibodies specific for IL-17 and IFN-γ. Contour plots in the upper row show production of IL-17 and IFN-γ by γδT cells. Histograms in the lower row show CD27 expression on either IL17+ γδT cells (blue trace) or IFN-γ+ γδT cells (red trace). Representative plots were chosen from n=4 biological replicates. B) 107 purified splenic T cells, containing αβT and either total γδT cells (grey traces) or purified CD27− γδT cells only (black traces), were transferred from 12- to 14-week old NOD donors into 4-5-week old NOD.SCID recipients. CD27− γδT cells were present in the inoculum at their orthtopic frequency (see Materials and Methods). On the same day, and twice a week thereafter, NOD.SCID recipients were injected i.p. with either anti-IL-17 neutralizing antibody or a rat IgG2a isotype control antibody. Antibody injection was continued for 10 weeks post-T cell transfer and recipients were monitored for T1D. All recipients of the isotype control antibody progressed to T1D within the 100-day observation period. Recipients of the IL-17 neutralizing antibody were protected from T1D (total γδT cells p=0.0006; CD27− γδT cells p<0.0001, n≥10 for each condition) compared to cohorts receiving isotype control antibody. P values represent pair-wise Log-rank Mantel-Cox tests of survival curves.
IL-17 neutralization attenuated adoptive transfer of T1D by CD27−γδT cells
In an adoptive transfer setting γδ T cells significantly potentiated the diabetogenic capacity of αβ T cells (Fig 1). In order to test the role of the minority CD27−CD44hi γδ T cell subset, which accumulates in islets preceding T1D (Fig 2D,E), we again used an adoptive transfer approach. T cell transfers into NOD.SCID recipients were performed with NOD splenic αβ T cells from 12-14-week old donors, supplemented with either highly purified total γδ T cells or with the CD27− γδT cell subset supplied in the cell inoculum at their orthotopic frequencies. Interestingly, CD27− γδT cells co-operated with αβ T cells to induce T1D in 100% of recipients, similar to the observations with total γδT cells (Fig 3B). These data do not preclude a possible role for CD27+ γδT cells in T1D pathogenesis, but indicated that the CD27− γδT cell subset was sufficient to potentiate T1D transfer by αβ T cells. Since we had shown that CD27− γδT cells were pre-programmed to produce IL-17 following activation (Fig. 3A) we asked whether their role in adoptive transfer of T1D was IL-17 dependent. To address this question, we performed adoptive transfers of αβ T cells supplemented with either total γδ T cells or only the CD27− γδT cell fraction into recipients that were given biweekly injections of either IL-17 neutralizing antibody, or an IgG2a isotype control antibody. Antibody treatment was initiated on the day of T cell transfer and continued for 100 days. Recipients of splenic αβ T cells supplemented with total γδT cells and IL-17 neutralizing antibody were dramatically protected from T1D, relative to the appropriate isotype control cohort (p=0.0006, Fig 3B). Similarly, recipients of splenic αβ T cells supplemented with the CD27− γδT cell subset together with IL-17 neutralizing antibody were protected from diabetes relative to their respective isotype control group (p<0.0001, Fig 3B). These data demonstrated that the CD27− γδT cell subset can co-operate with αβ T cells to efficiently transfer T1D, and that functional blockade of IL-17 ablated the pathogenic contribution of this γδT cell subset.
Genomic characterization of the NOD.TcRδ-KO strain
T1D in the NOD mouse is a polygenic trait resulting from the collective effects of many risk loci across the genome (31, 32, 39). To test whether γδ T cells make non-redundant contributions to T1D pathogenesis in the NOD mouse, we used a strain in which γδ T cell production was blocked by a Tcrd constant region mutation, NOD.129P2-Tcrdtm1Mom/J (NOD.TcRδ-KO) (40). We genotyped the NOD.TcRδ-KO strain to exclude genomic contamination by other (non-Tcrd) diabetes-risk loci to T1D-related phenotypes. Genome-wide analysis of the NOD.TcRδ-KO strain revealed that the only region of the genome showing non-identity between NOD and NOD.TcRδ-KO mice spanned positions 36200642 to 56445855 on chromosome 14 (see http://serrate.research.sickkids.ca:8081/Tcrd_Illumina_SNP_genotyping.xls) that carries the Tcra-d locus, the site of the TCRδ targeted mutation (32, 41). The original TCRδ-KO mutation, made in 129 strain embryonic stem cells, was bred onto a C57BL6 (B6) background (41), and then backcrossed to NOD for >10 generations (40). Thus it was possible that chromosome 14 could harbor B6- or 129-derived alleles in addition to the 129-derived Tcra-d locus. To further refine potential non-NOD-derived intervals on chromosome 14, we identified high density microsatellite markers using the B6 reference genome within this region, and genotyped DNA prepared from NOD, 129, B6, and NOD.TCRδ-KO mice to define the non-NOD genomic interval in NOD.TCRδ-KO as positions 35433125 – 57367153 (markers D14Gul503 – D14Gul2331, Table 1). We found that this interval did not include non-NOD alleles at two previously identified insulin dependent diabetes (Idd) risk loci on chromosome 14, Idd8 (42, 43) and Idd12 (44). Therefore the only non-NOD derived portion of the genome in the NOD.TCRδ-KO mice was the chromosome 14 interval containing the Tcra-d locus.
Table 1. High-density genotyping of the NOD.TCRδ-KO strain.
Novel microsatellite sequences which were polymorphic between NOD, C57BL/6, and 129/Sv inbred mouse strains were identified on Chromosome 14, and used in high-resolution genotyping of the NOD.TCRδ-KO strain (Materials and Methods). EnsEMBL start and end position of the novel microsatellites in the B6 reference sequence (‘ENS start’ and ‘ENS end’) were used to position these markers, and the PCR amplicon size used to distinguish the alleles. The NOD.TCRδ-KO strain carried a 22Mb interval including the Tcra/d locus and the TCRδ−/− mutation inherited from 129/Sv. This interval did not include any known Idd loci.
| Locus | ENS start | ENS end | Marker | Fwd primer | Rvs primer | Amplicon size | |||
|---|---|---|---|---|---|---|---|---|---|
| NOD | B6 | 129 | TcRd-KO | ||||||
| TCRd-KO=NOD | 35034545 | 35034904 | D14Gul453 | AGACAGACTGCTGTCAGGCT | GTGCCAGAGGCTGGAGAAAG | 181 | 183 | 183 | 181 |
| TCRd-KO=NOD | 35164265 | 35164624 | D14Gul470 | TGATGCCTGCTTGTCTTCCC | CCACAACACCTGTCAGCTCT | 211 | 211 | 214 | 211 |
| TCRd-KO=129 | 35433125 | 35433484 | D14Gul503 | CTGACCTCCATAAATGTGCAGTGG | CCAGAGGTGAGCTAGGCTCTT | 178 | 178 | 203 | 203 |
| TCRd-KO=129 | 35675405 | 35675764 | D14Gul537 | CACAGGTTTGCACCACCCTA | TGCGTGAGATGACCAACGAG | 214 | 214 | 242 | 242 |
| TCRd-KO=129 | 36121685 | 36122044 | D14Gul593 | TTGTTTGAACTGGCATGGTTGG | ATCAGACTTGAACTAAGCCCAAGAA | 266 | 266 | 291 | 291 |
| TCRd-KO=129 | 37679105 | 37679464 | D14Gul834 | GGTGAATGGTCTTGCAGCCA | TGCCTGGCAATGTATTTCTTTCCT | 232 | 228 | 187 | 187 |
| TCRd-KO=129 | 37848185 | 37848544 | D14Gul866 | GCACAGGCTCATGTCACCAT | ACTGCAAGCAGCACTTCCTT | 245 | 247 | 273 | 273 |
| TCRd-KO=129 | 37910645 | 37911004 | D14Gul879 | TGTGGGGACCTGGGCAATAT | CCACATGGTGACTCCCAACC | 285 | 286 | 293 | 293 |
| TCRd-KO=129 | 38039825 | 38040184 | D14Gul900 | GAGAGGTGCATGAGGGCAAC | AGCAAACCTGCACAACCTCC | 159 | 173 | 185 | 185 |
| TCRd-KO=129 | 41986565 | 41986924 | D14Gul1484 | AGCACTGTGGTACAGTTGGC | GGCTCACTGCACTGTGTCTT | 283 | 285 | 285 | 285 |
| TCRd-KO=129 | 42508325 | 42508684 | D14Gul1535 | AGTACAGGTCAGGGCTGCAA | ACCACAAACACTTACAAGGGCA | 263 | 268 | 268 | 268 |
| TCRd-KO=129 | 43692305 | 43692664 | D14Gul1695 | CCTTCCTTGCTTCCTCACTGT | TGTCAACAGCTGGAGTTGCC | 290 | 286 | 286 | 286 |
| TCRd-KO=129 | 44326805 | 44327164 | D14Gul1766 | TGCAATGTCTGCAGGCCATT | ACACAGGGAACAAAGTGCCC | 288 | 298 | 298 | 298 |
| TCRd-KO=129 | 44586605 | 44586964 | D14Gul1806 | ACATCCCTGACAAGAGGCCA | ATGACTGGGGATGTGGCTCA | 240 | 254 | 254 | 254 |
| TCRd-KO=129 | 45122225 | 45122584 | D14Gul1854 | TCAACAGCTGGAGTTGCCTG | AGTCAAGTCCTGTGCTCCTCA | 329 | 332 | 332 | 332 |
| TCRd-KO=129 | 56110875 | 56111234 | D14Gul2200 | ACCTACACATGCACAGTCACG | GGTGGGGTGAGAGAAGGGTT | 193 | 210 | 210 | 210 |
| TCRd-KO=129 | 56175015 | 56175374 | D14Gul2209 | ACGTGCAAAGTCCTGGGTTC | CCACATGGAAGCTCCCAACC | 188 | 210 | 210 | 210 |
| TCRd-KO=129 | 56216955 | 56217314 | D14Gul2212 | GGGCTCCACCTACTCTAGGTC | GGGCAACTGGCAATGGAGAA | 238 | 262 | 262 | 262 |
| TCRd-KO=129 | 56286315 | 56286674 | D14Gul2219 | AAGCATGTGCTACCATGCCC | AGCACAGCCTAGGATACACCT | 265 | 267 | 267 | 267 |
| TCRd-KO=129 | 56384115 | 56384474 | D14Gul2232 | AGTTGTGGTCTGACCTGCCT | GTCAAGGCCACCCTAGGCTA | 189 | 183 | 183 | 183 |
| TCRd-KO=129 | 56408535 | 56408894 | D14Gul2238 | TACATGGGCTACTGTCCCCAA | TTAGTGCATACTGCGTGCCTC | 233 | 225 | 225 | 225 |
| TCRd-KO=129 | 56716334 | 56716693 | D14Gul2264 | TCACAAGAACACAGCATTGGACA | GAGATCCTGCAGGCCCTAAC | 222 | 220 | 220 | 220 |
| TCRd-KO=129 | 56737934 | 56738293 | D14Gul2266 | CTGCTGGCTCTGACCTCTTG | ACAGGTGAGGAGTAGCCCAC | 197 | 195 | 195 | 195 |
| TCRd-KO=129 | 57011354 | 57011713 | D14Gul2305 | TCCCAGAGCATTCCCTGTTGT | GAGAGTGTAATGTTCTGCCACTGA | 182 | 186 | 182 | 182 |
| TCRd-KO=129 | 57366794 | 57367153 | D14Gul2331 | GAAGGCATTCGCTGAGCAAAC | TTCCTTTTCACACCATGCACCA | 267 | 267 | 263 | 263 |
| TCRd-KO=NOD | 57420794 | 57421153 | D14Gul2335 | CCAGTTTGAGGCAGCCCATATA | CTCTGGAGCAAACTAGATAGCCAG | 286 | 302 | 302 | 286 |
Frequency of γδT cells displayed TCRδ gene dosage effect
The frequencies of γδ and αβ T cells were measured by flow cytometry in various peripheral compartments of NOD, NOD.TCRδ-KO, and (NOD.TCRδ-KO x NOD/Jsd)F1 mice carrying one wild-type and one TCRδ-KO allele (NOD.TCRδ-HET) (Fig 4). Interestingly, the thymic CD4-CD8-population (thymic DN), pooled lymph node (LN), spleen and small intestinal intraepithelial (IEL) compartments of NOD.TCRδ-HET mice showed a γδ T cell frequency intermediate between that of NOD and NOD.TCRδ-KO mice in demonstrating a gene dosage effect of the Tcrd−/− mutation. (Fig 4, Fig S1). This result highlights an important feature of the γδ T cell lineages, where gene dosage determines the peripheral frequency of the γδ T cells.
Fig 4. NOD.TcRδ-HET mice had half the frequency of γδT cells compared to wild-type NOD mice.
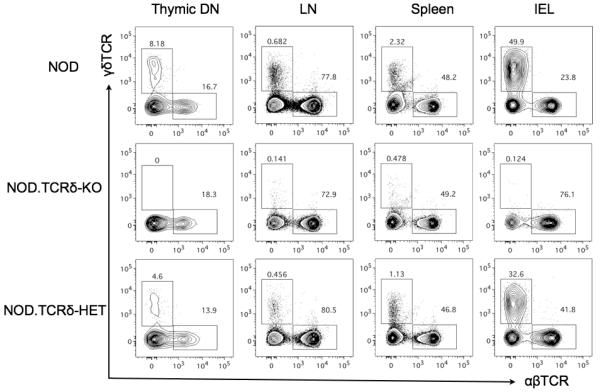
Multicolour flow cytometry was performed to define frequencies of γδT cells in CD4−CD8− thymocytes (thymic DN), pooled peripheral lymph nodes (LN), spleen, and small intestinal intraepithelial lymphocytes (IEL). NOD.TCRδ-HET mice displayed γδTCR+ cell frequencies intermediate between parental NOD and NOD.TCRδ-KO mice. Representative plots from n=3 per genotype.
TCRδ gene dosage correlates with T1D pathogenesis
Next, the impact of Tcrd gene dosage on the natural history of T1D was assessed by prospective longitudinal analysis of NOD, NOD.TCRδ-HET, and NOD.TCRδ-KO mouse cohorts over 250 days. As expected, NOD mice reached a high cumulative T1D incidence. In contrast, NOD.TCRδ-KO mice were dramatically protected from disease (p<0.0001, Fig 5A). Interestingly, NOD.TCRδ-HET mice showed an intermediate phenotype, more susceptible to T1D than NOD.TCRδ-KO (p=0.0118) but protected relative to parental NOD mice (p=0.0296, Fig 5A). To asses potential consequences of genetic γδ T cell ablation on αβ T cell diabetogenicity, we transferred αβ T cells from NOD or NOD.TCRδ-KO mice to NOD.SCID recipients, and found no evidence for impaired αβ T cell-mediated T1D transfer (Table S2). These data demonstrated a gene dose-dependent, non-redundant pathogenic role for γδ T cells in the NOD model of T1D. Work from our laboratory and from others has established that some Idd loci control distinct stages of insulitis progression in NOD mice (32, 45, 46). To identify the stage of insulitis most impacted by γδ T cells, longitudinal assessment of insulitis was performed in cohorts of non-diabetic NOD and NOD.TCRδ-KO mice at 80, 105, 120, and 240 days of age (Fig 5B). NOD mice showed progressively more severe islet infiltration over this period (Fig 5B). In contrast, insulitis severity in NOD.TCRδ-KO mice remained mild and stable after 80 days of age (Fig 5B), and the mice were protected from severe insulitis at 120 days and 240 days, relative to NOD (p<0.05). Thus, the absence of γδ T cells attenuated progression to invasive insulitis, consistent with the idea that γδ T cells function as effectors in this disease model.
Fig 5. Gene dosage at the TCRδ locus determines T1D susceptibility.
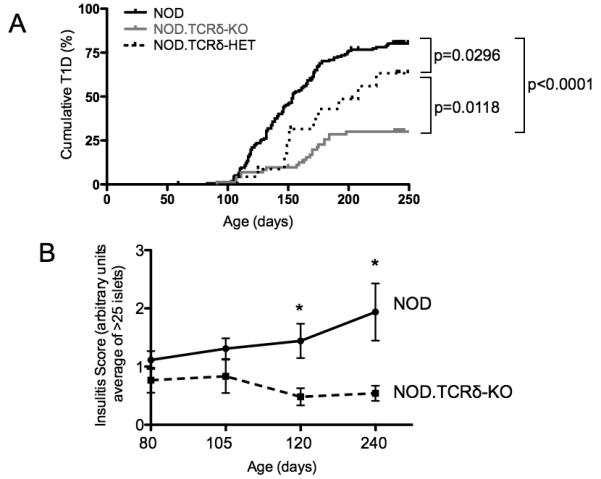
A) Longitudinal assessment of T1D was performed in cohorts of NOD, NOD.TCRδ-HET and NOD.TCRδ-KO mice. In contrast to NOD females, NOD.TCRδ-KO mice were protected from diabetes (p<0.0001), NOD.TCRδ-HET mice displayed an intermediate phenotype different from both parental NOD (p=0.0296) and NOD.TCRδ-KO (p=0.0118) animals. P values represent pair-wise Log-rank Mantel-Cox tests of survival curves, n>22 per genotype. B) Insulitis severity was assessed in the pancreata of non-diabetic female NOD and NOD.TCRδ-KO mice, at the ages indicated (see Materials and Methods). NOD mice showed a progressive increase in insulitis severity from ages 80 to 240 days. In contrast, NOD.TCRδ-KO mice showed a low and constant level of insulitis throughout this time frame. Graph depicts mean and SEM (≥7 biological replicates per condition). p<0.05, 2-tailed t-test comparing NOD to NOD.TCRδ-KO within the indicated time point.
DISCUSSION
Here we provide evidence that γδ T cells are a key component of T1D pathogenesis in the NOD mouse model. The CD27−CD44hi phenotype of the γδ T cells defines those that make IL-17 upon activation. We identified CD27−CD44hi γδ T cells infiltrating the islets of pre-diabetic NOD mice, suggesting they may participate directly or indirectly in islet destruction. Moreover, in an adoptive transfer model, we showed that CD27−CD44hi γδ T cells co-operate with αβT cells to induce T1D, and that the pathogenic role of this IL-17-producing γδ T subset can be blocked by in vivo neutralization of IL-17. Using NOD.TcRδ–KO and NOD.TcRδ–HET mice, we show that the TcRδ locus displays a gene dosage effect in terms of the frequency of γδ T cells in thymic and peripheral lymphoid compartments. Finally, frequency of γδ T cells determines differential susceptibility of these NOD-background mice to spontaneous T1D.
Previous studies have considered the possible roles of γδ T cells and γδ T cell-produced IL-17 in the NOD mouse model of T1D. For example, IL-17-producing γδ T cells were reported to play a protective role in T1D by secreting TGF-β in an adoptive transfer setting (28). However, neither the CD27/CD44 surface phenotype, nor IL-17 secreting capacity of the transferred γδ T cells were reported (28), and the splenocytes were isolated from young NOD mice at an age where we saw predominance of the CD27+CD44lo, IFN-γ secreting subset (Fig 2D, Fig 3A). IL-17 blockade in the adoptive transfer setting was reported to have no effect on γδ T cell-dependent diabetes pathogenesis but anti-IL-17 antibody treatment was given only during the first period of observation (28) and therefore differed from the approach presented here. In our study, IL-17 action was opposed over the full time course in which T1D development was scored, based on prior evidence that IL-17-producing cells participate in the effector phase rather than the initiation phase of T1D (34).
Another recent report described no effect on T1D among NOD mice in which lentiviral transgenesis was used to reduce IL-17 production (47). Although it was convincingly demonstrated that purified αβTcR+ CD4+ Th17 cells from these mice had reduced IL-17 production upon stimulation, IL-17 production was not reported for γδT cells (47). Evidence from multiple studies suggests that Th17 αβ T cells and CD27− CD44hi γδ T cells produce IL-17 by different mechanisms, with different kinetics, and in response to distinct stimuli, an in addition, γδT cells are committed to their cytokine transcriptional program upon thymic egress ((5), reviewed by (48)). It would be interesting to test how reduced IL-17 production by CD27−CD44hi γδ T cells impacts their ability to potentiate T1D. Finally, evidence for a tolerogenic role for γδTcR+ intraepithelial lymphocytes (IEL) has been reported in the NOD mouse model (26). When stimulated, γδTcR+ IEL secrete IFN-γ but not IL-17 (reviewed in (49)) so the finding that IFN-γ producing γδT IEL are tolerogenic is compatible with the data presented here that autoimmune pathogenesis is mediated by the IL-17 producing γδ T cell subset. Moreover, our demonstration that genetic γδT cell deficiency strongly protects NOD mice from T1D reveals the dominant diabetogenic effect of γδT cells in this model.
IL-17 has been implicated as an effector of other mouse models of autoimmune disease (reviewed in (9, 50)) and treatment with exogenous IL-17 can induce autoimmunity in otherwise disease-resistant mice. For example, infection of B6 mice with an adenoviral IL-17A expression construct induced a model of Sjorgren’s Syndrome (51). Injection of IL-17 into the joints induced pro-inflammatory TNF-α and IL-1β secretion, promoting rheumatoid arthritis (RA) (52). IL-17 produced by skin γδ T cells was shown to mediate epidermal inflammation in a model of psoriasis, which could be ameliorated by genetic deletion of Tcrd or Il17ra (53). Similarly, γδ T cell depletion or IL-17 neutralization ameliorated lung injury in a model of acute granulomatous disease (54).
While TCRαβ+ Th17 cells primarily reside in the intestine (55), γδ cells are a major IL-17 source in multiple tissues, particularly during infection (56-58). In contrast to conventional αβT cells, γδ T cell receptors (TCR) recognize currently poorly–defined ligands that do not require processing and presentation by specialized antigen-presenting cells. They can produce IL-17 without TCR ligation (reviewed in (59)) and far more rapidly than TCRαβ+ Th17 cells (20) due to developmental pre-programming of this function (8, 12). Interestingly, distinct from TCRαβ+ Th17 cells, activation of CD27− γδT cells does not require cognate ligand engagement (60). CD27− γδT cells respond poorly to TCR agonists under conditions that promote robust activation of the CD27+ γδT cell subset, and can respond to IL-23 without TCR engagement (12, 20). These unique characteristics of CD27− γδT cells more closely resemble innate lymphocytes. Thus, in the NOD mouse model, the pathogenic role of CD27− γδT cells may not require recognition of cognate γδTcR ligand, but instead depend on amplification of the antigen-specific αβT cell response in response to local cytokines.
One defined γδTcR ligand shown to regulate autoimmunity in the NOD mouse is the activating receptor NKG2D, expressed in the pancreatic islets of pre-diabetic animals. Moreover, monoclonal antibody blockade of NKG2D can prevent progression to overt T1D in this model (61). Interestingly, NKG2D is highly expressed by IL-17-producing γδ T cells (A. Hayday, personal communication). Upregulated expression of self-encoded “stress-ligands” for NKG2D can activate some murine γδ T cells in vivo and human γδ T cells in vitro (62-64). Such ligands might be evoked by macrophage or αβ T cell-mediated islet cell damage in NOD mice. Another study generated hybridomas from the NOD mouse spleen and PLN that included γδTCR+ clones responsive to purified islets, and to a processed insulin peptide (29). These clones used diverse TCR γ and δ chain sequences, indicative of a polyclonal population, but the cytokine expression or diabetogenic potential of these islet-reactive γδT cells were not reported. Further work will be required to resolve the relative contributions of γδT cells activated by TCR engagement and those activated by TCR-independent signals in T1D development in the NOD mouse.
Human IL-17 producing γδT cells have been difficult to isolate and characterize, owing to their low frequency in peripheral blood. However, recent work has shown that both murine and human CD27− γδT cells express high levels of IL-7R and can be expanded in vitro with IL-7, allowing for examination of their functional capacity (65). This recent study also clarifies the link between CD27 expression on human γδT cells and their cytokine potential. Specifically, 15-40% of CD27-γδT cells isolated from cord blood and expanded with IL-7, produced IL-17 following PMA/ionomycin stimulation (65). Moreover, IL-17-producing γδT cells were enriched in the skin lesions of psoriasis patients (53, 66). Together these studies suggest that the correlation between CD27 expression, cytokine program and contribution to inflammatory pathology is conserved between NOD mice and humans. A CD4−CD8− “double negative” IL17-secreting T cell population, potentially including γδT cells, is present in the blood and kidneys of patients with systemic lupus erythematosus (67). γδ T cell accumulations have also been reported in brain lesions and in the cerebrospinal fluid of MS patients (19, 68). Future studies are warranted to determine whether γδ T cells and their cytokine products contribute to the pathogenesis of human autoimmune diseases.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge significant contributions of Robert Tigelaar and Julie Lewis (Yale University) to the early derivation of TCRδ-deficient NOD mouse strain and of Marie-Laure Michel (LRI-CRUK) to data review. We thank Omid Gulban (Hospital for Sick Children) for bioinformatics analysis and Andrei Malko (Hospital for Sick Children) for technical assistance.
Grant Support: JGMM was funded by a Canadian Institutes of Health Research (CIHR) Doctoral Scholarship. AH acknowledges funding from the Wellcome Trust. The work was supported by grants to JSD from the CIHR and from Genome Canada with funds administered by the Ontario Genomics Institute.
Footnotes
AUTHOR CONTRIBUTIONS LG and AH began the introgression of the TCRδ deletion onto the NOD background, and initiated husbandry and analysis of the mice. AH provided mice to JGMM and JD who performed rigorous backcrossing and genomic analysis. JGMM, ASLW and SMT performed experiments and analyzed data. JGMM and JD designed experiments, analyzed data and wrote the manuscript. AH reviewed data and experimental design; and edited the manuscript.
CONFLICT OF INTEREST The authors declare no conflicts of interest.
REFERENCES
- 1.Hayday AC. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol. 2000;18:975–1026. doi: 10.1146/annurev.immunol.18.1.975. [DOI] [PubMed] [Google Scholar]
- 2.Schild H, Mavaddat N, Litzenberger C, Ehrich EW, Davis MM, Bluestone JA, Matis L, Draper RK, Chien YH. The nature of major histocompatibility complex recognition by gamma delta T cells. Cell. 1994;76:29–37. doi: 10.1016/0092-8674(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 3.Weintraub BC, Jackson MR, Hedrick SM. Gamma delta T cells can recognize nonclassical MHC in the absence of conventional antigenic peptides. J Immunol. 1994;153:3051–3058. [PubMed] [Google Scholar]
- 4.Sciammas R, Bluestone JA. HSV-1 glycoprotein I-reactive TCR gamma delta cells directly recognize the peptide backbone in a conformationally dependent manner. J Immunol. 1998;161:5187–5192. [PubMed] [Google Scholar]
- 5.Jensen KD, Su X, Shin S, Li L, Youssef S, Yamasaki S, Steinman L, Saito T, Locksley RM, Davis MM, Baumgarth N, Chien YH. Thymic selection determines gammadelta T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity. 2008;29:90–100. doi: 10.1016/j.immuni.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. 2001;1:177–186. doi: 10.1038/35105052. [DOI] [PubMed] [Google Scholar]
- 7.Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, Affaticati P, Gilfillan S, Lantz O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature. 2003;422:164–169. doi: 10.1038/nature01433. [DOI] [PubMed] [Google Scholar]
- 8.Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity. 2009;31:184–196. doi: 10.1016/j.immuni.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. 2010;10:467–478. doi: 10.1038/nri2781. [DOI] [PubMed] [Google Scholar]
- 10.Roark CL, Simonian PL, Fontenot AP, Born WK, O’Brien RL. gammadelta T cells: an important source of IL-17. Curr Opin Immunol. 2008;20:353–357. doi: 10.1016/j.coi.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermijlen D, Ellis P, Langford C, Klein A, Engel R, Willimann K, Jomaa H, Hayday AC, Eberl M. Distinct cytokine-driven responses of activated blood gammadelta T cells: insights into unconventional T cell pleiotropy. J Immunol. 2007;178:4304–4314. doi: 10.4049/jimmunol.178.7.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, Silva-Santos B. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol. 2009;10:427–436. doi: 10.1038/ni.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayday A, Geng L. Gamma delta cells regulate autoimmunity. Current opinion in immunology. 1997;9:884–889. doi: 10.1016/s0952-7915(97)80193-8. [DOI] [PubMed] [Google Scholar]
- 14.Gombert JM, Herbelin A, Tancrede-Bohin E, Dy M, Carnaud C, Bach JF. Early quantitative and functional deficiency of NK1+-like thymocytes in the NOD mouse. Eur J Immunol. 1996;26:2989–2998. doi: 10.1002/eji.1830261226. [DOI] [PubMed] [Google Scholar]
- 15.Wilson SB, Kent SC, Patton KT, Orban T, Jackson RA, Exley M, Porcelli S, Schatz DA, Atkinson MA, Balk SP, Strominger JL, Hafler DA. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature. 1998;391:177–181. doi: 10.1038/34419. [DOI] [PubMed] [Google Scholar]
- 16.Shi FD, Flodstrom M, Balasa B, Kim SH, Van Gunst K, Strominger JL, Wilson SB, Sarvetnick N. Germ line deletion of the CD1 locus exacerbates diabetes in the NOD mouse. Proc Natl Acad Sci U S A. 2001;98:6777–6782. doi: 10.1073/pnas.121169698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyazaki Y, Miyake S, Chiba A, Lantz O, Yamamura T. Mucosal-associated invariant T cells regulate Th1 response in multiple sclerosis. International immunology. 2011;23:529–535. doi: 10.1093/intimm/dxr047. [DOI] [PubMed] [Google Scholar]
- 18.Croxford JL, Miyake S, Huang YY, Shimamura M, Yamamura T. Invariant V(alpha)19i T cells regulate autoimmune inflammation. Nat Immunol. 2006;7:987–994. doi: 10.1038/ni1370. [DOI] [PubMed] [Google Scholar]
- 19.Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci U S A. 1992;89:4588–4592. doi: 10.1073/pnas.89.10.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Nanno M, Kanari Y, Naito T, Inoue N, Hisamatsu T, Chinen H, Sugimoto K, Shimomura Y, Yamagishi H, Shiohara T, Ueha S, Matsushima K, Suematsu M, Mizoguchi A, Hibi T, Bhan AK, Ishikawa H. Exacerbating role of gammadelta T cells in chronic colitis of T-cell receptor alpha mutant mice. Gastroenterology. 2008;134:481–490. doi: 10.1053/j.gastro.2007.11.056. [DOI] [PubMed] [Google Scholar]
- 22.Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol. 2007;179:5576–5583. doi: 10.4049/jimmunol.179.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spahn TW, Issazadah S, Salvin AJ, Weiner HL. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33 - 35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR delta chain gene. Eur J Immunol. 1999;29:4060–4071. doi: 10.1002/(SICI)1521-4141(199912)29:12<4060::AID-IMMU4060>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 24.Serreze DV, Leiter EH. Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Curr Dir Autoimmun. 2001;4:31–67. doi: 10.1159/000060527. [DOI] [PubMed] [Google Scholar]
- 25.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 26.Locke NR, Stankovic S, Funda DP, Harrison LC. TCR gamma delta intraepithelial lymphocytes are required for self-tolerance. J Immunol. 2006;176:6553–6559. doi: 10.4049/jimmunol.176.11.6553. [DOI] [PubMed] [Google Scholar]
- 27.Harrison LC, Dempsey-Collier M, Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J Exp Med. 1996;184:2167–2174. doi: 10.1084/jem.184.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han G, Wang R, Chen G, Wang J, Xu R, Wang L, Feng J, Li X, Guo R, Fu L, Shen B, Li Y. Interleukin-17-producing gammadelta+ T cells protect NOD mice from type 1 diabetes through a mechanism involving transforming growth factor-beta. Immunology. 2010;129:197–206. doi: 10.1111/j.1365-2567.2009.03166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Jin N, Nakayama M, O’Brien RL, Eisenbarth GS, Born WK. Gamma delta T cell receptors confer autonomous responsiveness to the insulin-peptide B:9-23. J Autoimmun. 2010;34:478–484. doi: 10.1016/j.jaut.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santamaria P, Lewis C, Jessurun J, Sutherland DE, Barbosa JJ. Skewed T-cell receptor usage and junctional heterogeneity among isletitis alpha beta and gamma delta T-cells in human IDDM [corrected] Diabetes. 1994;43:599–606. doi: 10.2337/diab.43.4.599. [DOI] [PubMed] [Google Scholar]
- 31.Ivakine EA, Gulban OM, Mortin-Toth SM, Wankiewicz E, Scott C, Spurrell D, Canty A, Danska JS. Molecular genetic analysis of the Idd4 locus implicates the IFN response in type 1 diabetes susceptibility in nonobese diabetic mice. J Immunol. 2006;176:2976–2990. doi: 10.4049/jimmunol.176.5.2976. [DOI] [PubMed] [Google Scholar]
- 32.Fox CJ, Paterson AD, Mortin-Toth SM, Danska JS. Two genetic loci regulate T cell-dependent islet inflammation and drive autoimmune diabetes pathogenesis. Am J Hum Genet. 2000;67:67–81. doi: 10.1086/302995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poussier P, Edouard P, Lee C, Binnie M, Julius M. Thymus-independent development and negative selection of T cells expressing T cell receptor alpha/beta in the intestinal epithelium: evidence for distinct circulation patterns of gut- and thymus-derived T lymphocytes. J Exp Med. 1992;176:187–199. doi: 10.1084/jem.176.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AM. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–1311. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nat Protoc. 2009;4:1649–1652. doi: 10.1038/nprot.2009.150. [DOI] [PubMed] [Google Scholar]
- 37.Jaakkola I, Jalkanen S, Hanninen A. Diabetogenic T cells are primed both in pancreatic and gut-associated lymph nodes in NOD mice. Eur J Immunol. 2003;33:3255–3264. doi: 10.1002/eji.200324405. [DOI] [PubMed] [Google Scholar]
- 38.Turchinovich G, Hayday AC. Skint-1 identifies a common molecular mechanism for the development of interferon-gamma-secreting versus interleukin-17-secreting gammadelta T cells. Immunity. 2011;35:59–68. doi: 10.1016/j.immuni.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 39.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, Chen SL, Rosa R, Cumiskey AM, Serreze DV, Gregory S, Rogers J, Lyons PA, Healy B, Smink LJ, Todd JA, Peterson LB, Wicker LS, Santamaria P. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Girardi M, Lewis J, Glusac E, Filler RB, Geng L, Hayday AC, Tigelaar RE. Resident skin-specific gammadelta T cells provide local, nonredundant regulation of cutaneous inflammation. J Exp Med. 2002;195:855–867. doi: 10.1084/jem.20012000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Itohara S, Mombaerts P, Lafaille J, Iacomini J, Nelson A, Clarke AR, Hooper ML, Farr A, Tonegawa S. T cell receptor delta gene mutant mice: independent generation of alpha beta T cells and programmed rearrangements of gamma delta TCR genes. Cell. 1993;72:337–348. doi: 10.1016/0092-8674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- 42.Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, Knight AM, Love JM, McAleer MA, Prins JB, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature. 1991;351:542–547. doi: 10.1038/351542a0. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh S, Palmer SM, Rodrigues NR, Cordell HJ, Hearne CM, Cornall RJ, Prins JB, McShane P, Lathrop GM, Peterson LB, et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet. 1993;4:404–409. doi: 10.1038/ng0893-404. [DOI] [PubMed] [Google Scholar]
- 44.Morahan G, McClive P, Huang D, Little P, Baxter A. Genetic and physiological association of diabetes susceptibility with raised Na+/H+ exchange activity. Proc Natl Acad Sci U S A. 1994;91:5898–5902. doi: 10.1073/pnas.91.13.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivakine EA, Fox CJ, Paterson AD, Mortin-Toth SM, Canty A, Walton DS, Aleksa K, Ito S, Danska JS. Sex-specific effect of insulin-dependent diabetes 4 on regulation of diabetes pathogenesis in the nonobese diabetic mouse. J Immunol. 2005;174:7129–7140. doi: 10.4049/jimmunol.174.11.7129. [DOI] [PubMed] [Google Scholar]
- 46.Lyons PA, Hancock WW, Denny P, Lord CJ, Hill NJ, Armitage N, Siegmund T, Todd JA, Phillips MS, Hess JF, Chen SL, Fischer PA, Peterson LB, Wicker LS. The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity. 2000;13:107–115. doi: 10.1016/s1074-7613(00)00012-1. [DOI] [PubMed] [Google Scholar]
- 47.Joseph J, Bittner S, Kaiser FM, Wiendl H, Kissler S. IL-17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. Journal of immunology. 2012;188:216–221. doi: 10.4049/jimmunol.1101215. [DOI] [PubMed] [Google Scholar]
- 48.Korn T, Petermann F. Development and function of interleukin 17-producing gammadelta T cells. Annals of the New York Academy of Sciences. 2012;1247:34–45. doi: 10.1111/j.1749-6632.2011.06355.x. [DOI] [PubMed] [Google Scholar]
- 49.Jensen KD, Chien YH. Thymic maturation determines gammadelta T cell function, but not their antigen specificities. Current opinion in immunology. 2009;21:140–145. doi: 10.1016/j.coi.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen CQ, Yin H, Lee BH, Carcamo WC, Chiorini JA, Peck AB. Pathogenic effect of interleukin-17A in induction of Sjogren’s syndrome-like disease using adenovirus-mediated gene transfer. Arthritis Res Ther. 2010;12:R220. doi: 10.1186/ar3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinto LG, Cunha TM, Vieira SM, Lemos HP, Verri WA, Jr., Cunha FQ, Ferreira SH. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain. 2010;148:247–256. doi: 10.1016/j.pain.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 53.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, Yan J. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti MC, Grohmann U, Segal BH, Puccetti P. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 55.Ivanov, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 56.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 57.Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- 58.Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 59.Kapsenberg ML. Gammadelta T cell receptors without a job. Immunity. 2009;31:181–183. doi: 10.1016/j.immuni.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Shibata K, Yamada H, Sato T, Dejima T, Nakamura M, Ikawa T, Hara H, Yamasaki S, Kageyama R, Iwakura Y, Kawamoto H, Toh H, Yoshikai Y. Notch-Hes1 pathway is required for the development of IL-17-producing gammadelta T cells. Blood. 2011;118:586–593. doi: 10.1182/blood-2011-02-334995. [DOI] [PubMed] [Google Scholar]
- 61.Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 62.Strid J, Roberts SJ, Filler RB, Lewis JM, Kwong BY, Schpero W, Kaplan DH, Hayday AC, Girardi M. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol. 2008;9:146–154. doi: 10.1038/ni1556. [DOI] [PubMed] [Google Scholar]
- 63.Strid J, Sobolev O, Zafirova B, Polic B, Hayday A. The intraepithelial T cell response to NKG2D-ligands links lymphoid stress surveillance to atopy. Science. 2011;334:1293–1297. doi: 10.1126/science.1211250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shafi S, Vantourout P, Wallace G, Antoun A, Vaughan R, Stanford M, Hayday A. An NKG2D-mediated human lymphoid stress surveillance response with high interindividual variation. Science translational medicine. 2011;3:113ra124. doi: 10.1126/scitranslmed.3002922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michel ML, Pang DJ, Haque SF, Potocnik AJ, Pennington DJ, Hayday AC. Interleukin 7 (IL-7) selectively promotes mouse and human IL-17-producing gammadelta cells. Proc Natl Acad Sci U S A. 2012;109:17549–17554. doi: 10.1073/pnas.1204327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laggner U, Di Meglio P, Perera GK, Hundhausen C, Lacy KE, Ali N, Smith CH, Hayday AC, Nickoloff BJ, Nestle FO. Identification of a novel proinflammatory human skin-homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. Journal of immunology. 2011;187:2783–2793. doi: 10.4049/jimmunol.1100804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimonkevitz R, Colburn C, Burnham JA, Murray RS, Kotzin BL. Clonal expansions of activated gamma/delta T cells in recent-onset multiple sclerosis. Proc Natl Acad Sci U S A. 1993;90:923–927. doi: 10.1073/pnas.90.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.