

Abstract
Autophagy has been shown to be protective against drug and alcohol-induced liver injury. CYP2E1 plays a role in the toxicity of ethanol, carcinogens and certain drugs. Inhibition of autophagy increased ethanol-toxicity and accumulation of fat in wild type and CYP2E1 knockin mice but not in CYP2E1 knockout mice as well as in HepG2 cells expressing CYP2E1 (E47 cells) but not HepG2 cells lacking CYP2E1 (C34 cells). The goal of the current study was to evaluate whether modulation of autophagy can affect CYP2E1-dependent cytotoxicity in the E47 cells. The agents used to promote CYP2E1 –dependent toxicity were a polyunsaturated fatty acid, arachidonic acid (AA), buthionine sulfoximine (BSO), which depletes GSH, and CCl4, which is metabolized to the CCl3 radical. These three agents produced a decrease in E47 cell viability which was enhanced upon inhibition of autophagy by 3-methyladenine (3-MA) or Atg 7 siRNA. Toxicity was lowered by rapamycin which increased autophagy and was much lower to the C34 cells which do not express CYP2E1. Toxicity was mainly necrotic and was associated with an increase in reactive oxygen production and oxidative stress; 3-MA increased while rapamycin blunted the oxidative stress. The enhanced toxicity and ROS formation produced when autophagy was inhibited was prevented by the antioxidant N-Acetyl cysteine. AA, BSO and CCl4 produced mitochondrial dysfunction, lowered cellular ATP levels and elevated mitochondrial production of ROS. This mitochondrial dysfunction was enhanced by inhibition of autophagy with 3-MA but decreased when autophagy was increased by rapamycin. The mitogen activated protein kinases p38 MAPK and JNK were activated by AA especially when autophagy was inhibited and chemical inhibitors of p38 MAPK and JNK lowered the elevated toxicity of AA produced by 3-MA. These results show that autophagy was protective against the toxicity produced by several agents known to be activated by CYP2E1. Since CYP2E1 plays an important role in the toxicity of ethanol, drugs and carcinogens and is activated under various pathophysiological conditions such as diabetes, NASH and obesity, attempts to stimulate autophagy may be beneficial in preventing/lowering CYP2E1/ethanol liver injury.
Abbreviations: CYP2E1, cytochrome P4502E1, E47 cells, HepG2 cells which express CYP2E1; C34 cells, HepG2 cells which do not express CYP2E1; AA, arachidonic acid; BSO, L-buthionine sulfoximine; CCl4, carbon tetrachloride; 3-MA, 3-methyadenine; Rap, rapamycin; NAC, N-acetyl-cysteine; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide; ROS, reactive oxygen species; DCFDA, 2′-7′-dichlorofluorescin-diacetate; SOD, superoxide dismutase; GSH, reduced glutathione; TBARs, thiobarbituric acid-reactive substances; Cox IV, cytochrome oxidase subunit 4
Keywords: CYP2E1, Autophagy, P38 MAPK, JNK, Mitochondria dysfunction, ROS, Cytotoxicity
Highlights
-
•
Inhibition of autophagy in HepG2 E47 cells which express CYP2E1 promotes AA, BSO and CCl4 toxicity.
-
•
Decreased autophagy in E47 cells increased reactive oxygen production and oxidative stress and potentiated mitochondrial dysfunction.
-
•
The inhibition of autophagy enhanced activation of p38 MAPK and JNK by AA and this activation contributed to the toxicity.
-
•
Autophagy is protective against the toxicity produced by agents known to be activated by CYP2E1.
Introduction
Autophagy is an intracellular pathway by which lysosomes degrade and recycle long-lived proteins and cellular organelles. This pathway degrades cellular components that are worn out or damaged or are needed to generate substrates that maintain cellular energy homeostasis under conditions of limited nutrients or stress [1–3]. The regulation of autophagy is complex and controlled by the coordinated actions of autophagy-related genes. A key regulator of autophagy is the mammalian target of rapamycin (mTOR) which senses cellular nutritional status and cell stress [4,5].
Removal of damaged mitochondria by mitophagy or of lipid droplets by lipophagy are selective forms of macroautophagy [6–11]. Removal of damaged mitochondria protects the cell against mitochondrial oxidative stress, while removal of lipid droplets limits the accumulation of lipids by hepatocytes. Defects in lipophagy can contribute to hepatic steatosis [10,11]. Autophagy is decreased in certain hepatic and pancreatic diseases e.g. α1-antitrypsin deficiency and non-alcoholic fatty liver disease, but increased in nutrient deficiency and hepatitis B infection [7,12]. In general, autophagy is considered as a cell survival pathway but one that can also mediate cell death under certain conditions or when over activated. Recent studies showed that autophagy protects cells against injury from alcohol, because chemical and genetic inhibition of autophagy increased the levels of injury in cultured hepatocytes and mouse liver [6,13–15].
CYP2E1 metabolizes and activates many toxicological important substrates including ethanol, to more toxic products [16–20]. CYP2E1 generates reactive oxygen radical species during its catalytic cycle and is induced by ethanol. [21–23]. Ethanol-induced liver pathology has been shown to correlate with CYP2E1 levels and lipid peroxidation [24–27]. Inhibitors of CYP2E1 prevented the elevation of lipid peroxidation and the ethanol-induced liver pathology [28,29]. The biochemical and toxicological properties of CYP2E1 have been studied in Rala hepatocytes [30], in HepG2 cell lines [31–33], in transgenic mice [34] and in mice infected with adenovirus expressing CYP2E1 [35]. CYP2E1 has been shown to contribute to ethanol-induced steatosis [36]. Autophagy can modulate CYP2E1-dependent ethanol toxicity in vitro and in vivo as inhibition of autophagy increased binge ethanol-induced steatosis in wild type and CYP2E1 knockin mice but not CYP2E1 knockout mice and increased ethanol-induced fat accumulation in E47 HepG2 cells which express CYP2E1 but not in C34 HepG2 cells which do not express CYP2E1 [37–39]. The rationale for these and the current study is that CYP2E1 plays a role in ethanol-induced oxidant stress, fatty liver and liver injury. Autophagy, in some settings is protective against cell injury, while in other settings, autophagy can promote cell toxicity. If autophagy is protective against ethanol/CYP2E1 toxicity, attempts to stimulate autophagy may prove to be helpful in lowering ethanol-/CYP2E1 induced liver injury. If autophagy promotes ethanol/CYP2E1 toxicity, inhibitors of autophagy may help to ameliorate this hepatotoxicity. In the current study we evaluated whether modulation of autophagy can affect CYP2E1 –dependent cytotoxicity in the E47 cells. Toxins used to promote toxicity included CCl4, which is activated by CYP2E1 to reactive intermediates [40], arachidonic acid as a representative polyunsaturated fatty acid which promotes ethanol-induced liver injury [25,28], and BSO which lowers GSH levels and impairs cellular antioxidant defense [41]. All three of these compounds have previously been shown to be toxic to the E47 cells but not to C34 cells [33,42] but the effects of autophagy on their toxicity has not been determined.
Previous studies, in the absence of modulation of autophagy, showed that activation of p38 MAPK played a role in AA, BSO and TNFα-induced toxicity to E47 cells [43–45]. JNK MAPK has been implicated in many models of chemical and drug-induced liver toxicity [46–51] and in binge ethanol-induced steatosis [38]. Induction of autophagy by free fatty acids was JNK-dependent [52]. These results suggest possible cross talk between autophagy and MAPK. A second goal of this study was to determine whether activation of MAPK occurs under these experimental conditions and whether MAPK play a role in the autophagy-modulated toxicity of AA, BSO and CCL4.
Materials and methods
Cell model and treatment
HepG2 E47 cells which express human CYP2E1 and control HepG2 C34 cells which do not express CYP2E1 [53] were treated with either 20 or 45 µM AA [43], or 300 µM BSO [41,44], or 300 µg/ml CCl4 [54] in the presence or absence of 3-MA (2.5 mM, Sigma), or rapacycin (0.2 µg/ml, LC Laboratories, MA) for 24 or 48 h respectively. The medium was replaced every 24 h with fresh reagents. Cell viability was determined by a MTT assay [53]. Cell morphology was observed under an inverted light contrast microscope. To determine the percentage of cells undergoing necrosis or apoptosis in the total cell population, 2×106 cells were harvested with trypsin and resuspended in 3 ml 1×PBS, and stained with 0.25 µg/ml annexin V and 1 µg/ml PI for 10 min. The percentage of cells undergoing necrosis and apoptosis was determined by flow cytometry according to the annexin V or PI positive cell staining. Cells after double staining were also observed under a fluorescence microscope.
Knockdown of Atg 7
Autophagy was blocked by knockdown of Atg 7 with Atg 7 siRNA. E47 cells, 5×105/well, were seeded into a 12 well plate with 1 ml medium and incubated overnight. Medium without serum, containing 25 nM Atg 7 siRNA, or scrambled siRNA (Cell Signaling) was mixed with 5ul transfection reagent for 30 min at room temperature and then added to the cells. The final volume was adjusted with medium to 0.5 ml per well and the mixture was incubated for 48 h. Cells were harvested and assays were carried out to evaluate Atg 7 content, cytotoxicity and ROS production.
ROS stress
ROS stress was evaluated by measuring lipid peroxidation (TBARS) and levels of GSH [41,43,44]. Cells were also incubated with 5 µM 2′-7′-dichlorofluorescin-diacetate (DCFDA, SERVA) for 10 min and green fluorescence was observed under a fluorescence microscope. Cells were also re-suspended in 1×PBS and the fluorescence was determined in a Perkin-Elmer fluorescence spectrophotometer at Ex488/Em530 nm. To evaluate ROS stress in the mitochondria, at the end of treatment cells were incubated with 5 µM Mitosox (Molecular Probes) as previously described [48] for 10 min and red fluorescence of the stained mitochondria was observed under a fluorescence microscope. The cells were also re-suspended in 1×PBS and the intensity of fluorescence was determined in the fluorescence spectrophotometer at Ex510/Em580 nm.
Autophagy evaluation
The modulation of autophagy by AA, BSO and CCl4 in the absence and presence of 3-MA or rapamycin was evaluated by immunoblots to determine the levels of LC3-Il and LC3-I (antibody from Thermo), the ratio of LC3-II/LC3-I and levels of autophagy regulatory proteins p62 and Beclin-1 (antibodies from Santa Cruz). Immunoblots were scanned and analyzed as the p62/β-actin or the Beclin-1/β-actin ratios using a LI-COR Odyssey densitometer and software Image J from NIH.
Mitochondrial dysfunction
Mitochondria membrane swelling
Cells were homogenized by 30 strokes with a glass Pyrex micro homogenizer. Fresh mitochondria were prepared by differential centrifugation in a buffer containing 0.25 M sucrose, 10 mM Tris–HCl, pH 7.4. Calcium-induced mitochondrial swelling was assayed as an index of the mitochondrial permeability transition [55,56]. In a 1 ml cuvette, 500 µg mitochondria protein was added into 1 ml of buffer containing 150 mM KCl, 10 mM Tris–MOPS, pH 7.4, 5 mM glutamic acid, 2.5 mM malic acid and 1 mM KPi. CaCl2 was added (100 µM final concentration) to trigger swelling which was recorded from the decrease in absorbance at 540 nm.
ATP levels
ATP levels were measured using the ATP Determination Kit (Molecular Probes, A22066). In the presence of ATP, luciferin and luciferase, the products oxyluciferin, AMP and light, are produced. The light was detected in a chemiluminescence detector and ATP levels were compared to a standard curve with ATP.
Cytochrome c release
The post mitochondrial supernatant was collected after homogenates were centrifuged at 8000g. Immunoblots were carried out to detect for the presence of cytochrome c in the cytosol fraction. To ensure the cytosol preparation was not contaminated with mitochondria, immunoblots were carried out to determine whether mitochondrial SOD2 was present in the cytosol Cytochrome c in the mitochondrial fraction was also determined by immunoblot.
Activation of p38 MAP kinase and JNK
E47 cells were treated with 20 µM AA in the presence and absence of 3-MA or rapamycin for 6, 12, 24 and 48 h. Cell lysates were collected and immunoblots were carried out to detect levels of pp38 MAPK, p38 MAPK, pJNK and JNK [38,43,44] (antibodies from Santa Cruz). Results are presented as the ratios of pp38 MAPK/p38 MAPK or pJNK/JNK. To evaluate whether the activation of p38 MAPK or JNK contribute to cytotoxicity, E47 cells were treated with 3-MA plus AA in the presence or absence of the p38 MAPK inhibitor SB203580 (5 µM, BioChem), or JNK inhibitor SP600125 (2.5 µM, BioChem), and cell viability determined by a MTT assay. Levels of pp38 and pJNK MAPK after treatment with the inhibitors were determined to validate their inhibitory effects.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance with subsequent post hoc comparisons by Scheffe. Values reflect means±standard error. The number of experiments is indicated in the figure legends.
Results
Inhibition of autophagy enhances CYP2E1-dependent AA, BSO and CCl4 cytotoxicity
To study whether autophagy promotes or protects against CYP2E1-mediated AA or BSO or CCl4 induced cytotoxicity, HepG2 E47 and C34 cells were treated with these compounds in the presence or absence of 3-MA or rapamycin to inhibit or induce autophagy, respectively. During the first 24 h of treatment, there was little or no toxicity to both cells lines by the various additions (data not shown). After 48 h of treatment, AA, BSO and CCl4 induced some toxicity in E47 cells but not in C34 cells (Fig. 1A). Inhibition of autophagy by 3-MA, significantly increased the toxicity of AA, BSO, and CCl4, whereas the induction of autophagy with rapamycin protected against this toxicity (Fig. 1A). Morphological observations also supported the potentiation of AA, BSO or CCl4 toxicity by 3-MA (Fig. 1B). Increases in cytotoxicity were found in E47 but not in C34 cells (Fig. 1A), suggesting that CYP2E1 plays a role in the 3-MA-potentiation of toxicity. To assess the mode of cell death, the E47 cells were incubated with AA or BSO or CCl4 in the absence and presence of 3-MA or rapamycin for 48 h, stained with annexin V and PI and analyzed by flow cytometry to determine the percentage of cells undergoing apoptosis and necrosis. The inhibition of cell autophagy with 3-MA primarily enhanced AA-, BSO-, and CCl4-induced necrosis (from 12–17% in the absence of 3-MA to 30–33% in the presence of 3-MA) (Fig. 1C). There was also a small increase in apoptosis produced by 3-MA (from 6-8% in the absence of 3-MA to 9–15% in the presence of 3-MA) (Fig. 1C). Rapamycin reduced the necrosis produced by AA, BSO and CCl4 (from 12–17% to 7–9%) but either had no effect (BSO) or slightly increased (AA, CCL4) apoptosis. Incubation of the E47 cells with AA resulted in some PI positive-staining cells over the no addition control (Fig. 1D). Treatment with 3-MA increased while incubation with rapamycin decreased the PI-positive-staining cells (Fig. 1D). Annexin V staining was low after AA incubation and was only slightly increased by 3-MA. Thus, cell death was primarily necrotic.
Fig. 1.
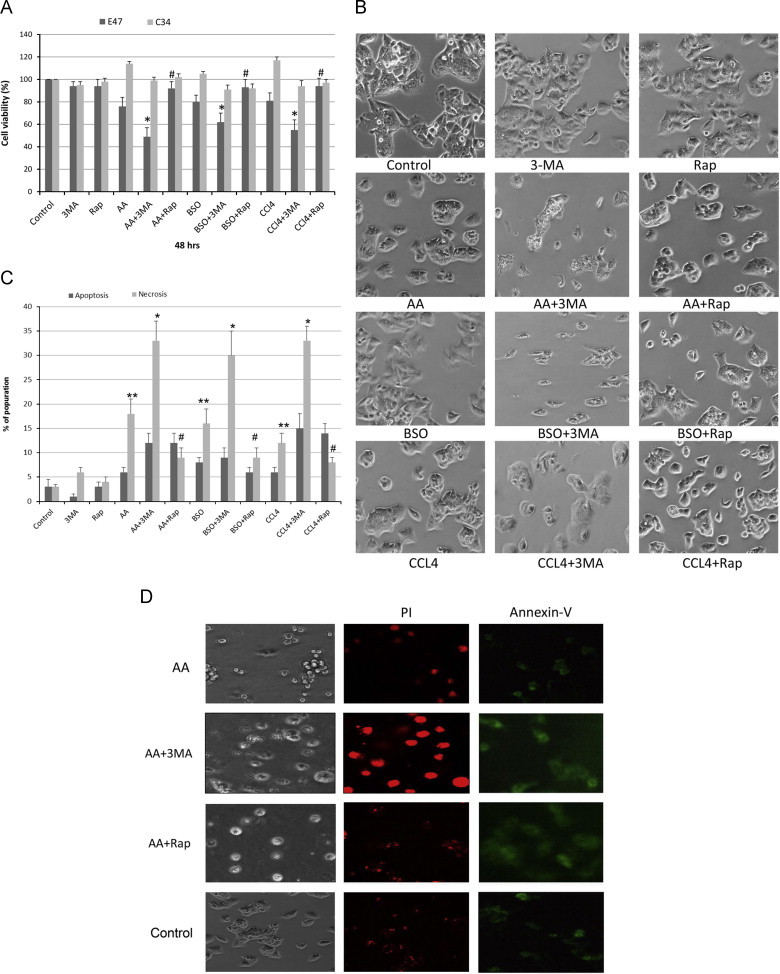
Autophagy protects against CYP2E1-mediated AA, CCl4 and BSO cell toxicity. HepG2 E47 and C34 cells were treated with AA (20 µM), CCl4 (300 ug/ml) or BSO (300 µM) for 48 h in the presence and absence of 3-MA (2.5 mM) or rapamycin (0.2 µg/ml). (A) Cell viability was determined by a MTT assay (*P<0.05 compared with AA or BSO or CCl4 alone; #P<0.05 compared to the AA+3-MA or BSO+ 3-MA or CCl4+3-MA groups; n=4). (B) Cell morphology was observed under an inverted light contrast microscope (magnification ×200). (C) E47 cells were treated as described above for 48 h. Cells were harvested with trypsin and resuspended in PBS and then were double stained with annexin V and PI and analyzed by flow cytometry to determine, the percentage of cells undergoing apoptosis and necrosis (**P<0.05 compared to control; &P<0.05 compared with AA alone or CCl4 alone group; ⁎, #P<0.05 compared with AA, BSO or CCl4 alone group; N=3). (D) E47 cells were treated with medium alone (control) or with AA in the presence or absence of 3-MA or rapamycin for 48 h, cells were stained with Annexin V or PI and were observed under a fluorescence microscope to determine the apoptosis (Annexin V staining, green fluorescence) and necrosis (PI staining, red fluorescence), (magnification ×200).
Inhibition of autophagy promotes ROS stress
The inhibition of autophagy with 3-MA in the E47 cells promoted AA, BSO or CCl4 induced ROS stress. Lipid peroxidation, assessed as formation of TBARs was increased by AA or BSO or CCl4 (Fig. 2). Treatment with 3-MA further elevated the level of TBARs whereas rapamycin decreased these levels (Fig. 2A). E47 cellular levels of GSH were decreased by AA or BSO or CCl4 (Fig. 2B). Inhibition of autophagy with 3-MA resulted in a further decline in GSH levels, whereas rapamycin either had no effect or increased GSH levels (Fig. 2B).
Fig. 2.
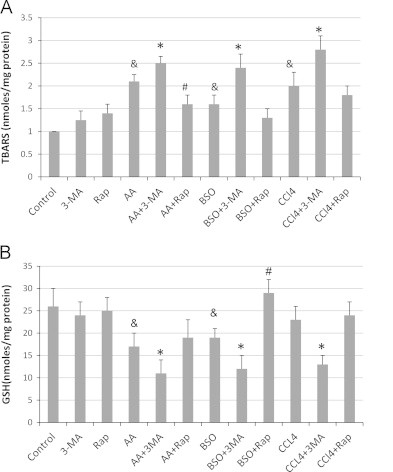
Pharmacological inhibition of autophagy enhances AA, BSO or CCl4 induced ROS stress. E47 cells were treated with AA, BSO or CCl4 in the presence or absence of 3-MA or rapamycin for 48 h. (A) Lipid peroxidation was determined by measuring the formation of TBARs products. (B) GSH levels were determined with GSH reductase. (&P<0.05 compared with control; ⁎, #P<0.05 compared with AA, BSO or CCl4 alone, N=3).
Inhibition of autophagy by knockdown of Atg 7
siRNA against Atg 7 was used as a genetic approach to decrease autophagy. E47 cells were incubated with Atg 7 siRNA or control scrambled siRNA for 48 h and levels of Atg 7 determined. When used at a final concentration of 25 nM, levels of Atg 7 were lowered by more than 90% as compared to the treatment with control siRNA (Fig. 3A). E47 cells were first incubated with Atg 7 (or control) siRNA for 48 h, AA was added and cell viability was assayed after 48 h incubation with AA. The knockdown of autophagy with Atg 7 siRNA significantly increased AA toxicity from a decline in cell viability of 38% in the absence of the Atg 7 siRNA to a decline of 62% in the presence of Atg 7 siRNA (Fig. 3B). The control siRNA had no effect on AA toxicity, whereas rapamycin afforded protection (decline in viability of 18% by AA) (Fig. 3B).
Fig. 3.
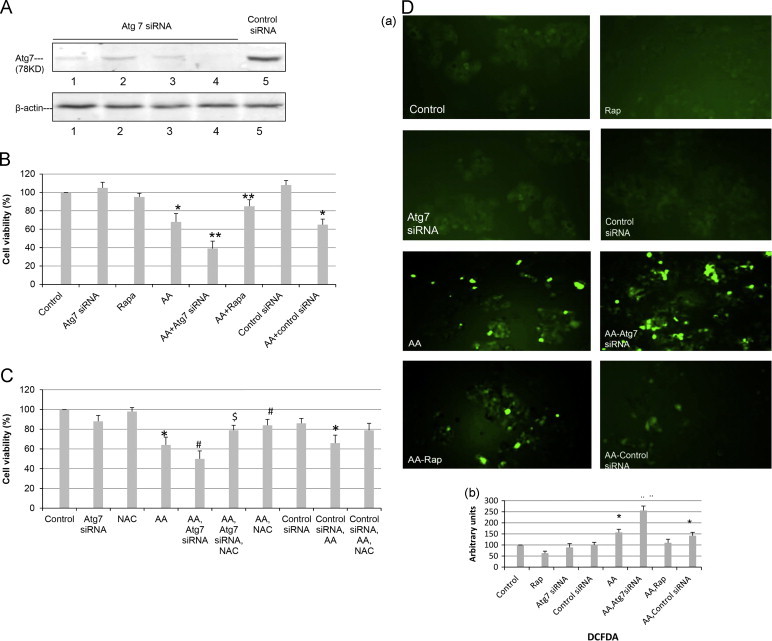
Knockdown of Atg 7 promotes AA induced ROS formation and cytotoxicity. (A) Transfection of E47 cells with Atg 7 siRNA but not control siRNA for 48 h lowers levels of Atg 7. (B) E47 cells were transfected with Atg 7 siRNA or control siRNA for 48 h. Cells were then treated with AA (20 µM) or rapamycin for an additional 48 h and cell viability determined by a MTT assay. (⁎P<0.05 compared with control; ⁎⁎P<0.05 compared with AA alone, N=3.) (C) NAC (5 mM) was added to evaluate the involvement of ROS in AA toxicity and the potentiation of this toxicity by Atg 7 siRNA. (⁎P<0.05 compared with control, #P<0.05 compared with AA alone, $P<0.05 compared with AA plus Atg 7 siRNA, N=3), (D) (a), E47 cells were treated as described in (B); DCFDA (5 µM) was added, and after 15 min incubation, ROS-activated fluorescence was detected under a fluorescence microscope (magnification ×100). (b), Cells were re-suspended in 1×PBS and the intensity of fluorescence determined in a Perkin-Elmer fluorescence spectrophotometer at Ex488/Em530 nm. ⁎P<0.05 compared with control; #P<0.05 compared with AA group, (N=3).
To evaluate whether increases in oxidative stress play a role in the increase in toxicity when autophagy is inhibited, the ability of the antioxidant NAC to protect against the AA-induced toxicity was determined. Toxicity by AA was partially prevented by NAC and importantly, the elevated toxicity of AA in the presence of Atg 7 siRNA was also blunted by NAC (Fig. 3C). The control siRNA did not block the AA toxicity but NAC again blocked AA toxicity in the presence of the control siRNA. ROS production by the E47 cells was measured using DCFDA as the probe. Incubation with AA produced an increase in DCF fluorescence (Fig. 3D (a), quantified in Fig. 3D (b)). This fluorescence was elevated by the Atg 7 siRNA but not control siRNA (Fig. 3D). Taken as a whole these results are supportive of a role for ROS in the potentiation of AA toxicity when autophagy is inhibited.
3-MA and rapamycin impact autophagy in E47 cells
To study the effect of the added toxins on autophagy and to validate that the treatment with 3-MA or rapamycin inhibited or induced autophagy, E47 cells were treated with AA, BSO or CCl4 in the absence and presence of 3-MA or rapamycin and the LC3-II/LC3-I ratio was measured along with levels of p62 and Beclin-1. Treatment with 3-MC alone or rapamycin alone or with AA, BSO or CCl4 had no effect on the LC3-II/LC3-I ratio but when the 3 toxins were incubated in the presence of 3-MA, the LC3-II/LC3-I ratio was decreased by 30–50%, (Fig. 4A). Treatment of the 3 toxins in the presence of rapamycin produced a 1.6–2.4 fold increase in the LC3-II/LC3-I ratio compared with AA, BSO or CCl4 alone (P<0.05) (Fig. 4). Lysosomal degradation of autophagosomes leads to a decrease in p62/SQSTM1 levels during autophagy and autophagy inhibitors stabilize and elevate p62/SQSTM1 levels. While AA, BSO, and CCl4 had no effect on p62 levels in the absence of 3-MA or rapamycin, p62 levels, were increased when either of the 3 toxins were incubated in the presence of 3-MA but decreased when incubated in the presence of rapamycin as compared with incubation with AA, BSO or CCl4 alone (Fig. 4B). Beclin-1 which regulates the formation of the autophagosome [1–3] was decreased by treatment with AA, BSO or CCl4 alone and further decreased when the toxins were incubated in the presence of 3-MA (Fig. 4C). These results show that in the E47 cells, incubated with either AA or BSO or CCl4, 3-MA inhibits, while rapamycin stimulates autophagy.
Fig. 4.
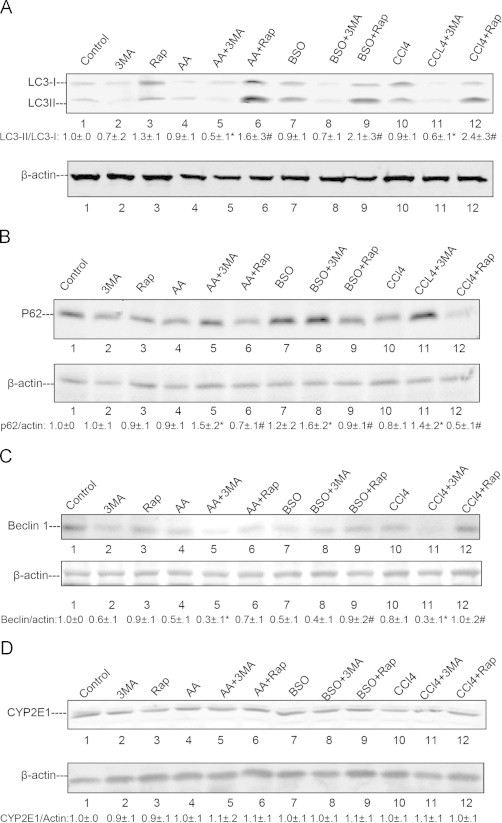
Effects of AA, BSO or CCl4 on autophagy-related proteins and CYP2E1. E47 cells were treated with AA, BSO or CCl4 in the presence or absence of 3-MA or rapamycin for 48 h as described in the legend to Fig. 1. (A) LC3 levels were evaluated by immunoblots and the LC3-II/LC3-I ratio calculated (⁎, #P<0.05 compared with AA, BSO or CCl4 alone groups, N=3). (B) and (C) Levels of autophagy regulation proteins p62 and Beclin-1 were determined by immunoblots and their relative ratios with β-actin are listed under the blots (⁎, #P<0.05 compared with AA or BSO or CCl4 alone groups, N=3). (D) The effect of AA, BSO or CCl4 with or without treatment with 3-MA or rapamycin on protein levels of CYP2E1.
Neither of the 3 toxins or 3-MA or rapamycin had an effect on CYP2E protein levels in the E47 cells (Fig. 4D) (or catalytic activity – data not shown).
Autophagy prevents CYP2E1-promoted mitochondrial dysfunction
Damaged and/or oxidized mitochondria are removed from the cell by autophagy (mitophagy) [8,11]. Experiments were carried out to evaluate whether mitochondrial dysfunction occurs when the 3 toxins are incubated in the E47 cells and how such dysfunction might be modulated by inhibiting or stimulating autophagy with 3-MA or rapamycin, respectively. Damage to the mitochondrial membrane promotes the mitochondrial permeability transition (MPT). Calcium-induced mitochondrial membrane swelling in the presence of glutamate–malate was determined as a reflection of the MPT. Mitochondria from E47 cells treated with AA, BSO or CCl4 underwent swelling as shown by the decrease in absorbance at 540 nm (Fig. 5A–C). 3-MA treatment enhanced the mitochondrial swelling while rapamycin treatment decreased the swelling (Fig. 5A–C). Cellular levels of ATP were determined as an index of the cellular energy state and because autophagy requires ATP [57,58]. Incubation with AA, BSO or CCl4 caused a 30% decline in ATP levels (Fig. 5D). ATP levels were further lowered to more than a 50% decline when AA, BSO or CCl4 were incubated in the presence of 3-MA. (Fig. 5D). Rapamycin had no effect on ATP levels or the decline in these levels produced by AA or BSO or CCl4. Mitochondrial membrane swelling and damage may cause cytochrome c release from the mitochondria to the cytosol. While the treatment with 3-MA or AA or BSO or CCl4 alone had little effect on cytosolic cytochrome c levels, incubation of the 3 toxins in the presence of 3-MA elevated cytosolic cytochrome c levels (Fig. 5E). Under the latter conditions, mitochondrial levels of cytochrome c were reduced (Fig. 5F), likely a reflection of cytochrome c release from the mitochondria to the cytosol.
Fig. 5.
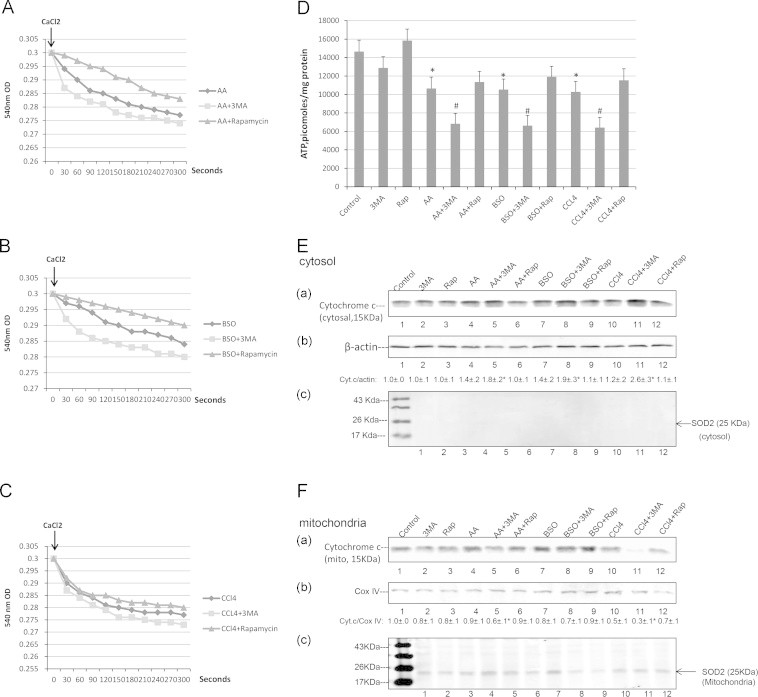
Autophagy modulates AA, BSO and CCl4 induced mitochondrial dysfunction. E47 cells were treated with AA, BSO or CCl4 with or without 3-MA or rapamycin for 48 h and mitochondria were freshly prepared. Mitochondrial membrane swelling (A–C) was evaluated by determining the decrease in absorbance at 540 nm after the addition of 100 µM CaCl2. (A) AA; (B) BSO; (C) CCl4; (D) Cellular ATP levels (⁎P<0.05 compared with control; #P<0.05 compared with AA, BSO or CCl4 alone, N=4). (E): Levels of cytochrome c in the cytosol. Numbers below the blots refer to the cytosolic cytochrome c/β-actin ratio as determined from blots (a) and (b) (⁎P<0.05 compared with AA, BSO or CCl4 alone, N=3). Blot (c) is an immunoblot of mitochondrial SOD2 to ensure that the cytosolic preparation was not contaminated with mitochondria. (F) levels of cytochrome c in the mitochondria. Numbers below the blots refer to the cytochrome c/COX IV ratio as determined from blots (a) and (b). (⁎P<0.05 compared with AA, CCl4 alone, N=3.) Blot (c) shows levels of SOD2 in the mitochondrial preparation.
Inhibition of autophagy with 3-MA or Atg 7 siRNA increased the AA- or BSO- or CCl4-induced total cellular ROS formation (TBARs, GSH, DCFDA fluorescence). We evaluated whether modulation of autophagy impacts on mitochondrial ROS formation. The E47 cells were incubated with Mitosox to evaluate mitochondrial ROS production. AA or BSO or CCl4 alone increased Mitosox fluorescence over the buffer control (Fig. 6A). Incubation of the 3 toxins in the presence of 3-MA strikingly increased the red fluorescence while incubation in the presence of rapamycin lowered the red staining (Fig. 6A (a), quantified in Fig. 6A (b)). Similarly, incubation of the E47 cells with AA plus Atg 7 siRNA, but not control siRNA, enhanced the Mitosox fluorescence over that induced by AA alone (Fig. 6B (a), quantified in Fig. 6B (b)). Both the AA-induced Mitosox fluorescence and the AA plus Atg 7 siRNA-enhanced Mitosox fluorescence were blunted by NAC (Fig. 6B).
Fig. 6.
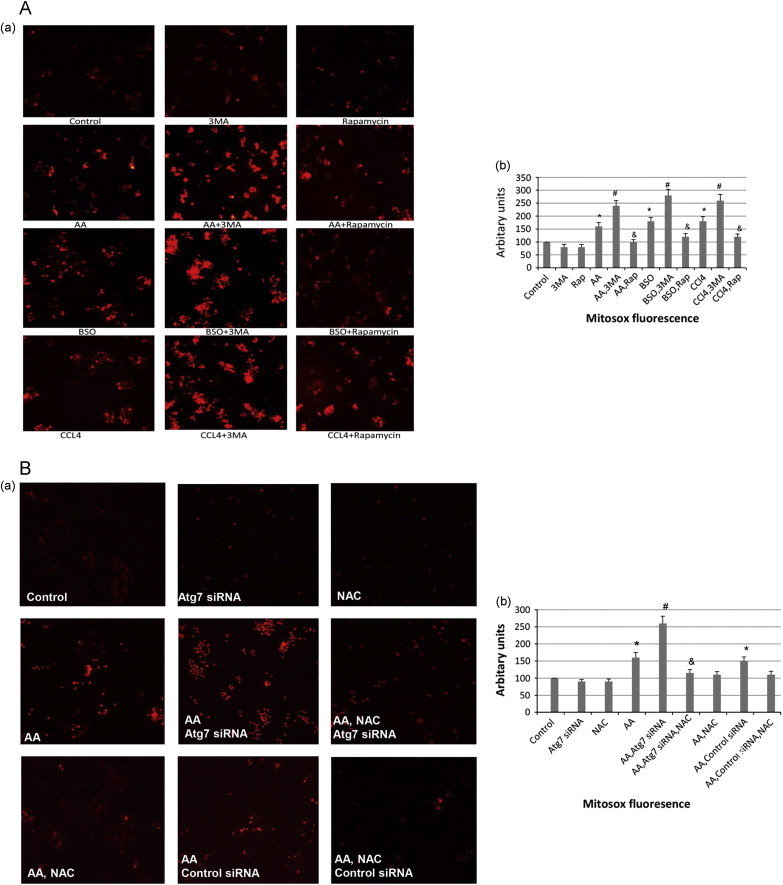
Inhibition of autophagy promotes AA, BSO and CCl4 induced mitochondrial ROS formation. (A) (a), E47 cells were treated with AA, BSO or CCl4 in the absence or presence of 3-MA or rapamycin for 48 h, and cells were then incubated with 5 µM MitoSox for 30 min. The red-stained mitochondria were observed under a fluorescence microscope (magnification ×100). (b), Cells were also re-suspended in 1×PBS and the intensity of fluorescence was determined in a Perkin-Elmer fluorescence spectrophotometer at Ex510/Em580 nm. ⁎P<0.05 compared with control; #, &P<0.05 compared with AA, BSO or CCl4 alone respectively, N=3; (B) (a), Genetic knockdown of Atg 7 with Atg 7 siRNA increases Mitosox fluorescence in E47 cells treated with AA for 48 h while treatment with NAC decreases Mitosox fluorescence. (Magnification ×100). (b), Determination of the fluorescence intensity as described in (a), ⁎P<0.05 compared with control; #P<0.05 compared with AA; &P<0.05 compared with AA, Atg 7 siRNA, N=3.
Activation of p38 MAPK and JNK
p38MAPK was shown to play a role in AA, BSO and TNFα toxicity in the E47 cells [43–45] while JNK was activated in many models of liver toxicity and steatosis [38,46–52]. We evaluated whether activation of p38 MAPK or JNK occurs under these experimental conditions and if activation of MAPK contributed to the potentiation of cytotoxicity when autophagy was blocked. AA alone activated p38MAPK about 2-fold after incubation for 24 and 48 h (Fig. 7A and B). Activation was enhanced to 3–4 fold when AA was incubated in the presence of 3-MA; there was also an early activation of p38 MAPK at 6 h (Fig. 7A and B). Rapamycin had no effect by itself or in the presence of AA or BSO or CCl4 on p38 MAPK activation. JNK was not activated by AA added alone. However, both JNK1 and JNK2 were activated by the combination of AA plus 3-MA after incubation for 24 and 48 h (Fig. 7C and D). To determine if activated p38 MAPK and/or JNK play a role in the enhanced toxicity when AA is incubated with 3-MA, E47 cells were treated with a p38 MAPK inhibitor SB203580 (SB) or a JNK inhibitor SP600125 (SP) for 24 and 48 h. SB and SP partially prevented the 3-MA enhanced AA toxicity elevating cell viability, especially at 24 h (Fig. 8A). SB, but not SP, was very effective in lowering the activation of p38 MAPK produced by AA plus 3-MA to below basal levels (Fig. 8B and C; lanes 3 and 4) while SP, but not SB, effectively blocked the 2–3 fold elevated ratio of pJNK1/JNK1 to below basal levels (Fig. 8D and E: lanes 3 and 5), validating their specific inhibitory effectiveness.
Fig. 7.
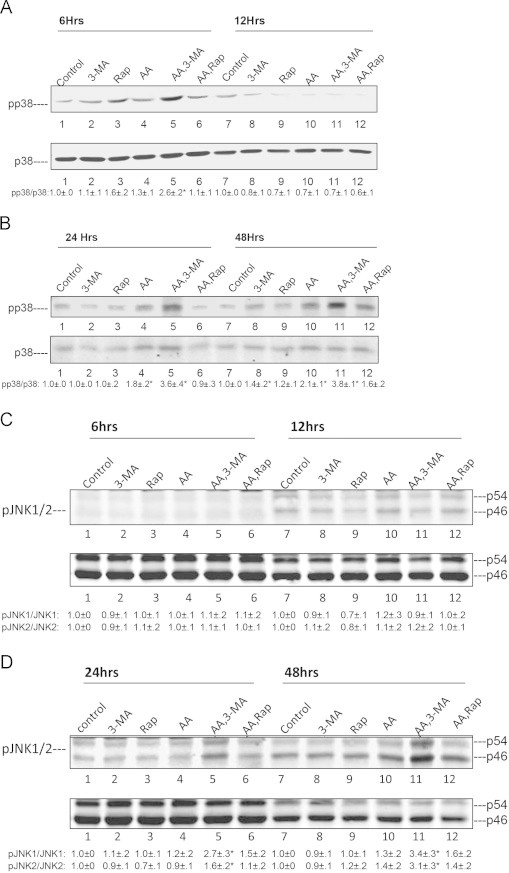
Inhibition of autophagy promotes AA activation of p38 MAPK and JNK. E47 cells were treated with 3-MA or rapamycin in the presence or absence of AA (45 µM) for 6, 12, 24 and 48 h. Cell lysates were prepared and immunoblots were carried out to determine the time course activation of p38 MAPK (A and B) or JNK (C and D). The ratio of pp38 MAPK/p38 MAPK is listed at the bottoms of blots A and B (⁎P<0.05 compared with AA alone or with control, N=3). The ratios of p-JNK/JNK (JNK 1 and JNK 2) are listed at the bottom of blots C and D (⁎P<0.05 compared with AA alone, N=3).
Fig. 8.
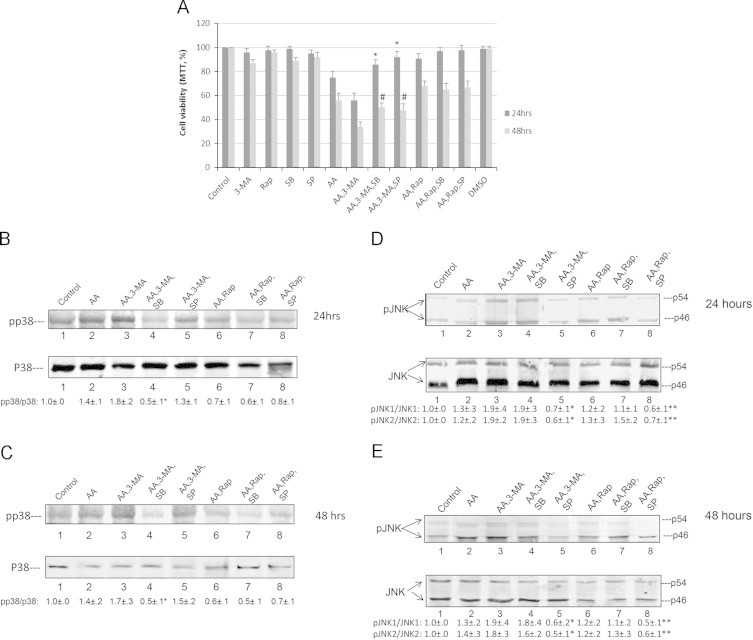
A p38MAPK inhibitor and a JNK inhibitor decrease 3-MA enhanced AA cytotoxicity. E47 cells were treated with 45 µM AA plus 3-MA or rapamycin in the presence or absence of SB203580 (5 µM) or SP600125 (2.5 µM) for 24 and 48 h. (A): cell viability was determined by a MTT assay (⁎, #P<0.05 compared with AA plus 3-MA for 24 or 48 h, N=3). (B) and (C): Effects of SB201580 and SP600125 on the activation of p38 MAPK. The inhibition of p38 MAPK by SB203580 at 24 h (B) and 48 h (C) was determined by Immunoblot analyses. The pp38 MAPK/p38 MAPK ratios are listed underneath the blots (⁎P<0.05 compared with AA plus 3MA, n=3). The inhibition of JNK activation by SP600125 at 24 h (D) and 48 h (E) was determined by immunoblot analyses. The ratios of pJNK/JNK are listed at the bottom of the blots. (⁎⁎P<0.05 compared with AA plus rapamycin; ⁎P<0.05 compared with AA plus 3-MA, n=3).
Discussion
Autophagy can either be increased or decreased by ethanol depending on the model used, the dose, the tissue evaluated and the experimental condition [6,7,12–15,37–39,59,60]. While the effects of ethanol on autophagy are complex and require further study, it is becoming clear that autophagy serves a protective function against ethanol-induced liver injury e.g. Ding et al. [13] showed that autophagy protects against liver injury in an acute mouse model of binge alcohol consumption. Donohue et al. proposed that chronic ethanol increases autophagosome formation but blocks autolysosome formation [14,61]. Chronic ethanol consumption was recently shown to increase autophagy [15,62] and in some cases this increase was associated with a decrease in activity of the other major cellular proteolyic system, the proteosome complex [63]. In response to alcohol, the liver might increase autophagy to selectively eliminate damaged mitochondria and limit lipid accumulation [15,62]. Indeed, inhibition of autophagy with chloroquin enhanced acute and chronic ethanol-induced steatosis and liver injury while stimulation of macroautophagy with either rapamycin or carbamazepine decreased acute and chronic ethanol-induced fatty liver [15], results consistent with autophagy serving a protective role against acute and chronic ethanol toxicity.
CYP2E1 metabolizes and activates many toxicological important substrates [16–20,40]. CYP2E1 plays a role in ethanol-induced oxidative stress, ethanol-induced hepatotoxcity [21–29] and ethanol-induced steatosis [36,64]. Understanding the effects of autophagy on CYP2E1-dependent metabolic and toxicological actions (promotion or protection) is important for many reasons, even besides the possible modulation of alcohol-induced liver injury since CYP2E1 is induced under a variety of pathophysiological conditions such as fasting, diabetes, obesity and high fat diet [65–69], by drugs [18,19,40] and in non alcohol-induced steatohepatitis [70,71]. Indeed, inhibition of autophagy potentiated high fat-induced steatohepatitis [15] and increased toxicity of acetaminophen, an effective substrate for metabolism by CYP2E1 [72]. Perhaps of major pathophysiological relevance is whether macroautophagy is protective against ethanol/CYP2E1 elevation of ROS, fatty liver and liver injury or promotes these responses by the liver to ethanol.
Inhibition of autophagy by treatment with 3-MA increased binge ethanol-induced hepatotoxicity, steatosis and oxidant stress in wild type mice and CYP2E1 knockin mice to as much greater extent than in CYP2E1 knockout mice [39]. Stimulation of autophagy by rapamycin blunted the binge ethanol toxicity [13,39]. Inhibition of autophagy in HepG2 E47 cells increased ethanol-induced fat accumulation and oxidant stress to a greater extent than that found in C34 cells which do not express CYP2E1 [37]. These results suggest that, autophagy is protective against ethanol-CYP2E1-dependent toxicity and fat accumulation.
The current report extended the previous studies of autophagy being protective against ethanol/CYP2E1-dependent steatosis to other CYP2E1-dependent toxicity models. We evaluated the toxicity of AA, a decline in hepatoprotective GSH and CCl4 since these have been either implicated in ethanol liver toxicity or are widely used to produce liver injury and fibrosis. All three were previously found to produce greater toxicity in the E47 cells compared to the C34 cells [43,44,54]. Inhibition of autophagy elevated the toxicity of the three agents in the E47 cells but not the C34 cells. The mode of elevated cell death was primarily necrotic, although there was a small increase in apoptotic cell death. Atg 7 siRNA, but not scrambled siRNA, elevated AA toxicity analogous to the chemical inhibition of autophagy by 3-MA. Rapamycin, in contrast to 3-MA or Atg 7 siRNA decreased the toxicity and the necrosis of AA or BSO or CCl4 in the E47 cells, consistent with autophagy being protective against CYP2E1-dependent toxicity. The cytoprotective effects of autophagy may be mediated, in part, by negative regulation of apoptosis [73,74], e.g. 50 mM ethanol decreased levels of Beclin-1 and LC3 II in HepG2 cells and activation of autophagy by globular adiponectin or rapamycin attenuated the ethanol-induced apoptosis by modulation of levels of Bax [75]. Most of the toxicity produced by AA or BSO or CCl4 in the E47 cells was necrotic although some apoptosis was also observed. Rapamycin, while lowering necrosis, increased the apoptosis induced by AA and CCl4, perhaps a reflection of the crosstalk between apoptosis and autophagy.
The increased loss of cell viability produced by AA, BSO or CCl4 in the E47 cells when autophagy was inhibited was associated with an increase in ROS. The increase in ROS is likely important for the increase in toxicity as the antioxidant NAC blunted the toxicity of AA alone and the elevated toxicity of AA in the presence of Atg 7 siRNA under conditions in which NAC also blocked the increase in mitochondrial ROS production produced by AA and AA plus Atg 7 siRNA. This would be consistent with CYP2E1 as an effective promoter of ROS, especially in the presence of AA or BSO or CCl4. There may be several explanations as to why ROS production by the toxins is elevated when autophagy is inhibited e.g. decreased lipophagy and mitophagy with the subsequent accumulation of oxidized lipids and damaged mitochondria. While there are reports that ROS can increase autophagy as a protective mechanism, there are also reports that elevated ROS can inhibit autophagy [76–78]. ROS-induction of autophagy has been implicated in cell survival but also cell death [79].
With respect to damaged mitochondria, AA or BSO or CCl4 produced toxicity to the mitochondria as evidenced by enhanced calcium-induced energized swelling, decreased levels of cellular ATP, and release of cytochrome c from the mitochondria to the cytosol. This mitochondrial dysfunction was elevated when autophagy was inhibited. This damage to the mitochondria is likely linked to elevated oxidative stress as the three toxins increased Mitosox fluorescence and these increases were further elevated when autophagy was inhibited by 3-MA but decreased by rapamycin. In addition, NAC blunted the increases in Mitosox fluorescence produced by AA and the further increase produced by the Atg 7 siRNA.
MAP kinases have been implicated in the regulation of autophagy [80–82]. p38MAPK appears to have a dual role acting as a positive or negative regulator of autophagy depending on conditions, cell type, cell stress [82,83]. For example, activation of p38MAPK in colon cancer cells induced cell survival autophagy [83] and p38 MAPK activation in hepatic stellate cells increased autophagy followed by stellate cell activation [84]. Activation of p38 MAPK induced autophagy in astrocytes and melanoma cells [85] and glucose induced autophagy under starvation conditions by a p38 MAPK mechanism independent of mTOR or AMPK in NIH 3T3 cells [82]. On the other hand, there are reports that activation of p38 MAPK decreased autophagy in colon cancer cells [86]. The maturation steps of autophagy were decreased by activation of p38 MAPK [87]. Activation of p38MAPK impaired autophagy in hepatocytes [88], as well as in macrophages with a resultant increase in accumulation of cholesterol esters and foam cell formation [89]. Possible mechanisms for the inhibition of autophagy by p38 MAPK activation might be due to decreased expression of ULK 1 [89] or increased binding of p62 to activated p38MAPK [81]. JNK MAPK also regulates autophagy [90] e.g. JNK was required for enhanced autophagy and cell survival in response to 4-hydroxynonenal treatment [91]. Menadione-induced ROS formation decreased autophagy in hepatocytes and this led to over activation of JNK/c-jun signaling and subsequent hepatocyte toxicity [92].
Previous studies showed that activation of p38 MAPK played an important role in the toxicity produced when AA or BSO or TNFα were added to the E47 cells [43–45]. The increase in liver injury when either LPS or TNFα were administered to CYP2E1 induced mice was associated with activation of p38 and JNK MAPK and could be blunted by inhibitors of these MAPK [55,93,94]. A JNK inhibitor blocked the decrease in autophagy produced by binge ethanol treatment [38]. These results led to studies as to whether p38 MAPK or JNK (no effects on ERK were observed) were activated by AA, especially when autophagy was inhibited, and if so, whether such activation played a role in the enhanced toxicity by AA when autophagy was inhibited. AA activated p38 MAPK about 2-fold after 24 and 48 h of incubation and inhibition of autophagy further elevated the activation of p38 MAPK by AA an additional 2-fold. In addition, an early activation of p38 MAPK at 6 h was observed when AA plus 3-MA were added together. Stimulation of autophagy with rapamycin decreased the AA alone activation of p38 MAPK. AA alone did not activate JNK but when autophagy was inhibited, AA activated JNK after 24 and 48 h of incubation. The elevated activation of p38 MAPK and JNK by AA when autophagy was inhibited contributes to the increased toxicity by AA when autophagy was inhibited as inhibitors of p38 MAPK and JNK largely (24 h) or partially (48 h) blunted the toxicity. We did not study the combined effects of SB and SP. The role of these MAPK in the toxicity by BSO or CCl4 and the potentiated toxicity of these agents when autophagy is inhibited remains to be determined. These results implicate p38 MAPK and JNK as contributing to the elevated toxicity by AA in the E47 cells when autophagy is inhibited. While the crosstalk between MAPK and autophagy requires further study, we speculate that the decrease in autophagy results in an increase in ROS which then results in an increase in, or an earlier activation of, or a more sustained activation of p38 MAPK or JNK. A scheme summarizing these results is depicted in Fig. 9. Increased production of ROS by cells expressing CYP2E1 occurs when GSH levels are lowered after treatment with BSO or when polyunsaturated fatty acids such as AA undergo lipid peroxidation or when halogenated hydrocarbon such as CCl4 are metabolized to CCl3 radical. ROS can activate p38 and JNK MAPK and activated p38 MAPK or JNK can further elevate ROS as there is a reciprocal relationship between ROS and activated MAPK. The combination of ROS and activated MAPK leads to mitochondrial dysfunction. Inhibition of autophagy elevates lipid accumulation (decreased lipophagy), lipid peroxidation and ROS while decreasing removal of damaged mitochondria by mitophagy thereby promoting the CYP2E1 toxicity. NAC is protective against this toxicity via its antioxidant activity against ROS.
Fig. 9.
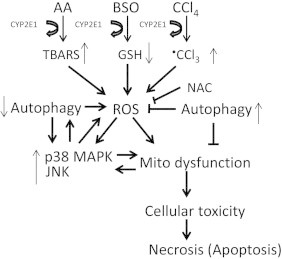
Increased production of ROS by cells expressing CYP2E1 occurs when GSH levels are lowered after treatment with BSO or when polyunsaturated fatty acids such as AA undergo lipid peroxidation or when halogenated hydrocarbon such as CCl4 are metabolized to CCl3 radical. ROS can activate p38 and JNK MAPK and the combination of ROS and activated MAPK lead to mitochondrial dysfunction. Inhibition of autophagy elevates lipid accumulation (decreases lipophagy), lipid peroxidation and ROS while decreasing removal of damaged mitochondria by mitophagy thereby promoting the CYP2E1 toxicity. NAC is protective against this toxicity via its antioxidant activity against ROS.
In summary, autophagy is protective against toxicity of several agents known to be either directly activated by CYP2E1 (e.g. CCl4) or whose toxicity is elevated after induction of CYP2E1 (e.g. AA, BSO). Autophagy has also been shown to protect against CYP2E1-dependent toxicity of acute ethanol in vivo [38,39] and chronic ethanol treatment (Wu and Cederbaum, unpublished observations). Since CYP2E1 plays an important role in the liver injury produced by ethanol, many chemicals and procarcinogens and under various pathophysiological conditions, efforts to maintain and/or activate autophagy would likely be beneficial and important for therapeutic considerations.
Acknowledgments
These studies were supported by USPHS Grants R21 AA021362 and RO1 AA018790 from the National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Defeng Wu, Email: defeng.wu@mssm.edu.
Arthur I. Cederbaum, Email: Arthur.cederbaum@mssm.edu.
References
- 1.Klionsky D.J., Emr S.D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shintanl T., Klionsky D.J. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Jung C.H., Ro S.H., Cao J., Otto N.M., Kim D.H. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendelsohn A.R., Larrick J.W. Rapamycin as an antiaging therapeutic? Rejuvenation. Res. 2011;14:437–441. doi: 10.1089/rej.2011.1238. [DOI] [PubMed] [Google Scholar]
- 6.Ding W.X., Manley S., Ni H.N. The emerging role of autophagy in alcoholic liver disease. Exp. Biol. Med. 2011;236:546–556. doi: 10.1258/ebm.2011.010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin X.M., Ding W.X., Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 8.Czaja M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–1980. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amir M., Czaja M.J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011;52:159–166. doi: 10.1586/egh.11.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H., Czaja M.J. Regulation of lipid droplets by autophagy. Trends Endocr. Metab. 2011;22:234–240. doi: 10.1016/j.tem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Czaja M.J. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implication. Am. J. Physiol. Cell Physiol. 2010;298:C973–C978. doi: 10.1152/ajpcell.00527.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rautou P.E., Mansouri A., Lebrec D., Durand F., Valla D., Moreau R. Autophagy in liver diseases. J. Hepatol. 2010;53:1123–1134. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Ding W.X., Li M., Chen X., Ni H.N., Lin C.W., Gao W., Lu B., Stolz D.B., Clemens D.L., Yin X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osna N.A., Thomes P.G., Donohue T.M., Jr. Involvement of autophagy in alcoholic liver injury and hepatitis c pathogenesis. World J. Gastroenterol. 2011;17:2507–2514. doi: 10.3748/wjg.v17.i20.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin C.W., Zhang H., Li M., Xiong X., Chen X., Chen X., Dong X.C., Yin X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and nonalcoholic fatty liver conditions in mice. J. Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raucy J.L., Kraner J.C., Lasker J.M. Bioactivation of halogenated hydrocarbons by cytochrome P4502E1. Crit. Rev. Toxicol. 1993;2:1–20. doi: 10.3109/10408449309104072. [DOI] [PubMed] [Google Scholar]
- 17.Bolt M., Koos P.H., Their R. The cytochrome P450 isoenzyme CYP2E1 in the biological processing of industrial chemicals. Int. Arch. Occup. Environ. Health. 2003;76:174–185. doi: 10.1007/s00420-002-0407-4. [DOI] [PubMed] [Google Scholar]
- 18.Koop D.R. Oxidative and reductive metabolism by cytochrome P4502E1. FASEB J. 1992;6:724–730. doi: 10.1096/fasebj.6.2.1537462. [DOI] [PubMed] [Google Scholar]
- 19.Song B.J., Cederbaum A.I., Koop D.R., Ingelman-Sundberg M., Nanji A. Ethanol-inducible cytochrome P450 (CYP2E1): Biochemistry, molecular biology and clinical relevance. Alcohol.: Clin. Exp. Res. 1996;20(Suppl):138A–146A. doi: 10.1111/j.1530-0277.1996.tb01764.x. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka E., Terada M., Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J. Clin. Pharm. Ther. 2000;25:165–175. doi: 10.1046/j.1365-2710.2000.00282.x. [DOI] [PubMed] [Google Scholar]
- 21.Lieber C.S. Cytochrome p4502E1: its physiological and pathological role. Physiol. Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 22.Lu Y., Cederbaum A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radical Biol. Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cederbaum A.I., Lu Y., Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009;83:519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 24.French S.W., Morimoto M., Reitz R.C., Koop D., Klopfenstein B., Estes K., Clot P., Ingelman-Sundberg M., Albano E. Lipid peroxidation, CYP2E1 and arachidonic acid metabolism in alcoholic liver disease in rats. J. Nutr. 1997;127:907S–911S. doi: 10.1093/jn/127.5.907S. [DOI] [PubMed] [Google Scholar]
- 25.Castillo T., Koop D.R., Kamimura S., Triadafilopoulos G., Tsukamoto H. Role of cytochrome P4502E1 in ethanol, carbon tetrachloride and iron-dependent microsomal lipid peroxidation. Hepatology. 1992;16:992–996. doi: 10.1002/hep.1840160423. [DOI] [PubMed] [Google Scholar]
- 26.Morimoto M., Zern M.A., Hagbjork A.L., Ingelman-Sundberg M., French S.W. Fish oil, alcohol and liver pathology: role of cytochrome P450 2E1. Proc. Soc. Exp. Biol. Med. 1994;207:197–205. doi: 10.3181/00379727-207-43807. [DOI] [PubMed] [Google Scholar]
- 27.Nanji A.A., Zhao S., Sadrzadeh S.M.H., Dannenberg A.J., Tahan S.R., Waxman D.J. Markedly enhanced cytochrome P4502E1 induction and lipid peroxidation is associated with severe liver injury in fish oil–ethanol-fed rats. Alcohol.: Clin. Exp. Res. 1994;18:1280–1285. doi: 10.1111/j.1530-0277.1994.tb00119.x. [DOI] [PubMed] [Google Scholar]
- 28.Morimoto M., Hagbjork A.L., Wan Y.J., Fu P.C., Clot P., Albano E., Ingelman-Sundberg M., French S.W. Modulation of experimental alcohol-induced liver disease by cytochrome P450 2E1 inhibitors. Hepatology. 1995;21:1610–1617. [PubMed] [Google Scholar]
- 29.Gouillon Z., Lucas D., Li J., Hagbjork A.L., French B.A., Fu P., Fang C., Ingelman-Sundberg M., Donohue T.M., Jr, French S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proc. Soc. Exp. Biol. Med. 2000;224:302–308. doi: 10.1046/j.1525-1373.2000.22435.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu H., Jones B.E., Bradham C., Czaja M.J. Increased cytochrome p4502E1 expression sensitizes hepatocytes to c-Jun-mediated cell death from TNF-alpha. Am. J. Physiol. GI. Liver Physiol. 2002;282:G257–G266. doi: 10.1152/ajpgi.00304.2001. [DOI] [PubMed] [Google Scholar]
- 31.Donohue T.M., Jr, Osna N.A., Clemens D.L. Recombinant HepG2 cell that overexpress alcohol dehydrogenase and cytochrome P4502E1 as a model of ethanol-elicited cytotoxicity. Int. J. Biochem. Cell Biol. 2006;38:92–101. doi: 10.1016/j.biocel.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Clemens D.L. Use of cultured cells to study alcohol metabolism. Alcohol. Res. Health. 2006;29:291–295. [PMC free article] [PubMed] [Google Scholar]
- 33.Caro A.A., Cederbaum A.I. Oxidative stress, Toxicology and Pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 34.Butura A., Nilsson K., Morgan K., Morgan T.R., French S., Johanssson I., Schuppe-Koistinen I., Ingelman-Sundberg M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2008;50:572–583. doi: 10.1016/j.jhep.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Bai J., Cederbaum A.I. Adenovirus-mediated expression of CYP2E1 produces liver toxicity in mice. Toxicol. Sci. 2006;91:365–371. doi: 10.1093/toxsci/kfj165. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y., Zhuge J., Wang X., Bai J., Cederbaum A.I. Cytochrome P4502E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008;47:1483–1494. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 37.Wu D., Wang X., Zhou R., Cederbaum A.I. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem. Biophys. Res. Commun. 2010;402:116–122. doi: 10.1016/j.bbrc.2010.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L., Wu D., Wang X., Cederbaum A.I. Cytochrome P4502E1, oxidative stress, JNK and autophagy in acute alcohol-induced fatty liver. Free Radic. Biol. Med. 2013;53:1170–1180. doi: 10.1016/j.freeradbiomed.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu D., Wang X., Zhou R., Yang L., Cederbaum A.I. Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy. Free Radic. Biol. Med. 2012;53:1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat. Res. 2005;569:101–110. doi: 10.1016/j.mrfmmm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 41.Wu D., Cederbaum A.I. Removal of glutathione produces apoptosis and necrosis in HepG2 cells overexpressing CYP2E1. Alcohol.: Clin. Exp. Res. 2001;25:619–628. [PubMed] [Google Scholar]
- 42.Jimenez-Lopez J.M., Cederbaum A.I. CYP2E1-dependent oxidative stress and toxicity: role in ethanol-induced liver injury. Expert Opin. Drug Metab. Toxicol. 2001;1:671–685. doi: 10.1517/17425255.1.4.671. [DOI] [PubMed] [Google Scholar]
- 43.Wu D., Cederbaum A.I. Role of p38 MAPK in CYP2E1-dependent arachidonic acid toxicity. J. Biol. Chem. 2003;278:1115–1124. doi: 10.1074/jbc.M207856200. [DOI] [PubMed] [Google Scholar]
- 44.Wu D., Cederbaum A.I. Glutathione depletion in CYP2E1-expressing liver cells induces toxicity due to the activation of p38 MAPK and reduction of NF-KB DNA binding activity. Mol. Pharmacol. 2004;66:749–760. doi: 10.1124/mol.104.002048. [DOI] [PubMed] [Google Scholar]
- 45.Pastorino J.G., Shulga N., Hoek J.B. TNF-alpha-induced cell death in ethanol-exposed cells depends on p38 MAPK signaling but is independent of Bid and caspase-8. Am. J. Physiol. GI Liver Physiol. 2003;285:G505–G516. doi: 10.1152/ajpgi.00442.2002. [DOI] [PubMed] [Google Scholar]
- 46.Schattenberg J.M., Singh R., Wang Y., Lefkowitch J.H., Rigoli R.M., Scherer P.E., Czaja M.J. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 47.Czaja M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol. Metab. 2010;21:707–713. doi: 10.1016/j.tem.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X., Wu D., Yang L., Cederbaum A.I. Hepatotoxicity mediated by pyrazole (CYP2E1) plus TNF-alpha treatment occurs in jnk2(−/−) but not in jnk1(−/−) Mice. Hepatology. 2011;54:1753–1766. doi: 10.1002/hep.24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh R., Wang Y., Xiang Y., Tanaka K.E., Gaarde W.A., Czaja M.J. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J., Minemoto Y., Lin A. C-Jun N-terminal kinase1 (JNK1) but not JNK2 is essential for TNFα-induced c-Jun kinase activation and apoptosis. Mol. Cell Biol. 2004;24:10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y., Singh R., Leflkowitch J.H., Rigoli R.M., Scherer P.E., Czaja M.J. Tumor necrosis factor induced liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J. Biol. Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komiya K., Uchida T., Ueno T., Koike M., Abe H., Hirose T., Kawamori R., Uchiyama Y., Kominami E., Fujitani Y., Watada H. Free fatty acids stimulate autophagy in pancreatic beta-cells via JNK pathway. Biochem. Biophys. Res. Commun. 2010;401:561–567. doi: 10.1016/j.bbrc.2010.09.101. [DOI] [PubMed] [Google Scholar]
- 53.Wu D., Cederbaum A.I. Development and properties of HepG2 cells that constitutively express CYP2E1. Methods Mol. Biol. 2008;447:137–150. doi: 10.1007/978-1-59745-242-7_11. [DOI] [PubMed] [Google Scholar]
- 54.Dai Y., Cederbaum A.I. Inactivation and degradation of human cytochrome P4502E1 by CCl4 in a transfected HepG2 cell line. J. Pharmacol. Exp. Ther. 1995;275:1614–1622. [PubMed] [Google Scholar]
- 55.Wu D., Cederbaum A.I. Cytochrome P450 2E1 sensitizes to tumor necrosis factor alpha-induced liver injury through activation of mitogen-activated protein kinases in mice. Hepatology. 2008;47:1005–1017. doi: 10.1002/hep.22087. [DOI] [PubMed] [Google Scholar]
- 56.Wu D., Xu C.J., Cederbaum A.I. Role of nitric oxide and nuclear factor kappa mice in the CYP2E1 potentiation of tumor necrosis factor α hepatotoxicty in mice. Free Radic. Biol. Med. 2009;46:480–491. doi: 10.1016/j.freeradbiomed.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Schellens J.P., Vreeling-Sindelarova H., Plomp P.J., Meijer A.J. Hepatic autophagy and intracellular ATP. A morphometric study. Exp. Cell Res. 1988;42:103–108. doi: 10.1016/0014-4827(88)90028-6. [DOI] [PubMed] [Google Scholar]
- 58.Zalckvar E., Yosef N., Ber Y., Rubinstein A.D., Mor I., Sharan R., Ruppin E., Kimchi A. A systems level strategy for analyzing the cell death network; implication in exploring the apoptosis/autophagy connection. Cell Death Differ. 2010;17:1244–1253. doi: 10.1038/cdd.2010.7. [DOI] [PubMed] [Google Scholar]
- 59.Von Haefen C., Sifringer M., Menk M., Spies C.D. Ethanol enhances susceptibility to apoptotic cell death via down regulation of autophagy-related proteins. Alcohol.: Clin. Exp. Res. 2011;35:1381–1391. doi: 10.1111/j.1530-0277.2011.01473.x. [DOI] [PubMed] [Google Scholar]
- 60.Noh B., Lee J., Jun H., Lee H.J., Jia Y., Hoang M., Kim J., Park K., Lee S. Restoration of autophagy by puerarin in ethanol—treated hepatocytes via the activation of AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 2011;414:361–366. doi: 10.1016/j.bbrc.2011.09.077. [DOI] [PubMed] [Google Scholar]
- 61.Donohue T.M., Jr. Autophagy and ethanol-induced liver injury. World J Gastroenterol. 2009;15:1178–1185. doi: 10.3748/wjg.15.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eid N., Ito Y., Maemura K., Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J. Mol. Histol. 2013;44:311–326. doi: 10.1007/s10735-013-9483-x. [DOI] [PubMed] [Google Scholar]
- 63.Thomes P.C., Trambly C.S., Thiele G.M., Duryee M.J., Fox H.S., Haorah J., Donohue T.M., Jr. proteosome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem. Biophys. Res. Commun. 2012;417:262–267. doi: 10.1016/j.bbrc.2011.11.097. [DOI] [PubMed] [Google Scholar]
- 64.Lu Y., Wu D., Wang X., Ward S.C., Cederbaum A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knockin mice. Free Radic. Biol. Med. 2010;49:1406–1416. doi: 10.1016/j.freeradbiomed.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raucy J.L., Lasker J.M., Kramer J.C., Salazer D.E., Lieber C.S., Corcoran G.B. Induction of P-450IIE1 in the obese rat. Mol. Pharmacol. 1991;39:275–280. [PubMed] [Google Scholar]
- 66.Yun Y., Casazza J.P., Sohn D.H., Veech R.L., Song B.J. Pretranslational activation of cytochrome P450IIE during ketosis induced by a high fat diet. Mol. Pharmacol. 1992;41:474–479. [PubMed] [Google Scholar]
- 67.Woodcroft K.J., Hafner M.S., Novak R.F. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology. 2002;35:263–273. doi: 10.1053/jhep.2002.30691. [DOI] [PubMed] [Google Scholar]
- 68.Hong J.Y., Pan J., Gonzalez F.J., Gelboin H.V., Yang C.S. The induction of a specific form of cytochrome P-450 (P-450j) by fasting. Biochem. Biophys. Res. Commun. 1987;142:1077–1083. doi: 10.1016/0006-291x(87)91525-7. [DOI] [PubMed] [Google Scholar]
- 69.Weltman M.D., Farrell G.C., Liddle C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis and inflammation. Gastroenterology. 1996;111:1645–1653. doi: 10.1016/s0016-5085(96)70028-8. [DOI] [PubMed] [Google Scholar]
- 70.Weltman M.D., Farrell G.C., Hall P., Ingelman-Sundberg M., Liddle C. Hepatic Cytochrome P4502E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–133. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 71.Abdelmegeed M.A., Banerjee A., Yoo S., Jang S., Gonzalez F.J., Song B. Critical role of cytochrome P450E1(CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ni H.M., Bockus A., Boggess N., Jaeschke H., Ding W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–231. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gordy C., Her Y.W. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3:17–27. doi: 10.1007/s13238-011-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maiuri M.C., Zalckvar E., Kimchi A., Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 75.Nepal S., Park P. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cell: involvement of AMPK/FoxO3A axis. Biochim. Biophys. Acta. 2013;1833:2111–2125. doi: 10.1016/j.bbamcr.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 76.Shouval-Scherz R., Shuets E., Fass E., Shorer H., Gil L., Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulates the activity of Atg 4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shouvel-Scherz R., Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 78.Liu Z., Lenardo M.J. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev. Cell. 2007;12:484–485. doi: 10.1016/j.devcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 79.Li L., Ishdorj G., Gibson S.B. Reactive oxygen species regulation of autophagy in cancer: implications for cancer treatment. Free Radic. Biol. Med. 2012;53:1399–1410. doi: 10.1016/j.freeradbiomed.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 80.McClung J.M., Judge A.R., Powers S.K., Yan Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2010;298:C542–C549. doi: 10.1152/ajpcell.00192.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qiang L., Wu C.M., Ming W., Viollert B., He Y. Autophagy controls p38 activation to promote cell survival under genotoxic stress. J. Biol. Chem. 2013;288:1603–1611. doi: 10.1074/jbc.M112.415224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moruno J.F., Perez-Jimenez E., Knecht E. Glucose-induced autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem. J. 2013;449:497–506. doi: 10.1042/BJ20121122. [DOI] [PubMed] [Google Scholar]
- 83.Paillas S., Causse A., Marzi L., De Medina P., Poirot M., Denis V., Vezzio-Vie N., Espert L., Arzouk H., Coquelle A., Martineau P., Del Rio M., Pattingre S., Gongora C. MAPK14/p38α confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy. 2012;8:1098–1112. doi: 10.4161/auto.20268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hernandez-Gea V., Hilscher M., Rozenfeld R., Lim M.P., Nieto N., Werner S., Devi L.A., Friedman S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013;59:98–104. doi: 10.1016/j.jhep.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lui B., Cheng Y., Zhang B., Bian H.G., Bao J.K. Ploygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondrial-mediated ROS-p38-p53 pathway. Cancer Lett. 2009;275:54–60. doi: 10.1016/j.canlet.2008.09.042. [DOI] [PubMed] [Google Scholar]
- 86.Thyagarajan A., Jedinak A., Nguyen H., Terry C., Baldridge L.A., Jiang J., Sliva D. Triterpenes from G. Lucidum induce autophagy in colon cancer through the inhibition of p38 mitogen activated kinase. Nutr. Cancer. 2010;62:630–640. doi: 10.1080/01635580903532390. [DOI] [PubMed] [Google Scholar]
- 87.Corcelle E., Djerbi N., Mari M., Nebout M., Fiorini C., Fenichel P., Hofman P., Poujeol P., Mograbi B. Control of autophagy maturation step by the MAPK ERK and p38. Autophagy. 2007;3:57–59. doi: 10.4161/auto.3424. [DOI] [PubMed] [Google Scholar]
- 88.Webber J.L., Tooze S.A. Coordinated regulation of autophagy by p38α MAPK through mATG9 and p38IP. EMBO J. 2010;29:27–40. doi: 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mei S., Gu H., Ward A., Yang X., Guo H., He K., Liu Z., Cao W. p38MAPK promotes cholesterol ester accumulation in macrophages through inhibition of autophagy. J. Biol. Chem. 2012;287:11761–11768. doi: 10.1074/jbc.M111.333575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jia G., Cheng G., Gangahar D.M., Agrawal D.K. Insulin-like growth factor-1 and TNFα regulate autophagy through c-jun-N terminal kinase and AKT pathways in human atherosclerotic vascular smooth muscle cells. Immunol. Cell Biol. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 91.Haberzettl P., Hill B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013;1:53–64. doi: 10.1016/j.redox.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y., Singh R., Xiang Y., Czaja M.J. Macroautophagy and chaperone –mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 2010;52:266–277. doi: 10.1002/hep.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lu Y., Wang X., Cederbaum A.I. Lipopolysaccharide-induced liver injury in rats treated with the CYP2E1 inducer pyrazole. Am. J. Physiol. GI Liver Physiol. 2005;289:G308–G319. doi: 10.1152/ajpgi.00054.2005. [DOI] [PubMed] [Google Scholar]
- 94.Wu D., Cederbaum A.I. Activation of ASK-1 and downstream MAP kinases in cytochrome P4502E1 potentiated TNFα liver injury. Free Radic. Biol. Med. 2010;49:348–360. doi: 10.1016/j.freeradbiomed.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]