

Abstract
Vascular smooth muscle cells (VSMCs) develop a highly proliferative and synthetic phenotype in arterial diseases. Because such phenotypic changes are likely integrated with the energetic state of the cell, we hypothesized that changes in cellular metabolism regulate VSMC plasticity. VSMCs were exposed to platelet-derived growth factor-BB (PDGF) and changes in mitochondrial morphology, proliferation, contractile protein expression, and mitochondrial metabolism were examined. Exposure of VSMCs to PDGF resulted in mitochondrial fragmentation and a 50% decrease in the abundance of mitofusin 2. Synthetic VSMCs demonstrated a 20% decrease in glucose oxidation, which was accompanied by an increase in fatty acid oxidation. Results of mitochondrial function assays in permeabilized cells showed few changes due to PDGF treatment in mitochondrial respiratory chain capacity and coupling. Treatment of VSMCs with Mdivi-1—an inhibitor of mitochondrial fission—inhibited PDGF-induced mitochondrial fragmentation by 50% and abolished increases in cell proliferation; however, it failed to prevent PDGF-mediated activation of autophagy and removal of contractile proteins. In addition, treatment with Mdivi-1 reversed changes in fatty acid and glucose oxidation associated with the synthetic phenotype. These results suggest that changes in mitochondrial morphology and bioenergetics underlie the hyperproliferative features of the synthetic VSMC phenotype, but do not affect the degradation of contractile proteins. Mitochondrial fragmentation occurring during the transition to the synthetic phenotype could be a therapeutic target for hyperproliferative vascular disorders.
Abbreviations: ADP, adenine dinucleotide phosphate; ATP5A1, ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1; ATP5B, ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide; CPT1, carnitine palmitoyl transferase 1; DMEM, Delbucco's Eagle Modified Medium; Drp1, dynamin-related protein 1; EDTA, ethylenediaminetetraacetic acid; EGTA, ethylene glycol tetraacetic acid; MOPS, 3-(N-morpholino)propanesulfonic acid; Fis1, mitochondrial fission 1 protein; FCCP, Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; FBS, fetal bovine serum; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; NDUFB8, NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8; NP-40, noniodet P40; LC3, (microtubule-associated protein 1 light chain 3); Opa1, optic atrophy 1; PCNA, proliferating cell nuclear antigen; PDGF-BB, platelet-derived growth factor-BB; PVDF, polyvinylidene fluoride; SDS, sodium dodecyl sulfate; SDHB, succinate dehydrogenase subunit B; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine; VSMC, vascular smooth muscle cells
Keywords: Metabolism, Oxidative phosphorylation, Restenosis, Atherosclerosis, Fusion, Extracellular flux
Graphical abstract
Highlights
-
•
PDGF promotes mitochondrial fragmentation in vascular smooth muscle cells.
-
•
PDGF increases metabolic reliance on fatty acids.
-
•
Mitochondrial fragmentation regulates proliferation and bioenergetics.
-
•
PDGF-induced bioenergetic and autophagic responses regulate de-differentiation.
Introduction
Vascular smooth muscle cells (VSMCs) are essential in regulating vessel tone and blood flow [1]. In vascular diseases such as atherosclerosis or after percutaneous coronary interventions, VSMCs lose their contractile function and convert to a highly proliferative phenotype capable of producing elevated levels of extracellular matrix. This VSMC phenotype, typically described as the synthetic phenotype, can dramatically affect the composition and stability of vascular lesions, or, in the case of restenosis, promote vessel occlusion [2–7]. While several factors are involved in mediating this phenotype transition, platelet-derived growth factor-BB (PDGF) appears to be one of the most robust phenotype-modulating agents. Its importance in vascular disease is adduced by multiple studies showing that inhibition of either PDGF binding to its cognate receptor or its downstream signaling inhibits smooth muscle accumulation in the intima after arterial injury [8–15]. Furthermore, VSMCs lacking PDGFR-β demonstrate a resistance to neointimal accumulation after carotid artery injury [16]. Therefore, understanding how PDGF regulates the transition to the synthetic phenotype may be essential for developing strategies for preventing or ameliorating diseases characterized by VSMC hyperproliferation.
Increasingly, mitochondrial metabolism is being recognized as a critical regulator of cell growth and proliferation. Rapidly dividing cells demonstrate enhanced glycolysis and glutamine utilization, which helps provide energy, reducing equivalents, and the carbon and nitrogen building blocks required for synthesis of daughter cells [17]. In VSMCs, growth factors such as PDGF have been shown to increase glycolysis and mitochondrial activity [18]. Recently, it was also shown that, in the context of pulmonary artery hypertension, VSMCs have higher rates of glycolysis [19] and develop a fragmented morphology [19,20]. Such changes in mitochondrial shape have been shown to impact cell differentiation and proliferation directly [21]; however, little is known regarding how mitochondrial structure regulates the phenotype and metabolic characteristics of VSMCs. The goal of this study was to determine how PDGF affects mitochondrial shape and metabolism in VSMCs and identify whether such changes regulate the contractile-to-synthetic phenotype transition. We find that PDGF-induced conversion to the synthetic phenotype is accompanied by mitochondrial fragmentation, which regulates glucose and fat oxidation. This increase in the fragmented mitochondrial phenotype was found to be critical to the hyperproliferative response induced by PDGF.
Materials and methods
Reagents
Antibodies against ATP5A1, ATP5B, SDHB, and NDUFB8 were part of an antibody cocktail purchased from Mitosciences (MitoProfile® Total OXPHOS Rodent WB Antibody Cocktail). The Mito ID red dye, Hoechst stain, and Mdivi-1 were obtained from Enzo life sciences (Farmingdale, NY, USA). PDGF-BB, palmitoylcarnitine, ADP, pyruvate, malate, succinate, protease inhibitor cocktail, etomoxir, FCCP, antimycin A and rotenone were obtained from Sigma-Aldrich (St. Louis, MO, USA). Mouse monoclonal antibody against rat Mfn2 was obtained from Abcam (ab56889). Mouse monoclonal antibodies against α-smooth muscle actin, calponin and α-tubulin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rabbit monoclonal antibody against microtubule associated light chain 3 (LC3) was purchased from Cell Signaling Technologies (Danvers, MA, USA). Antibodies for PCNA and cyclin D1 were from Santa Cruz.
Cell culture
All animal procedures were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Louisville Institutional Animal Care and Use Committee. Rat aortic smooth muscle cells were isolated from the aortas of six week-old, male Sprague-Dawley rats and grown in Dulbecco's Modified Eagle's Medium (DMEM; Life Technologies–Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS) and 0.1% streptomycin/penicillin, as previously described [22,23]. Cells used in experiments were from between passages 2–7 and were maintained in a humidified atmosphere of air and 5% CO2 at 37 °C. At approximately 70% confluency, the VSMCs were serum-starved in DMEM containing 0.1% FBS for 24 h.
Western blotting
After desired treatments, cells were rinsed twice with sterile phosphate buffered saline and then lysed in a protein lysis buffer (pH 7.0) containing 25 mM HEPES, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 1% NP-40, and 1×protease and phosphatase inhibitors. Protein concentration in the cell lysates was measured using the Lowry DC assay (Biorad). For Western blotting, between 0.5 and 25 μg of cell protein (depending on target protein) was applied to each lane of a 10.5–14% Bis–Tris gel and electroblotted onto a PVDF membrane. The membrane was then incubated overnight at 4 °C using appropriate dilutions of primary antibodies. The PVDF membranes were incubated at room temperature with horseradish peroxidase-conjugated secondary antibodies for 1.5 h. After washing steps, immunoreactive bands were detected with enhanced chemiluminescent detection reagent (GE Healthcare) using a Typhoon 9400 Variable Mode Imager (GE Healthcare).
Confocal imaging
Cells plated on glass-bottom dishes were serum-starved for 24 h in 0.1% FBS DMEM. Cells were then stimulated with vehicle or PDGF and/or Mdivi-1 for the indicated times. For visualization, mitochondria were stained using the MitoID Red kit (Enzo life sciences; Farmingdale, NY, USA) following the manufacturer's protocol. Briefly, MitoID Red and Hoechst stains were diluted in phenol red–free DMEM supplemented with 25 mmol/L HEPES (pH 7.4) and then added to cells for 10 min. For imaging, cells were visualized using a Nikon TE-2000E2 microscope interfaced with a Nikon A1 confocal system and images were acquired using a 60×objective lens [24].
Measurement of cellular energetics using the XF24 extracellular flux analyzer
Cellular energetics was measured in intact VSMCs using a Seahorse Bioscience XF24 extracellular flux analyzer as described previously [18,25], with the following modifications. Fifty-thousand cells were seeded in 10 wells of a 24-well XF plate (control group), while 25,000 cells were seeded in the remaining 10 wells (PDGF-treated group); 4 wells were left unseeded for use as temperature control wells. This cell seeding density was chosen based on the expected doubling times of cells treated without or with PDGF [22]. Cells were then serum-starved in 0.1% FBS DMEM for 24 h and subsequently stimulated with 20 ng/ml PDGF for 48 h. After PDGF stimulation, the medium was replaced with DMEM containing 100 µM BSA-palmitate, 5 mM d-glucose, and 100 µM l-carnitine. No l-glutamine or pyruvate was included in the medium. After equilibration in the medium for 1 h, baseline oxygen consumption rates (OCRs) of the cells were recorded. Etomoxir (100 μM) was then injected and changes in the OCR were recorded. Following these measurements, FCCP (1 μM), and antimycin A and rotenone (10 μM and 1 μM, respectively) were sequentially injected, with OCR measurements recorded after each injection. Data were normalized to total protein in each well. For normalization, cells were lysed in the 24-well XF plates using 20 μl/well of protein lysis buffer. Protein concentration was then measured using the Lowry DC assay kit (Biorad).
For measuring electron transport chain activity, the medium was changed to MAS medium (220 mM mannitol, 70 mM sucrose, 5 mM MOPS, and 4% fatty acid-free BSA, pH 7.2), and cells were permeabilized during the XF assay protocol via injection of the following from port A: 25 μg/ml saponin in MAS medium, essentially as described previously [26,27]. Port A also contained substrates to support Complex I-mediated respiration (5 mM pyruvate, 2.5 mM malate, and 1 mM ADP) or Complex II-mediated respiration (10 mM succinate, 1 μM rotenone, and 1 mM ADP). In addition, synthetic substrates such as duroquinol and TMPD+ascorbate were used as described before [28]. Palmitate oxidation was similarly measured in permeabilized cells by including palmitoylcarnitine (50 μM) and malate (0.5 mM) and ADP (1 mM). After measurement of State 3 rates, oligomycin (1 μg/ml) was administered and State 4ο rates were measured. Finally, antimycin A (10 μM)±rotenone (1 μM) was injected to inhibit all consumption of oxygen by mitochondria.
Cell proliferation assays
After serum starvation (24 h), VSMCs were treated for the indicated durations. For cell harvest, cells were rinsed twice with PBS and then trypsinized with a 0.25% trypsin solution (Gibco) for 3 min at 37 °C. Cells were then re-suspended in DMEM containing 10% FBS and cell numbers were determined using a C6 Accuri flow cytometer.
Statistical analysis
Data are mean±SEM. Experimental groups were compared using one-way ANOVA, followed by Bonferroni post-tests. Unpaired Student's t-test was used for direct comparisons. A p-value <0.05 was considered significant.
Results
PDGF promotes mitochondrial fragmentation
We first assessed how treatment of contractile smooth muscle cells with PDGF affects the mitochondrial network. For this, VSMCs were serum-starved for 24 h followed by treatment with PDGF or vehicle. As shown in Fig. 1A and B, 93% of the cells treated with vehicle alone harbored vermiform, filamentous mitochondria. Exposure of the cells to PDGF, however, led to a remarkable loss of this mitochondrial network, with nearly all of the cells acquiring a fragmented (globular) mitochondrial structure. This was associated with a 50% decrease in the abundance of Mfn2 (Fig. 1C and D), with no significant changes noted in the abundance of other regulators of the mitochondrial network, such as Opa1, Drp1, and Fis1. Because of these remarkable changes in mitochondrial structure, we then examined the abundance of some of the key subunits of the oxidative phosphorylation machinery. As shown in Fig. 2A–D, there were no remarkable changes in NDUFB8, SDHB, or ATP5A or ATP5B, which are key proteins in Complexes I, II, and V, respectively. These results suggest that exposure of VSMCs to PDGF results in remarkable mitochondrial remodeling in the absence of gross changes in mitochondrial abundance.
Fig. 1.
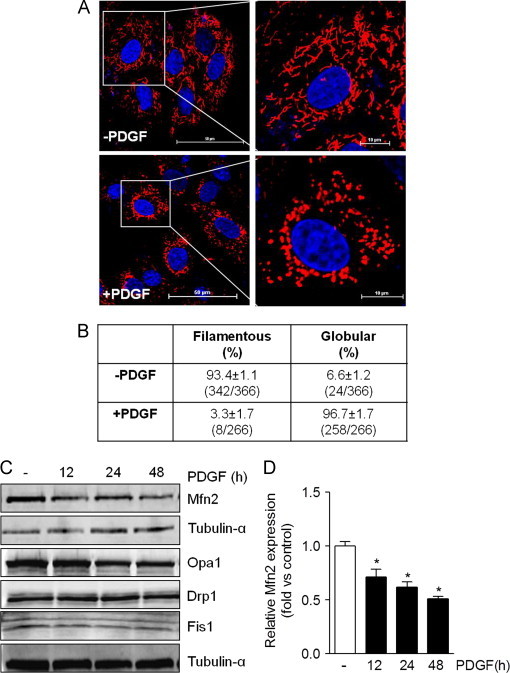
PDGF induces mitochondrial fragmentation in VSMCs. Mitochondria morphology in contractile and synthetic VSMCs: (A) Representative live-cell confocal images of VSMCs treated with vehicle (–PDGF) or PDGF (20 ng/ml) for 48 h. Data shown are representative of four independent experiments. Blue=Hoechst, Red=MitoID red. (Scale bars: left panels, 50 μm; right panels, 10 μm). (B) Mitochondria lengths were measured using NIS Elements software and scored as follows: Fragmented (globular), mainly small and round (<2 μm in diameter); and filamentous, long and higher interconnectivity (> 2 μm in length). The percentage of cells with indicated mitochondrial morphologies was determined as a percentage of the total number of stained cells counted. (C) Representative Western blots of mitofusin 2 (Mfn2), optic atrophy 1 (Opa1), dynamin-related protein 1 (Drp1), and mitochondrial fission 1 protein (Fis1) in control and PDGF-stimulated cells. (D) Quantification of C. Results are representative of three independent experiments. ⁎p<0.05 vs. control (vehicle-treated, − ) cells. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.
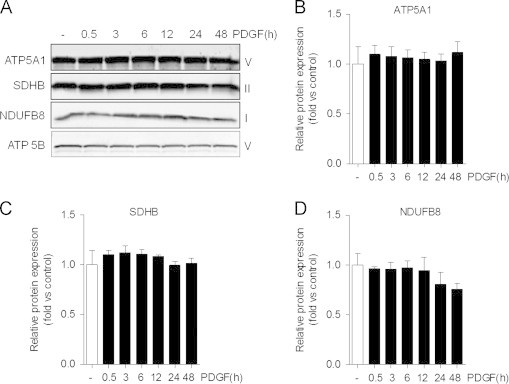
Protein abundance of critical electron transport chain subunits is not affected by PDGF treatment. Immunoblots of subunits of electron transport chain complexes: (A) After serum-starvation for 24 h, VSMCs were treated with PDGF (20 ng/ml) for up to 48 h. The abundance of ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1 (ATP5A1); succinate dehydrogenase subunit B (SDHB), NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8 (NDUFB8); and ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide (ATP5B) were measured by Western blotting. The respective complexes that comprise each subunit are shown to the left of each representative immunoblot. (B–D) Quantification of immunoblots shown in panel A. Results are representative of three independent experiments.
Inhibition of mitochondrial fission prevents VSMC hyperproliferation
Next, we examined how inhibition of mitochondrial fission affects PDGF-induced cell responses. Cells co-treated with PDGF and Mdivi-1, an inhibitor of mitochondrial fission [29], showed a largely preserved filamentous mitochondrial morphology compared with cells treated with PDGF alone (Fig. 3A and B). In addition, treatment with Mdivi-1 completely prevented VSMC proliferation induced by PDGF (Fig. 3C) and inhibited induction of PCNA and cyclin D1 (Fig. 3D–F). However, Mdivi-1 failed to prevent PDGF-induced degradation of the contractile proteins α-smooth muscle actin and calponin (Fig. 4A–C), which were shown previously to be removed via autophagy [22]. In line with this observation, increases in the abundance of LC3-I and II were not affected by the presence of Mdivi-1 (Fig. 4A and D). These data insinuate a causal role of mitochondrial fragmentation in the hyperproliferative VSMC phenotype, which is independent of PDGF-induced autophagy and loss of contractile proteins.
Fig. 3.
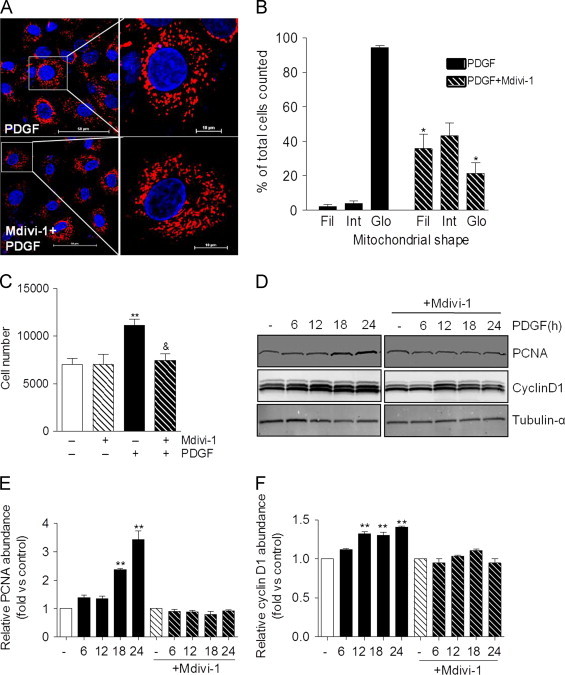
Inhibition of mitochondrial fission prevents PDGF-induced VSMC hyperproliferation. (A) Representative confocal images of VSMC treated with PDGF (20 ng/ml) for 24 h in the absence or presence of Mdivi-1 (10 μm). Blue=Hoechst nuclear stain; Red=mitoID red. (Scale bars: left panels, 50 μm; right panels, 10 μm) (B) Quantification of mitochondrial structures shown in panel A: Mitochondria lengths were measured using NIS Elements software and scored as follows: Fragmented (globular;<2 μm in diameter); intermediate (2–4 μm in length); and filamentous (>4 μm in length). The percentage of cells with the indicated mitochondrial morphologies was determined as a percentage of the total number of stained cells counted. (n=3 per treatment group, ⁎p<0.05 vs. PDGF-treated cells not exposed to Mdivi-1). (C) Equal numbers of VSMCs were seeded. Following serum-starvation, the cells were treated without or with PDGF (20 ng/ml) in the absence or presence of Mdivi-1 (10 μm) for 48 h. After trypsinization, cells were counted using an Accuri flow cytometer. n=3 (experiments) per group, ⁎⁎p<0.01 vs. untreated cells, ⁎p<0.05 vs. PDGF-treated cells. (D) Western blots showing the relative abundances of PCNA and cyclin D1 in cells treated for up to 24 h with PDGF in the absence and presence of Mdivi-1. (E,F) Quantification of the blots from panel D (n=3 per treatment group, ⁎⁎p<0.01 vs. untreated, control cells). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.
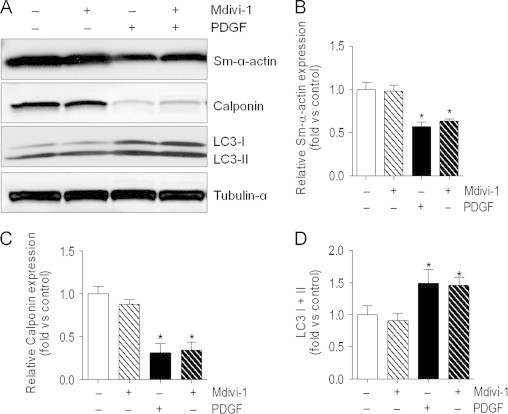
Inhibition of mitochondrial fragmentation does not affect PDGF-induced changes in autophagy and loss of contractile proteins. Abundance of contractile proteins and LC3: (A) Representative immunoblot of α-smooth muscle actin (Sm-α-actin), calponin, and microtubule-associated light chain 3 (LC3): VSMCs were serum-starved for 24 h and then treated with PDGF (20 ng/ml)±Mdivi-1 (10 μM) for 48 h. (B–D) Quantification of immunoblots in panel A (n=3 per group, ⁎p<0.05 vs. untreated cells).
PDGF treatment results in a mitochondrial substrate switch
To determine how PDGF affects mitochondrial metabolism, we developed a substrate selection assay to delineate metabolic reliance on glucose and fatty acids. For this, the cell medium was changed to DMEM containing only glucose, palmitate, and l-carnitine. After three baseline OCR measurements, etomoxir was injected. The decrease in basal OCR and the non-mitochondrial rates of OCR were then used to determine the basal bioenergetic reliance on fatty acid, versus glucose, oxidation (Fig. 5A). This assay was then used to interrogate substrate selectivity in contractile and synthetic VSMCs. As shown in Fig. 5B–D, ~85% of the OCR in intact contractile VSMCs was associated with glucose oxidation, whereas ~15% was related with the oxidation of fat. Treatment of the cells with PDGF-BB for 48 h resulted in a 2-fold increase in apparent fat oxidation and a concomitant diminishment in glucose oxidation. The FCCP-stimulated rate of oxygen consumption was not changed in PDGF-treated cells compared with control cells; however, as shown in the OCR traces (Fig. 5B), PDGF-treated cells exposed to etomoxir showed a lower maximal OCR (Fig. 5B). To determine if inhibiting mitochondrial fragmentation affects substrate selectivity, VSMCs were treated with vehicle, Mdivi-1, PDGF or PDGF+Mdivi-1, and substrate selectivity of the cells was assessed after 48 h. As shown in Fig. 5E and F, treatment with Mdivi-1 completely prevented PDGF-induced increases in fatty acid oxidation.
Fig. 5.
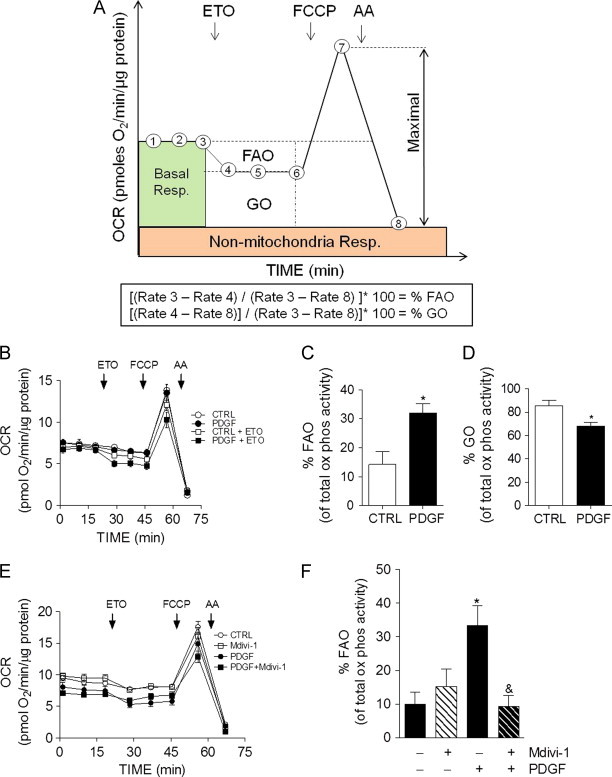
PDGF-mediated increases in mitochondrial fragmentation promote fatty acid oxidation in VSMCs. Measurements of glucose oxidation (GO) and fatty acid oxidation (FAO) in VSMCs using extracellular flux analysis: (A) Diagrammatic example of metabolic reliance assay: After three baseline measurements, etomoxir (ETO; 100 μm) is injected. The change in OCR is then recorded, and FCCP (1 μm) and antimycin A (AA; 10 μm) is injected to establish maximal and non-mitochondrial rates of oxygen consumption, respectively. The equations below the diagram are used to calculate the percent reliance on fatty acid oxidation and glucose oxidation. (B) The assay illustrated in panel A was used to measure fuel selection in contractile and synthetic cells. VSMCs were treated with vehicle or PDGF (20 ng/ml) for 48 h prior to the assay. (C and D) The %FAO and %GO were calculated based upon the equations shown in Panel A. (E) VSMCs were treated without or with PDGF for 48 h in the absence or presence of Mdivi-1 (10 μM). These cells were then subjected to the metabolic reliance assay shown in panel A. (F) The %FAO was calculated from data in panel E. Data shown represent three independent experiments; ⁎p<0.05 vs. vehicle-treated control cells, ⁎p< 0.05 vs. cells treated with PDGF alone.
PDGF treatment does not directly affect mitochondrial electron transport chain or substrate utilization capacity
To determine if conversion to the synthetic phenotype affected the ability of mitochondria to respire directly, we assessed State 3 and State 4ο mitochondrial respiration in permeabilized cells. For this, cells were permeabilized with saponin, allowing for the controlled delivery of substrates to the mitochondrion (Fig. 6A). Using this assay, we first tested whether the synthetic VSMC phenotype has an increased capacity to utilize fatty acids. The cell medium was changed to MAS medium and three baseline oxygen consumption rates were measured. A mixture of saponin, palmitoylcarnitine, and ADP was then injected from port A, and OCRs were measured. Oligomycin and antimycin A/rotenone mix were then injected sequentially with two OCR measurements recorded after each injection (Fig. 6B). As shown in Fig. 6C,D, the State 3 and State 4o values were not significantly different between control (contractile) and PDGF-treated (synthetic) VSMCs. To determine if Complex I-mediated respiration was different between the groups State 3 and State 4ο respiration rates were measured in the presence of pyruvate and malate (Fig. 6E). As shown in Fig. 6F,G, there was no difference in pyruvate-supported respiration between the groups. Succinate-supported respiration was not different between the cell phenotypes either (Fig. 6H–J). Similarly, we did not find changes in duroquinol- or TMPD+ascorbate-supported respiration in the PDGF-treated cells, suggesting that synthetic cells do not have any difference in mitochondrial capacity to support electron flow through Complexes III and IV (data not shown).
Fig. 6.
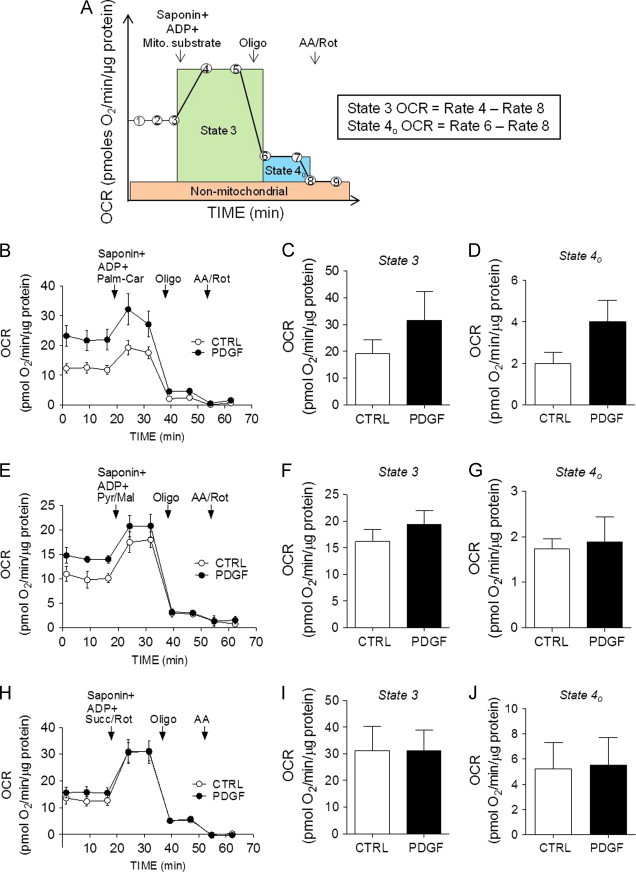
Permeabilized cell assays in contractile and synthetic VSMCs. OCR traces and indices of mitochondrial respiration in permeabilized cells. VSMCs were treated with vehicle or PDGF (20 ng/ml) for 48 h followed by permeabilized cell assays. (A) Illustration demonstrating the permeabilized cell, extracellular flux assay: Cell medium is changed to MAS buffer. After three baseline measurements, saponin (25 μg/ml), mitochondrial substrates, and ADP (1 mM) are injected. Following two more OCR recordings, oligomycin (Oligo, 1 μg/ml) is injected and two measurements are recorded. Then, antimycin A and rotenone (AA/Rot, 10 and 1 μM, respectively) are co-injected and two more OCR measurements are recorded. From this assay, the State 3 and State 4ο rates of oxygen consumption are measured. After normalization to total protein, the equations shown to the right are used for calculations. (B) OCR traces showing palmitoylcarnitine-supported respiration in VSMCs: after three baseline measurements, saponin was co-injected with ADP and palmitoylcarnitine (50 μm) and the assay shown in panel A was completed. (C,D) Quantification of State 3 and State 4ο OCRs from panel B. (E) Complex I-mediated respiration: after three baseline measurements, saponin was co-injected with ADP and pyruvate+malate (pyr/mal; 5/2.5 mM) and the assay shown in panel A was completed. (F,G) Quantification of State 3 and State 4ο OCRs from panel E. (H) Complex II-mediated respiration: after three baseline measurements, saponin was co-injected with ADP and succinate+rotenone (Succ, 10 mM; Rot, 1 μM). (I,J) Quantification of State 3 and State 4ο OCRs from panel H. Results represent 3–4 independent experiments.
Discussion
The present findings demonstrate a novel dichotomy in the acquisition of the synthetic VSMC phenotype. PDGF-induced conversion of VSMCs into the synthetic phenotype was accompanied by mitochondrial fragmentation, decreased abundance of Mfn2, a decrease in apparent glucose oxidation and a concomitant increase in fatty acid oxidation. Inhibition of mitochondrial fragmentation with Mdivi-1 did not prevent PDGF-induced activation of autophagy or loss of contractive elements, but it did prevent PDGF-induced cell proliferation and the disposition to oxidize fatty acids. Thus, the hyperproliferative responses caused by PDGF appear to be due, in part, to its effects on mitochondrial morphology and bioenergetic function, which is discrete from the loss of contractile elements that occurs with conversion to the synthetic VSMC phenotype. These studies are consistent with previous studies showing a role of mitochondrial fission in VSMC hyperproliferation. For example, the hyperproliferation of pulmonary artery smooth muscle cells has been associated with a fragmented mitochondrial phenotype [20], and inhibition of Drp-1-mediated mitochondrial fission with Mdivi-1 prevented not only cell proliferation, but cobalt-induced pulmonary artery hypertension in vivo as well [19]. These results are in harmony also with previous studies showing that overexpression of Mfn2 inhibits VSMC proliferation [30].
The findings of this study contribute to the understanding of how mitochondrial structure regulates VSMC metabolism and hyperproliferative responses. Using a PDGF-induced model of the synthetic VSMC phenotype [7,18,22], we demonstrate that the attainment of the synthetic phenotype is associated with remarkable changes in mitochondrial morphology. With conversion to the synthetic phenotype, VSMC mitochondria assume a fragmented (or globular) phenotype. The synthetic cells demonstrate a greater capacity to oxidize fatty acids and a decreased propensity to utilize glucose. As a limitation, it should be noted that the conclusions drawn from intact cell studies were based on the inhibition of OCR when cells were exposed to etomoxir, an inhibitor of carnitine palmitoyl transferase (CPT) [31]. Hence, off-target effects of etomoxir, which may change with cell phenotype, cannot be ruled out. Nevertheless, it would appear that the preference for lipid oxidation was increased in synthetic cells.
These studies also facilitated the use of permeabilized cells to examine metabolic changes intrinsic to the mitochondrion. VSMCs permeabilized with saponin showed no differences in pyruvate- or succinate-supported respiration between treatment groups, indicating that pyruvate and succinate uptake and oxidation in mitochondria is unaffected by conversion to the synthetic phenotype. Similarly, palmitoylcarnitine- and glutamine-supported respiration were not different between contractile and synthetic cells, and PDGF treatment had no effect on Complex III and IV activities, as indicated by identical rates in State 3 respiration compared with contractile cells.
Although it is unclear how bioenergetic reprogramming may affect the hyperproliferative VSMC response, it could be that VSMCs maximize their ability to generate energy by increasing mitochondrial β-oxidation. Lipid oxidation provides more ATP than does oxidation of carbohydrates (per molecule), although it does diminish energy efficiency [32]. While fatty acid oxidation is known to result in the generation of FADH2 (which is also generated by succinate dehydrogenase and similarly transfers electrons to ubiquinone), it generates NADH as well, which transfers electrons to more thermodynamically efficient reactions commencing at Complex I [33]. Nevertheless, smooth muscle cells, both in vivo and ex vivo, would be thought to have adequate levels of oxygen and thus could afford a lower P:O stoichiometry. Based on our results, we speculate that metabolism is restructured in synthetic cells to support the maximum production of ATP, which is utilized for cell proliferation. It is also possible that glucose or its metabolites may be diverted into pathways that support the biosynthesis of phospholipids and DNA, an abundant supply of which are required in rapidly dividing cells. This would suggest that increases in fatty acid oxidation could be a consequence of decreased pyruvate availability. That fatty acid oxidation is not different between permeabilized synthetic and contractile cells would appear to support this hypothesis. It is also possible that increased reliance of synthetic VSMCs on mitochondrial β-oxidation is due to altered allosteric regulation of β-oxidation enzymes [34].
Perhaps most remarkable is the dichotomous nature of the conversion to the synthetic cell phenotype. Our previous studies show that PDGF activates autophagy, which is required for removal of contractile myofilaments such as α-smooth muscle cell actin and calponin [22]. Such a conversion is associated with a higher rate of cell proliferation [7]. The results of this study suggest that the de-differentiation process occurs via two separate processes, i.e., proliferative responses driven by coupled transcriptional and bioenergetic processes, and loss of contractile proteins largely mediated by autophagy and downregulation of myofilament genes. Interestingly, inhibition of fission with Mdivi-1 prevented PDGF-induced upregulation of the cell cycle proteins PCNA and cyclin D1 as well. This could support the hypothesis that mitochondrial structure and activity regulate the transcriptional machinery associated with proliferation.
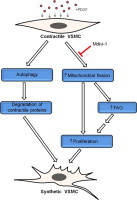
Collectively, these studies provide novel insights into the role of mitochondria in PDGF-induced proliferation. Mitochondrial morphology appears to be a critical determinant of substrate usage and cell proliferation. Furthermore, when combined with our previous studies [22,35], these studies suggest a new model of VSMC de-differentiation in which removal of contractile elements occurs in parallel, but not in series, with the coordinated bioenergetic and transcriptional changes that support proliferation (Fig. 7). Compounds that inhibit mitochondrial fission could be promising candidates for preventing diseases associated with aberrant VSMC hyperproliferation.
Fig. 7.
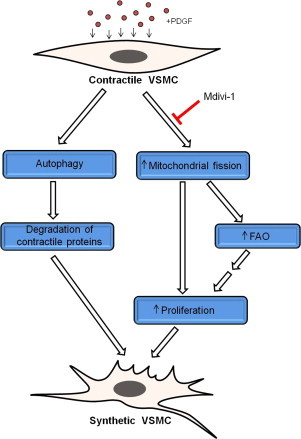
Model of PDGF-induced VSMC de-differentiation. Combined with previous findings [1,22], the current study suggests that PDGF promotes the full development of the VSMC synthetic phenotype by activating parallel processes. Exposure of VSMCs to PDGF results in activation of autophagy, which is required for prompt removal of the contractile apparatus. In addition, PDGF induces mitochondrial fragmentation, which regulates fuel selection and is required for hyperproliferation. It is hypothesized that higher levels of fatty acid oxidation (FAO) provide additional energy for the synthetic cells to rapidly divide, migrate, and synthesize and secrete extracellular matrix.
Competing financial interests
The authors declare that they have no competing financial interests.
Author contributions
JKS performed experiments and JKS and BGH were involved in all parts of manuscript preparation.
Acknowledgments
This work was supported by the National Institutes of Health [grants GM103492 and HL078825]. The authors acknowledge the expert technical assistance of Steven Jones (Imaging and Physiology Core of the Diabetes and Obesity Center).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Lacolley P., Regnault V., Nicoletti A., Li Z., Michel J.B. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc. Res. 2012;95:194–204. doi: 10.1093/cvr/cvs135. [DOI] [PubMed] [Google Scholar]
- 3.Lee R.T., Libby P. The unstable atheroma. Arterioscler. Thromb. Vasc. Biol. 1997;17:1859–1867. doi: 10.1161/01.atv.17.10.1859. [DOI] [PubMed] [Google Scholar]
- 4.Gomez D., Owens G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orford J.L., Selwyn A.P., Ganz P., Popma J.J., Rogers C. The comparative pathobiology of atherosclerosis and restenosis. Am. J. Cardiol. 2000;86:6H–11H. doi: 10.1016/s0002-9149(00)01094-8. [DOI] [PubMed] [Google Scholar]
- 6.Pyle A.L., Young P.P. Atheromas feel the pressure: biomechanical stress and atherosclerosis. Am. J. Pathol. 2010;177:4–9. doi: 10.2353/ajpath.2010.090615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salabei J.K., Hill B.G. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radic. Biol. Med. 2013;65C:693–703. doi: 10.1016/j.freeradbiomed.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferns G.A., Raines E.W., Sprugel K.H., Motani A.S., Reidy M.A., Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 9.Jackson C.L., Raines E.W., Ross R., Reidy M.A. Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler. Thromb. 1993;13:1218–1226. doi: 10.1161/01.atv.13.8.1218. [DOI] [PubMed] [Google Scholar]
- 10.Lewis C.D., Olson N.E., Raines E.W., Reidy M.A., Jackson C.L. Modulation of smooth muscle proliferation in rat carotid artery by platelet-derived mediators and fibroblast growth factor-2. Platelets. 2001;12:352–358. doi: 10.1080/09537100120071013. [DOI] [PubMed] [Google Scholar]
- 11.Noiseux N., Boucher C.H., Cartier R., Sirois M.G. Bolus endovascular PDGFR-beta antisense treatment suppressed intimal hyperplasia in a rat carotid injury model. Circulation. 2000;102:1330–1336. doi: 10.1161/01.cir.102.11.1330. [DOI] [PubMed] [Google Scholar]
- 12.Leppanen O., Janjic N., Carlsson M.A., Pietras K., Levin M., Vargeese C., Green L.S., Bergqvist D., Ostman A., Heldin C.H. Intimal hyperplasia recurs after removal of PDGF-AB and -BB inhibition in the rat carotid artery injury model. Arterioscler. Thromb. Vasc. Biol. 2000;20:E89–95. doi: 10.1161/01.atv.20.11.e89. [DOI] [PubMed] [Google Scholar]
- 13.Myllarniemi M., Calderon L., Lemstrom K., Buchdunger E., Hayry P. Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J. 1997;11:1119–1126. doi: 10.1096/fasebj.11.13.9367346. [DOI] [PubMed] [Google Scholar]
- 14.Yamasaki Y., Miyoshi K., Oda N., Watanabe M., Miyake H., Chan J., Wang X., Sun L., Tang C., McMahon G., Lipson K.E. Weekly dosing with the platelet-derived growth factor receptor tyrosine kinase inhibitor SU9518 significantly inhibits arterial stenosis. Circ. Res. 2001;88:630–636. doi: 10.1161/01.res.88.6.630. [DOI] [PubMed] [Google Scholar]
- 15.Yu J.C., Lokker N.A., Hollenbach S., Apatira M., Li J., Betz A., Sedlock D., Oda S., Nomoto Y., Matsuno K., Ide S., Tsukuda E., Giese N.A. Efficacy of the novel selective platelet-derived growth factor receptor antagonist CT52923 on cellular proliferation, migration, and suppression of neointima following vascular injury. J. Pharmacol. Exp. Ther. 2001;298:1172–1178. [PubMed] [Google Scholar]
- 16.Buetow B.S., Tappan K.A., Crosby J.R., Seifert R.A., Bowen-Pope D.F. Chimera analysis supports a predominant role of PDGFRbeta in promoting smooth-muscle cell chemotaxis after arterial injury. Am. J. Pathol. 2003;163:979–984. doi: 10.1016/s0002-9440(10)63457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moncada S., Higgs E.A., Colombo S.L. Fulfilling the metabolic requirements for cell proliferation. Biochem. J. 2012;446:1–7. doi: 10.1042/BJ20120427. [DOI] [PubMed] [Google Scholar]
- 18.Perez J., Hill B.G., Benavides G.A., Dranka B.P., Darley-Usmar V.M. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochem. J. 2010;428:255–267. doi: 10.1042/BJ20100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsboom G., Toth P.T., Ryan J.J., Hong Z., Wu X., Fang Y.H., Thenappan T., Piao L., Zhang H.J., Pogoriler J., Chen Y., Morrow E., Weir E.K., Rehman J., Archer S.L. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012;110:1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnet S., Michelakis E.D., Porter C.J., Andrade-Navarro M.A., Thebaud B., Haromy A., Harry G., Moudgil R., McMurtry M.S., Weir E.K., Archer S.L. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 21.Mitra K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. Bioessays. 2013;35:955–964. doi: 10.1002/bies.201300011. [DOI] [PubMed] [Google Scholar]
- 22.Salabei J.K., Cummins T.D., Singh M., Jones S.P., Bhatnagar A., Hill B.G. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem. J. 2013;451:375–388. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill B.G., Haberzettl P., Ahmed Y., Srivastava S., Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem. J. 2008;410:525–534. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 24.Cummins T.D., Higdon A.N., Kramer P.A., Chacko B.K., Riggs D.W., Salabei J.K., Dell'Italia L.J., Zhang J., Darley-Usmar V.M., Hill B.G. Utilization of fluorescent probes for the quantification and identification of subcellular proteomes and biological processes regulated by lipid peroxidation products. Free Radic. Biol. Med. 2013;59:56–68. doi: 10.1016/j.freeradbiomed.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill B.G., Higdon A.N., Dranka B.P., Darley-Usmar V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta. 2010;1797:285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clerc P., Polster B.M. Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS One. 2012;7:e34465. doi: 10.1371/journal.pone.0034465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Divakaruni A.S., Wiley S.E., Rogers G.W., Andreyev A.Y., Petrosyan S., Loviscach M., Wall E.A., Yadava N., Heuck A.P., Ferrick D.A., Henry R.R., McDonald W.G., Colca J.R., Simon M.I., Ciaraldi T.P., Murphy A.N. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. USA. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye F., Hoppel C.L. Measuring oxidative phosphorylation in human skin fibroblasts. Anal. Biochem. 2013;437:52–58. doi: 10.1016/j.ab.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 29.Cassidy-Stone A., Chipuk J.E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M.J., Shaw J.T., Hinshaw J.E., Green D.R., Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K.H., Guo X., Ma D., Guo Y., Li Q., Yang D., Li P., Qiu X., Wen S., Xiao R.P., Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell. Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 31.Fillmore N., Lopaschuk G.D. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim. Biophys. Acta. 2013;1833:857–865. doi: 10.1016/j.bbamcr.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 32.Leverve X., Batandier C., Fontaine E. Choosing the right substrate. Novartis Found Symp. 2007;280:108–121. (discussion 121–107, 160–104) [PubMed] [Google Scholar]
- 33.Nicholls D.G., Ferguson S.J. Vol. 3. Academic Press; Amsterdam; Boston: 2002. (Bioenergetics). [Google Scholar]
- 34.Schreurs M., Kuipers F., van der Leij F.R. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev. 2010;11:380–388. doi: 10.1111/j.1467-789X.2009.00642.x. [DOI] [PubMed] [Google Scholar]
- 35.Haberzettl P., Hill B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013;1:56–64. doi: 10.1016/j.redox.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]