

Abstract
Recent investigations have documented that constitutively activated phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian Target of Rapamycin (mTOR) signaling is a common feature of T-cell acute lymphoblastic leukemia (T-ALL) where it strongly influences growth and survival. These findings lend compelling weight for the application of PI3K/Akt/mTOR inhibitors in T-ALL. However, our knowledge of PI3K/Akt/mTOR signaling in T-ALL is limited and it is not clear whether it could be an effective target for innovative therapeutic strategies. Here, we have analyzed the therapeutic potential of the dual PI3K/mTOR inhibitor, PI-103, a small synthetic molecule of the pyridofuropyrimidine class, on both T-ALL cell lines and patient samples, which displayed constitutive activation of PI3K/Akt/mTOR signaling. PI-103 inhibited the growth of T-ALL cells, including 170-kDa glycoprotein overexpressing cells. PI-103 cytotoxicity was independent of p53 gene status. PI-103 was more potent than inhibitors which are selective only for PI3K (Wortmannin, LY294002) or for mTOR (rapamycin). PI-103 induced G0/G1 phase cell cycle arrest and apoptosis which was characterized by activation of caspase-3 and -9. PI-103 caused Akt dephosphorylation, accompanied by dephosphorylation of the Akt downstream target, glycogen synthase kinase-3β. Also mTOR downstream targets were dephosphorylated in response to PI-103, including p70S6 kinase, ribosomal S6 protein, and 4E-BP1. PI-103 strongly synergized with vincristine. These findings indicate that multi-targeted therapy towards PI3K and mTOR, alone or with existing drugs, may serve as an efficient treatment towards T-ALL cells which require upregulation of PI3K/Akt/mTOR signaling for their survival and growth.
Keywords: PI3K/Akt/mTOR signaling, apoptosis, caspases, drug resistance, combination therapy
INTRODUCTION
Activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway plays a central role in cancer biology, conferring resistance both in vivo and in vitro to therapeutic treatments of various types of malignancies which include leukemias (1–3). Upon activation, PI3K generates phosphatidylinositol 3,4,5 (PIP3), which then activates a number of downstream targets, including Akt. A fundamental negative regulator of the PI3K/Akt pathway is the lipid phosphatase PTEN (phosphatase and tensin deleted on chromosome 10), which removes the 3-phosphate from PIP3, thus downmodulating PI3K/Akt signaling (2, 3). A PI3K/Akt downstream target which plays an important role in tumorigenesis and drug resistance is mammalian Target of Rapamycin (mTOR) (4). mTOR phosphorylates p70 ribosomal S6 kinase (p70S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1). By doing so, mTOR increases synthesis of proteins which are important for cell cycle regulation. T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive neoplastic disorder of lymphoblasts committed to the T-cell lineage (5). Over the past 20 years, survival rates of T-ALL patients have improved, due to advances in therapeutic protocols (6). Survival rates at five years for children and adolescents with T-ALL are 70–75%, whereas, for adults they are 35–40% (7). Nevertheless, novel therapies aimed at improperly activated signaling pathways are needed to combat induction failure, relapse rate, and the development of drug-resistance in T-ALL patients (7). Aberrant Notch-1 receptor signaling is a hallmark of T-ALL (8). Fifty-percent of T-ALL patients display Notch-1 activating mutations (7). As a consequence, the receptor is activated independently of the ligand (9). Other Notch-1 mutations impair its degradation and result in a longer receptor half-life (9, 10). When the intracellular portion of Notch-1 is released through cleavage mediated by γ-secretase (GSI), it translocates to the nucleus where it affects gene expression (11). Therefore, GSI inhibition could be exploited to abrogate aberrant Notch-1 signaling in T-ALL. However, the results of the first clinical trial testing the GSI inhibitor, MK-0752, in relapsed/refractory T-ALL patients, showed no clinical responses and a high incidence of gastrointestinal toxicity (9). Recently, it was demonstrated that in T-ALL activated Notch-1 leads to PI3K/Akt signaling upregulation by HES1 (hairy and enhancer of split 1) -mediated transcriptional repression of PTEN (12). Other reports have highlighted that PTEN posttranslational inactivation through phosphorylation and oxidation, could result in PI3K/Akt/mTOR activation in T-ALL (13). These findings lend compelling weight for the application of PI3K/Akt/mTOR inhibitors in T-ALL (14). mTOR inhibitors are being evaluated for treatment of several types of tumors, including leukemias (15). Rapamycin or its analogues, used alone or in combination with traditional chemotherapeutic drugs, have shown some promising effects in pre-clinical models of TALL (e.g. 14). However, mTOR inhibitors are mainly cytostatic (16) and could hyperactivate Akt due to the existence of feedback loops between mTOR, PI3K, and Akt (17). Recently, dual PI3K/mTOR inhibitors have been synthesized, including PI-103, a small molecule of the pyridofuropyrimidine class (18). Since PI-103 targets class IA PI3K in addition to mTOR, it should not lead to Akt activation which is seen when only mTOR is targeted by inhibitors (18). Moreover, PI-103 should affect mTOR signaling also when it is activated independently from PI3K/Akt, as described in some T-ALL cases (19). Here, we have analyzed the therapeutic potential of the novel, dual PI3K/mTOR inhibitor, NVP-BEZ235, an orally bioavailable imidazoquinoline derivative, which has entered clinical trials for solid tumors, on both T-ALL cell lines and patient samples.
MATERIALS AND METHODS
Materials
PI-103 was purchased from Alexis Biochemicals (Lausen, Switzerland). Rapamycin, LY294002, and Wortmannin, were from Sigma-Aldrich (St. Louis, MO, USA). P110α PI3K inhibitor [compound 15e or [3-[4-(4-Morpholinyl)thieno[3,2-d]pyrimidin-2-yl]-phenol] was from Alexis, while p110β (TGX–221) and p110γ [5-(2,2--Difluoro-benzo[1,3]dioxol-5-ylmethylene)-thiazolidine-2,4-dione] PI3K inhibitors and Akt 1/2 inhibitor were from Calbiochem-Novabiochem (La Jolla, CA, USA). For western blots, the following primary antibodies were from Cell Signaling Technology (Danvers, MA, USA): β-actin, Akt, Ser 473 p-Akt (which recognizes both Akt1 and Akt2 when phosphorylated on Ser 473 and Ser 474, respectively), p70S6K, Thr 389 p-p70S6K, 4E-BP1, Thr 37/46 p-4E-BP1, glycogen synthase kinase (GSK) -3β, Ser 21/9 p-GSK-3α/β, S6 ribosomal protein (S6RP), Ser 235/236 p-S6RP, p110γ PI3K, caspase-3, and caspase-9. Antibodies to p110α, p110β, and p110δ PI3K were from Upstate Biotechnology/Millipore Corporation. Antibodies to Akt1 (raised in goat) and Akt2 (raised in mouse) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture and patient samples
The T-ALL cell lines Jurkat, MOLT-4, CEM-S, CEM-R [CEM VBL100, drug-resistant cells overexpressing 170-kDa P-glycoprotein (P-gp) (20)] were grown in RPMI 1640, supplemented with 10% heat-inactivated fetal bovine serum (FBS). Patient samples or peripheral blood lymphocytes from healthy donors were obtained with informed consent according to institutional guidelines and isolated by Ficoll-Paque (Amersham Biosciences, Uppsala, Sweden).
Cell viability analysis
MTT (3–[4,5-Dimethylthythiazol-2-yl]-2,5-Diphenyltetrazolium Bromide) assays were performed to assess the sensitivity of cells to PI-103 and other inhibitors, as previously reported (21).
Annexin V-FITC/PI staining and cell cycle analysis
To assess the extent of apoptosis induction after treatments, flow cytometric analysis of Annexin V-FITC/PI-stained samples was performed, as reported elsewhere (21). Samples were analyzed by an EPICS XL flow cytometer (Beckman Coulter, Miami, FL, USA). At least 15,000 events per sample were acquired. For cell cycle analysis, samples were stained with propidium iodide (PI) and analyzed by flow cytometry, as described elsewhere (21). At least 10,000 events per sample were acquired
Western blot analysis
Cells were collected by centrifugation, washed twice in PBS and lysed as previously reported (21). Samples were separated on SDS-polyacrylamide gels and electro-transferred to nitrocellulose membranes. Western blot analysis was performed as detailed elsewhere (20). Each blot is representative of three separate experiments. Antibody to β-actin served as a loading control.
Immunoprecipitation
This was carried out according to others (22). Cell extracts were incubated for 2 h at 4°C under constant agitation with 5 µg of the antibody to either Akt1 or Akt2. Ten micrograms of 50% protein A/G agarose was then added and incubation proceeded for 1 h at 4°C under constant agitation.
Combined drug effect analysis
The combination effect and a potential synergy were evaluated from quantitative analysis of dose-effect relationships as described previously (22). For each PI-103/vincristine drug combination experiment, a CI (combination index) number was calculated using the Biosoft CalcuSyn software (Cambridge, UK). This method of analysis generally defines CI values of 0.9 to 1.1 as additive, 0.3 to 0.9 as synergistic, and <0.3 as strongly synergistic, whereas values >1.1 are considered antagonistic.
Flow cytometric analysis of p-Akt, p-4E-BP1, and cleaved caspase-3 levels in T-ALL patient samples
Blasts from 7 pediatric patients with T-ALL were fixed in reagent 1 of the Intraprep Kit and permeabilized with saponin-based reagent 2 (Beckman Coulter) as reported elsewhere (23, 24). Cells were incubated with primary antibody to Ser473 p-Akt, Thr 37/46 p-4E-BP1, or cleaved caspase-3. All the antibodies were from Cell Signaling and were conjugated to AlexaFluor® 488. A rabbit IgG conjugated to AlexaFluor® 488 was used as an irrelevant antibody. Cells were analyzed on a FC500 flow cytometer (Beckman Coulter). At least 10,000 events per sample were acquired
RESULTS
PI-103 has cytotoxic proapoptotic effects on T-ALL cell lines
The effect of the dual PI3K/mTOR inhibitor PI-103 on PTEN-negative T-ALL cell lines was examined first. While Jurkat and MOLT-4 cells do not express p53, CEM cells display nonfunctional p53 (25 and data not shown). Moreover, both Jurkat and MOLT-4 cells have aberrant Notch-1 signaling (26). T-ALL cell lines were treated with different concentrations of PI-103. After 24 and 48 h, the rates of survival were measured using MTT assays. Cell lines displayed an IC50 for PI-103 ranging from 0.25 to 1.0 µM at 24 h and from 0.25 to 0.40 µM at 48 h. The vehicle alone (DMSO) did not affect cell survival (not shown). Remarkably, also the drug-resistant CEM-R cell line, a subclone overexpressing 170 kDa P-gp, showed sensitivity to PI-103 (IC50= 0.6 µM at 24 h and 0.25 µM at 48 h) (Fig. 1A). Next, a comparison was made between PI-103 and inhibitors of either mTOR (rapamycin) or PI3K (Wortmannin, LY294002). Rapamycin was less effective than PI-103 in negatively affecting cell growth of T-ALL cell lines, also when used at equimolar concentrations, (Fig. 1B). Wortmannin was much less effective than PI-103 and the same was true of LY294002, which approached PI-103 cytotoxicity only when used at 25–50 µM (Fig. 1B).
Figure 1. PI-103 induces cytotoxicity in T-ALL cell lines.

A: MTT assays of T-ALL cell lines treated with PI-103 for 24 and 48 h. Results are the mean of at least three different experiments ± SD. The asterisk indicates statistically significant difference (p<0.001) with respect to untreated cells. B: Effect of mTOR (rapamycin) or PI3K (Wortmannin, LY294002) inhibitors. The results of MTT assays performed at 24 h are shown. Results are the mean of at least three different experiments ± SD.
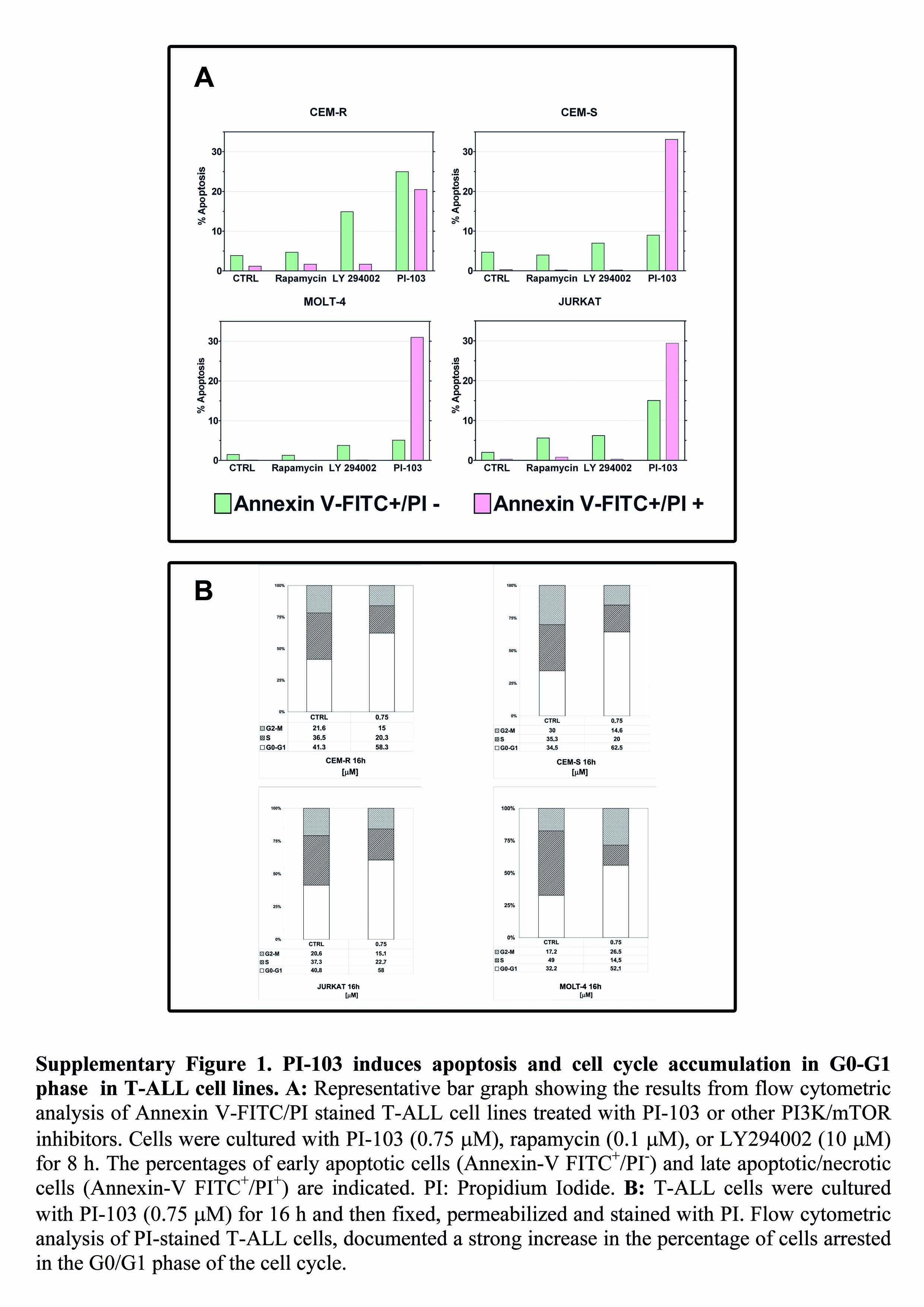
It was next investigated whether decreased cell proliferation was related to apoptosis, using Annexin V-FITC/PI staining and flow cytometry. After 9 h of treatment, approximately 45% of CEM-R cells were apoptotic with 25% in early apoptosis (Annexin V-FITC positive only) and 20% mid-late apoptosis (Annexin V-FITC/PI positive), while approximately 9% of CEM-S cells were positive for early apoptosis (Annexin V-FITC only) and another 33% were positive for both Annexin-V and PI, indicating mid-late apoptotic cells. The degree of apoptosis induced in MOLT-4 and Jurkat cells by PI-103 was similar to that detected in CEM-S cells (Supplementary Fig. 1A). Both rapamycin and LY294002 were much less effective than PI-103 in inducing apoptosis (Supplementary Fig. 1A). Given the fundamental role played by PI3K/Akt/mTOR signaling in cell proliferation, the effects of PI-103 on cell cycle progression were also investigated. Flow cytometric analysis of PI-stained T-ALL cells treated with PI-103 for 16 h documented an increase in G0/G1 phase cells and a decrease in S and G2/M phase cells (Supplementary Fig. 1B). As rapamycin is mainly cytostatic, we reasoned that MTT assay could not be the most appropriate technique to calculate its IC50. Therefore, we also performed flow cytometric analysis of cell cycle. However, as shown in Supplementary Fig.2, also this technique documented that the IC50 was not reached even at 3 µM rapamycin for CEM-R, CEM-S, and MOLT-4 cells, while for Jurkat cells the decrease in cell proliferation was around 50% at 3 µM.
Overall, these findings demonstrated the PI-103 potently reduced the growth of T-ALL cell lines and that this effect was due to both apoptosis and cell cycle arrest.
PI-103 induces caspase activation
A distinguishing feature of apoptotic cell death is the activation of a family of proteases referred to as caspases, which are responsible for most of the biochemical and morphological changes which characterize apoptosis (27). Western blotting analysis of extracts from both CEM-R and MOLT-4 cells treated with PI-103, documented that cleavage of procaspase-9 and -3 was detectable at 8 h of treatment. In contrast, rapamycin activated caspase-3 and -9 only in MOLT-4 cells, and at a later time (16 h) (Fig. 2).
Figure 2. PI-103 activates caspase-9 and -3 in CEM-R and MOLT-4 cells, while rapamycin activates caspase-9 and -3 in MOLT-4 cells only.

CEM-R and MOLT-4 cells were treated with PI-103 (0.75 µM) or rapamycin (0.1 µM) for the indicated times, collected, and then lysed. Fifty micrograms of each lysate were electrophoresed on SDS-PAGE gels followed by transfer to nitrocellulose membrane.
PI-103 affects PI3K/Akt/mTOR signaling in T-ALL cell lines
Western blot analysis with an antibody to Ser 473 p-Akt, demonstrated a marked decrease in this p-Akt form in response to 0.75 µM PI-103 already after 8 h of treatment (Fig. 3A). Rapamycin (0.1 µM) treatment, transiently activated Ser 473 p-Akt at 8 h, in agreement with others (28). The p-Akt levels then decreased but at 48 h they increased again. Total Akt levels were unaffected by either PI-103 or rapamycin. Akt inhibition by PI-103 had functional consequences, because the well established Akt downstream target, GSK3-β, showed a decreased phosphorylation level, whereas the expression of total GSK3-β was unaffected. p-GSK-3β behavior in response to rapamycin was similar to p-Akt in that it increased at 8 h, then decreased and increased again at 48 h. mTOR downstream substrates (p70S6K, 4E-BP1, and S6RP) were efficiently dephosphorylated by both PI-103 and rapamycin, with the exception of 4E-BP1 which was resistant to rapamycin (Fig. 3A). A dose-dependent investigation carried on CEM-R cells demonstrated that PI-103 induced Akt dephosphorylation already at 125 nM, whereas 4E-BP1 dephosphorylation was detectable at 375 nM (Fig. 3B).
Figure 3. Effect of PI-103 and rapamycin on critical components of the PI3K/AKT/mTOR signaling pathway.

A: cells were treated with PI-103 (0.75 µM) or rapamycin (0.1 µM), collected, and then lysed. Fifty micrograms of each lysate were electrophoresed on SDS-PAGE gels. B: Treatment with PI-103 for 8 h induced a dose-dependent dephosphorylation of Akt on Ser473 and of 4E-BP1 in CEM-R cells treated for 8 h. In A and B one representative of three different experiments is shown
Characterization of PI3K/Akt signaling in T-ALL cells
No information is available regarding the class I PI3K catalytic subunits and Akt isoforms which are active in PTEN-negative T-ALL cell lines. This issue is relevant to cancer therapy, as Akt isoform specific inhibitors are actively sought (29). Moreover, inhibitors selective for class I PI3K catalytic subunits are being tested in clinical trials (30). Immunoprecipitation experiments were performed on extracts from T-ALL cells using antibodies specific for either Akt1 or Akt2. The immunoprecipitates were blotted to nitrocellulose membranes which were then probed with the antibody used for the immunoprecipitation, or an antibody which recognizes both Akt1 or Akt2 when phosphorylated on Ser 473 and Ser 474, respectively. We found that T-ALL cell lines expressed both Akt1 and Akt2 which were phosphorylated (Fig. 4A). As a control, the blots with the Akt1 immunoprecipitates were probed with the antibody to Akt2, and vice versa. The antibody to Akt2 picked up some bands in the Akt1 blots, however, the molecular weight of these bands did not correspond to Akt1 molecular weight (60-kDa). In contrast, the antibody to Akt1, did not pick up any bands in the Akt2 blots (Fig. 4A).
Figure 4. Expression of Akt1 and Akt2 and involvement of PI3K isoforms in T-ALL cell survival.

A: Immunoprecipitation analysis. Whole cell extracts were incubated with either Akt1 or Akt2 antibody followed by protein A/G-agarose immunoprecipitation. The immunoprecipitates (IP) were probed with Ab (WB) to Akt1, to p-Akt (which recognizes both Ser 473 p-Akt1 and Ser 474 p-Akt2), and Akt2. B: T-ALL cell lines expressed p110α PI3K, p110β PI3K, p110γ PI3K, and p110δ PI3K, as documented by western blots. MTT assays of T-ALL cell lines, performed after 24 h treatment, demonstrated that only a p110α PI3K inhibitor (compound 15e) displayed a marked dose-dependent cytotoxicity. Results are the mean of at least three different experiments ± SD. C: Western blot analysis demonstrating that only compound 15e resulted in a marked dose-dependent downregulation of p-Akt in both Jurkat and CEM-R cells. D: Immunoprecipitation analysis performed on Jurkat cells documented that compound 15e and PI-103 dephosphorylated both Akt1 and Akt2. Experimental design is as in Panel A. Input demonstrates that the same amount of protein was present in lysates prior to immunoprecipitation.
T-ALL cell lines expressed p110α, p110β, p110γ, and p110δ PI3K, as demonstrated by western blot analysis (Fig. 4B). We next examined the effects of p110 PI3K isoform selective inhibitors, to establish which isoforms were involved in T-ALL cell survival. We employed compound 15e, which inhibits p110α PI3K with an IC50 of 0.58 µM (31); TGX-221, which displays an IC50 of around 40 nM for p110β PI3K (32); and 5-(2,2--Difluoro-benzo[1,3]dioxol-5-ylmethylene)-thiazolidine-2,4-dione, which has an IC50 of approximately 250 nM against p110γ PI3K (33). MTT assays were performed after 24 h of treatment. A dose-dependent decrease in cell survival was obtained with the p110α PI3K selective inhibitor. In contrast, inhibitors of β and γ p110 PI3K had either much lower (β) or no (γ) effects (Fig. 4B). No effects were observed with a p110 δ PI3K-selective inhibitor (data not shown) (32). Consistently, inhibition of Ser 473 p-Akt phosphorylation was maximally observed with the α inhibitor, and to a much lower extent with β inhibitor, while the γinhibitor actually increased Akt phosphorylation when used at 2 µM (Fig. 4C). The p110δ PI3K inhibitor was ineffective in dephosphorylating Akt (not shown). Immunoprecipitation experiments documented that the PI3K p110α inhibitor dephosphorylated both Akt1 and Akt2 in Jurkat cells. Also PI-103 dephosphorylated both Akt isoforms (Fig. 4D). However, when the p110α PI3K selective inhibitor was combined with rapamycin, the growth inhibition effect was not as strong as with PI-103. An allosteric Akt 1/2 inhibitor (34) was less effective than PI-103, when employed at equimolar concentrations (Supplementary Fig. 3A–B). Also, this inhibitor was much less cytotoxic than PI-103 when employed against CEM-R cells. Overall, these experiments demonstrated that T-ALL cell lines display both p-Akt1 and p-Akt2 expression and that p110α PI3K is the most important isoform for their growth.
PI-103 synergizes with vincristine
It was investigated whether PI-103 could synergize with vincristine, a drug commonly used for treating T-ALL patients (35). Previous reports have indicated that mTOR activation specifically confers resistance to chemotherapeutic drugs which target the microtubules, including vincristine (36). T-ALL cell lines were incubated for 24 h with vincristine alone, PI-103 alone, or the drugs together at a fixed ratio (PI-103:vincristine, 25:1). MTT assays were then performed. The combined treatment was much more cytotoxic than either of the two drugs alone (Fig. 5A). Analysis of the CI, documented the existence of a strong synergism (CI<0.3) at PI-103 concentrations which were well below the IC50 (Fig. 5B).
Figure 5. A PI-103 and vincristine combination is synergistic in MOLT-4, JURKAT, and CEM-S cell lines.

A: Cells were cultured in the presence of vincristine (VCR) and PI-103 alone, or in combination, at a fixed ratio (1:25). The combined treatment was highly effective in inducing cytotoxicity, as demonstrated by MTT assays at 24 h. Results are the mean of at least three different experiments ± SD. B: The combination index value for each data point was calculated with the appropriate software for dose effect analysis (CalcuSyn).
T-ALL blasts are sensitive to PI-103
To better assess the effectiveness of PI-103 as a potential therapeutic agent in T-ALL, we examined pediatric T-ALL patient samples isolated from bone marrow or peripheral blood, for the levels of Ser 473 p-Akt, and Thr 37/46 p-4E-BP1, as well as for their sensitivity to PI-103, using flow cytometry and MTT assays. The clinical features of T-ALL patients are presented in Supplementary Table 1. All patient samples (7/7) had higher levels of p-Akt and p-4E-BP1 than those detected in peripheral blood lymphocytes from healthy donors (Fig. 6A and data not shown). Flow cytometric analysis documented a decrease in the levels of Ser 473 p-Akt in samples treated with PI-103 (Fig. 6B). The same technique also demonstrated increased levels of cleaved caspase-3 after treatment with PI-103, which were higher than in samples exposed to rapamycin (Fig. 6C). In order to determine the susceptibility of T-ALL lymphoblasts to inhibition of PI3K/Akt/mTOR signaling, the samples were treated with increasing concentrations of PI-103 for 96 h, and cell survival was analyzed by MTT assays. A strong reduction of cell viability at 96 h was detected. The IC50 for patient samples ranged between 0.18 and 0.63 µM PI-103. Overall, these findings demonstrated that PI-103 has a potent cytotoxic activity and activated caspase-3 also in primary cells from T-ALL patients with upregulated PI3K/Akt/mTOR signaling.
Figure 6. PI-103 is cytotoxic to primary lymphoblasts from pediatric patients with T-ALL, displaying constitutive phosphorylation of AKT and 4E-BP1 and induces caspase-3 activation.

A: Samples from T-ALL patients were analyzed by flow cytometry for the levels of Ser 473 p-Akt and Thr 37/46 p-4E-BP1. Four representative patients are shown. Black histograms: negative control (irrelevant Ab); grey histograms: cells positive for the specific Ab. B: Patient samples were analyzed by flow cytometry for the levels of Ser 473 p-Akt prior to (CTRL) and after treatment with PI-103 (0.5 µM for 72 h). Black histograms: negative control (irrelevant Ab); grey histograms: cells positive for the specific Ab. C: Patient samples were treated with 0.1 µM rapamycin or 0.5 µM PI-103 or DMSO for 72 h, then analyzed by flow cytometry for cleaved caspase-3. Black histograms: DMSO-treated samples; grey histograms: drug-treated samples. D: MTT assay of TALL blasts treated with PI-103 for 96 h. Results are the mean of at least two different experiments ± SD, and asterisk indicates statistically significant difference (p<0.0001) with respect to untreated cells. Four representative patients are shown.
DISCUSSION
The PI3K/Akt/mTOR pathway is a recently identified potential target for therapeutic intervention in T-ALL. Here, we have demonstrated that PI-103 has a strong cytotoxic activity against T-ALL cell lines and lymphoblasts derived from T-ALL patients. So far, the antiproliferative effects of PI-103 have been mainly documented in glioblastomas (18) and in acute myeloid leukemia (AML) cells (37, 38). PI-103 cytototoxicity in T-ALL cell lines was independent of the p53 gene status. It should be emphasized that p53 mutations are associated with a much worse prognosis in T-ALL patients (39). Our findings are different to those reported for AML cell lines, where the modest cytotoxic effect of PI-103 could be enhanced by the murine double minute-2 inhibitor, Nutlin-3, in a wild-type p53-dependent fashion (38). In this respect, our results are in agreement with those reported for glioblastoma cell lines, where PI-103 was cytototoxic independently of the p53 gene status (18).
Moreover, PI-103 was effective also against a CEM cell subclone, CEM-R, which overexpresses high levels of 170-kDa P-gp, one of the main determinants of drug resistance. This observation is relevant, as 170 kDa P-gp is detected in about 24% of T-ALL patients and negatively correlates with the achievement of a complete remission (40, 41). In T-ALL cell lines, PI-103 induced both G0/G1 phase cell cycle arrest and apoptosis, which was characterized by activation of caspase-9 and -3. In contrast, in AML cell lines, PI-103 was mainly cytostatic and did not induce apoptosis or only had a modest apoptogenic activity (37, 38). PI-103 was capable of dephosphorylating Akt and its downstream target, GSK-3β, as well as mTOR downstream targets. PI-103 strongly synergized with vincristine, a drug used in treating T-ALL patients. Remarkably, PI-103 displayed cytotoxic activity against lymphoblasts from patients with pediatric T-ALL characterized by enhanced levels of p-Akt and p-4E-BP1. Also in patient samples, PI-103 dephosphorylated Akt and activated caspase-3.
In our hands, PI-103 was more effective than rapamycin alone in negatively affecting growth of T-ALL cells, even when used on an equimolar basis. PI-103 was also more effective than Wortmannin, an irreversible PI3K inhibitor. LY294002, a reversible PI3K inhibitor, displayed a potency which approached that of PI-103 only when used at extremely high concentrations.
Since rapamycin and its analogues only target mTORC1 and not mTORC2 (which is responsible for Ser 473 p-Akt phosphorylation (17)), it has been demonstrated that the use of these inhibitors could indeed result in Akt hyperactivation (28). Consistently, we have observed that rapamycin treatment of T-ALL cell lines resulted in Akt hyperactivation at 8 h and 48 h. Also the Akt substrate GSK-3β displayed hyperphosphorylation at the same times, implying an activation of Akt by rapamycin. In contrast, PI-103, which has been documented to inhibit both mTORC1 and mTORC2 activity (18), did not hyperactivate Akt in T-ALL cells, even at later incubation times and this might be related to mTORC2 inhibition. A previous report highlighted that in Jurkat cells prolonged treatment with rapamycin inhibited mTORC2 at 24 h (42). However, that study did not examine later times. At present, we do not know if the hyperphosphorylation of Akt we observed after 48 h treatment with rapamycin was due to a low stability of the drug in vitro or to other reason(s). Interestingly, PI-103, but not rapamycin, dephosphorylated 4E-BP1. This might be due to the fact that, in T-ALL cells, 4E-BP1 is downstream of PIM kinases and not of mTORC1. This would not be unprecedented, as it has been recently demonstrated to occur in AML cells (43). Therefore, we could hypothesize that PI-103, by blocking PI3K/Akt, would result also in PIM kinase downregulation, as PIM kinases have been reported to be downstream of PI3K/Akt (44). Lack of 4E-BP1 dephosphorylation by rapamycin, could also explain why the drug is less efficacious than PI-103 against T-ALL cell lines.
We also have demonstrated for the first time that T-ALL cell lines express Akt1 and Akt2. Both of these isoforms were constitutively phosphorylated and PI-103 downregulated their phosphorylation levels. Another new finding emerging from our study is that in T-ALL cell lines, p110α PI3K seems to be the most important isoform for cell growth and Akt1/Akt2 phosphorylation. PI-103 was originally described as a compound specifically targeting p110α PI3K (18), even though subsequent investigations have highlighted that it also inhibited other class IA PI3Ks (37). However, a combination of rapamycin plus the p110α PI3K inhibitor, 15e, was less effective than PI-103. Also, an Akt1/Akt2 allosteric inhibitor was less effective than PI-103.
In conclusion, we have provided evidence that PI-103, either alone or in combination with vincristine, is a highly effective drug against T-ALL cells, also derived from patients. It displayed cytotoxic activity against cells with activating Notch-1 mutations, such as Jurkat and MOLT-4, which are resistant to GSI (26).
PI-103 has proven efficacy also in vivo against models of human tumors xenografted in mice, where it displayed low toxicity and was well tolerated (18, 28). Interestingly, in a mouse model of Bcr-Abl+ B-ALL, PI-103 was less effective than rapamycin (45). This could reflect important biological differences between T-ALL and Bcr-Abl+ B-ALL which, however, remain to be elucidated. In any case, hypersensitivity of Bcr-Abl+ B-ALL cell lines to rapamycin has been recently reported also by another group (46).
PI-103 did not induce apoptosis in normal human CD34+ cells and had moderate effects on their clonogenicity and proliferation (37). This could indicate that dual targeting of PI3K/mTOR signaling would not detrimentally affect normal hematopoiesis in humans.
PI-103 did not enter clinical trials mainly because of its rapid in vivo metabolism (47). Nevertheless, we feel that our findings strongly support the hypothesis that longitudinal inhibition at two nodes of the PI3K/Akt/mTOR pathway in T-ALL cells could ensue in more effective results than the use of a single inhibitor targeting either PI3K or mTOR.
Recently, a new orally available dual PI3K/mTOR inhibitor, NVP-BEZ235, has entered phase I clinical trials (48). It will be interesting to test if NVP-BEZ235 is as effective as PI-103 against T-ALL cells. These investigations could pave the way for using dual PI3K/mTOR inhibitors to improve the therapeutic outcome of T-ALL patients displaying activation of PI3K/Akt/mTOR signaling.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by grants from: Fondazione CARISBO and Progetti Strategici Università di Bologna EF2006 (to A.M.M.), National Institutes of Health (USA) (RO1CA091025 to J.A.M.).
REFERENCES
- 1.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708–722. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- 3.Steelman LS, Abrams SL, Whelan J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 4.Jiang BH, Liu LZ. Role of mTOR in anticancer drug resistance: perspectives for improved drug treatment. Drug Resist Updat. 2008;11:63–76. doi: 10.1016/j.drup.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 6.Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6:347–359. doi: 10.1038/nrc1880. [DOI] [PubMed] [Google Scholar]
- 7.Demarest RM, Ratti F, Capobianco AJ. It's T-ALL about Notch. Oncogene. 2008;27:5082–5091. doi: 10.1038/onc.2008.222. [DOI] [PubMed] [Google Scholar]
- 8.Palomero T, McKenna K, O-Neil J, et al. Activating mutations in NOTCH1 in acute myeloid leukemia and lineage switch leukemias. Leukemia. 2006;20:1963–1966. doi: 10.1038/sj.leu.2404409. [DOI] [PubMed] [Google Scholar]
- 9.Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res. 2008;14:5314–5317. doi: 10.1158/1078-0432.CCR-07-4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calzavara E, Chiaramonte R, Cesana D, Basile A, Sherbet GV, Comi P. Reciprocal regulation of Notch and PI3K/Akt signalling in T-ALL cells in vitro. J Cell Biochem. 2008;103:1405–1412. doi: 10.1002/jcb.21527. [DOI] [PubMed] [Google Scholar]
- 11.Weerkamp F, van Dongen JJ, Staal FJ. Notch and Wnt signaling in T-lymphocyte development and acute lymphoblastic leukemia. Leukemia. 2006;20:1197–1205. doi: 10.1038/sj.leu.2404255. [DOI] [PubMed] [Google Scholar]
- 12.Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118:3762–3774. doi: 10.1172/JCI34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown VI, Seif AE, Reid GS, Teachey DT, Grupp SA. Novel molecular and cellular therapeutic targets in acute lymphoblastic leukemia and lymphoproliferative disease. Immunol Res. 2008;42:84–105. doi: 10.1007/s12026-008-8038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radhakrishnan SK, Halasi M, Bhat UG, Kurmasheva RT, Houghton PJ, Gartel AL. Proapoptotic compound ARC targets Akt and N-myc in neuroblastoma cells. Oncogene. 2008;27:694–699. doi: 10.1038/sj.onc.1210692. [DOI] [PubMed] [Google Scholar]
- 16.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–6446. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 17.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3KAKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmcol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood. 2007;110:278–286. doi: 10.1182/blood-2006-08-039883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiarini F, Del Sole M, Mongiorgi S, et al. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22:1106–1116. doi: 10.1038/leu.2008.79. [DOI] [PubMed] [Google Scholar]
- 21.Papa V, Tazzari PL, Chiarini F, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008;22:147–160. doi: 10.1038/sj.leu.2404980. [DOI] [PubMed] [Google Scholar]
- 22.Fala F, Blalock WL, Tazzari PL, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor (2S)-1-(1H-Indol-3-yl)-3-[5-(3-methyl-2H-indazol-5-yl)pyridin-3-yl]oxyprop an2-amine (A443654) in T-cell acute lymphoblastic leukemia. Mol Pharmacol. 2008;74:884–895. doi: 10.1124/mol.108.047639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tazzari PL, Cappellini A, Ricci F, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia. 2007;21:427–438. doi: 10.1038/sj.leu.2404523. [DOI] [PubMed] [Google Scholar]
- 24.Tazzari PL, Tabellini G, Bortul R, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007;21:886–896. doi: 10.1038/sj.leu.2404643. [DOI] [PubMed] [Google Scholar]
- 25.Gong B, Almasan A. Apo2 ligand/TNF-related apoptosis-inducing ligand and death receptor 5 mediate the apoptotic signaling induced by ionizing radiation in leukemic cells. Cancer Res. 2000;60:5754–5760. [PubMed] [Google Scholar]
- 26.De Keersmaecker K, Lahortiga I, Mentens N, et al. In vitro validation of γ-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica. 2008;93:533–542. doi: 10.3324/haematol.11894. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 28.Chaisuparat R, Hu J, Jham BC, Knight ZA, Shokat KM, Montaner S. Dual inhibition of PI3Kalpha and mTOR as an alternative treatment for Kaposi's sarcoma. Cancer Res. 2008;68:8361–8368. doi: 10.1158/0008-5472.CAN-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 30.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–5496. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayakawa M, Kaizawa H, Moritomo H, et al. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2006;14:6847–6858. doi: 10.1016/j.bmc.2006.06.046. [DOI] [PubMed] [Google Scholar]
- 32.Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008;27:2561–2574. doi: 10.1038/sj.onc.1210918. [DOI] [PubMed] [Google Scholar]
- 33.Bilancio A, Okkenhaug K, Camps M, et al. Key role of the p110δisoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110δ function in B cells. Blood. 2006;107:642–650. doi: 10.1182/blood-2005-07-3041. [DOI] [PubMed] [Google Scholar]
- 34.DeFeo-Jones D, Barnett SF, Fu S, et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther. 2005;4:271–279. [PubMed] [Google Scholar]
- 35.Kantarjian H, Thomas D, O'Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101:2788–2801. doi: 10.1002/cncr.20668. [DOI] [PubMed] [Google Scholar]
- 36.VanderWeele DJ, Zhou R, Rudin CM. Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther. 2004;3:1605–1613. [PubMed] [Google Scholar]
- 37.Park S, Chapuis N, Bardet V, et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia. 2008;22:1698–1706. doi: 10.1038/leu.2008.144. [DOI] [PubMed] [Google Scholar]
- 38.Kojima K, Shimanuki M, Shikami M, et al. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia. 2008;22:1728–1736. doi: 10.1038/leu.2008.158. [DOI] [PubMed] [Google Scholar]
- 39.Diccianni MB, Yu J, Hsiao M, Mukherjee S, Shao LE, Yu AL. Clinical significance of p53 mutations in relapsed T-cell acute lymphoblastic leukemia. Blood. 1994;84:3105–3112. [PubMed] [Google Scholar]
- 40.Tafuri A, Gregorj C, Petrucci MT, et al. MDR1 protein expression is an independent predictor of complete remission in newly diagnosed adult acute lymphoblastic leukemia. Blood. 2002;100:974–981. doi: 10.1182/blood-2001-12-0371. [DOI] [PubMed] [Google Scholar]
- 41.Vitale A, Guarini A, Ariola C, et al. Adult T-cell acute lymphoblastic leukemia: biologic profile at presentation and correlation with response to induction treatment in patients enrolled in the GIMEMA LAL 0496 protocol. Blood. 2006;107:473–479. doi: 10.1182/blood-2005-04-1754. [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Tamburini J, Green AS, Bardet V, et al. Protein sythesis escapes mTORC1 control and constitutes a promising therapeutic target in acute myeloid leukemia. Blood. 2008;112:934. doi: 10.1182/blood-2008-10-184515. (abstract). [DOI] [PubMed] [Google Scholar]
- 44.Muraski JA, Rota M, Misao Y, et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13:1467–1475. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 45.Kharas MG, Janes MR, Scarfone VM, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008;118:3038–3050. doi: 10.1172/JCI33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirase C, Maeda Y, Takai S, Kanamaru A. Hypersensitivity of Ph-positive lymphoid cell lines to rapamycin: Possible clinical application of mTOR inhibitor. Leuk Res. 2009;33:450–459. doi: 10.1016/j.leukres.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 47.Raynaud FI, Eccles S, Clarke PA, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 48.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.