

Abstract
Recent studies demonstrate that mitochondrial dysfunction is a mediator of acute kidney injury (AKI). Consequently, restoration of mitochondrial function after AKI may be key to the recovery of renal function. Mitochondrial function can be restored through the generation of new, functional mitochondria in a process called mitochondrial biogenesis (MB). Despite its potential therapeutic significance, very few pharmacological agents have been identified to induce MB. To examine the efficacy of phosphodiesterase (PDE) inhibitors (PDE3: cAMP and cGMP activity; and PDE4: cAMP activity) in stimulating MB, primary cultures of renal proximal tubular cells (RPTCs) were treated with a panel of inhibitors for 24 hours. PDE3, but not PDE4, inhibitors increased the FCCP-uncoupled oxygen consumption rate (OCR), a marker of MB. Exposure of RPTCs to the PDE3 inhibitors, cilostamide and trequinsin, for 24 hours increased peroxisome proliferator–activated receptor γ coactivator-1α, and multiple mitochondrial electron transport chain genes. Cilostamide and trequinsin also increased mRNA expression of mitochondrial genes and mitochondrial DNA copy number in mice renal cortex. Consistent with these experiments, 8-Br-cGMP increased FCCP-uncoupled OCR and mitochondrial gene expression, whereas 8-Br-cAMP had no effect. The cGMP-specific PDE5 inhibitor sildenafil also induced MB in RPTCs and in vivo in mouse renal cortex. Treatment of mice with sildenafil after folic acid–induced AKI promoted restoration of MB and renal recovery. These data provide strong evidence that specific PDE inhibitors that increase cGMP are inducers of MB in vitro and in vivo, and suggest their potential efficacy in AKI and other diseases characterized by mitochondrial dysfunction and suppressed MB.
Introduction
Mitochondrial dysfunction is increasingly recognized as an important pathophysiological mediator of a variety of disease states, including neurodegeneration, cardiovascular disease, metabolic syndrome, and acute organ injury (Choumar et al., 2011; Pundik et al., 2012; Andreux et al., 2013; Bayeva et al., 2013; Cheng and Ristow, 2013; Cooper, 2013; Hwang, 2013; Yan et al., 2013). Mitochondrial dysfunction is an established component of the pathogenesis of acute kidney injury (AKI) and a cause of renal tubular dysfunction and cell death (Jassem et al., 2002; Jassem and Heaton, 2004; Hall and Unwin, 2007; Weinberg, 2011; Venkatachalam and Weinberg, 2012). Our group has demonstrated persistent disruption of mitochondrial homeostasis and inhibition of mitochondrial biogenesis (MB) after ischemia-reperfusion (I/R), rhabdomyolysis-induced AKI (Funk and Schnellmann, 2012), and folic acid (FA)–induced AKI (unpublished data). Restoration of mitochondrial number and function is thought to be required for recovery from AKI due to the high energy requirements of tissue repair. These data provide support for development of pharmacological agents that induce MB for treatment of AKI and other pathologies characterized by mitochondrial dysfunction.
Mitochondria are dynamic organelles that are continuously regenerated through the processes of biogenesis, mitophagy, fission, and fusion (Brooks et al., 2009; Shaw and Winge, 2009; Cho et al., 2010; Funk and Schnellmann, 2012; Kubli and Gustafsson, 2012). MB is the assembly of new mitochondria from existing mitochondria, occurring under basal conditions to replace damaged mitochondria, but is rapidly induced in response to both physiologic and pathophysiological stimuli, including sepsis, exercise, fasting, hypoxia, and cellular injury (Puigserver and Spiegelman, 2003; Tran et al., 2011; Kang and Li Ji, 2012; Wenz, 2013). The primary regulator of MB is the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). PGC-1α exerts its functions by activating the transcription factors, nuclear respiratory factors 1 and 2 (Nrf1 and Nrf2). Nrf1 controls the expression of mitochondrial transcription factor A (Tfam), which regulates the transcription of mitochondrial DNA (mtDNA) (Puigserver et al., 1998; Wu et al., 1999; Scarpulla, 2008; Scarpulla et al., 2012). PGC-1α is enriched in tissues with high metabolic demand, including heart, muscle, and kidneys (Liang and Ward, 2006). The ability of PGC-1α to respond to a variety of stimuli and its importance in cellular bioenergetics make it an ideal target for pharmacological intervention in disease states characterized by mitochondrial disruption.
Despite the promise of PGC-1α and MB as a therapeutic target, there is a paucity of pharmacological agents capable of stimulating PGC-1α expression and activity. Activators of silent mating type information regulation 2 homolog 1 (SIRT1)—including isoflavones, resveratrol, and N-[2-[3-(piperazin-1-ylmethyl)imidazo[2,1-b][1,3]thiazol-6-yl]phenyl]quinoxaline-2-carboxamide (SRT1720) —have been demonstrated to induce PGC-1α and promote increased mitochondrial number and function (Rasbach and Schnellmann, 2008; Funk et al., 2010; Menzies et al., 2013). Our laboratory also identified the 5-hydroxytryptamine type 2 agonist, 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane hydrochloride (DOI), and the β2-adrenergic receptor agonist, formoterol, as potent inducers of PGC-1α and MB (Rasbach et al., 2010; Wills et al., 2012). Stimulation of MB after injury accelerates recovery of cellular morphology and function (Rasbach and Schnellmann, 2007; Funk et al., 2010; Rasbach et al., 2010). These data demonstrate the importance of MB in recovery of renal tubular epithelial cells after injury and suggest that agents that stimulate MB could serve as viable therapies after AKI.
Because of the importance of the cAMP/protein kinase A (PKA)/cAMP-response element-binding protein (CREB) axis in PGC-1α regulation, drugs that increase cellular cAMP levels may induce MB. The β2-adrenergic signaling cascade, which upon activation increases intracellular cAMP through Gs-mediated activation of adenylyl cyclase, was shown to regulate oxidative metabolism and energy expenditure (Tadaishi et al., 2011; Muller et al., 2013). Formoterol induces MB in renal proximal tubular cells (RPTCs), and mice treated with formoterol demonstrated increased mitochondrial gene expression and mtDNA copy number in renal cortex and heart (Wills et al., 2012). cGMP levels have also been shown to regulate PGC-1α expression and MB. Pharmacologically induced generation of nitric oxide (NO) via endothelial nitric-oxide synthase (eNOS) and subsequent NO-dependent activation of guanylyl cyclase led to MB in U937, L6, and PC12 cells. (Nisoli et al., 2004).
Both cAMP and cGMP levels are tightly regulated through cleavage to AMP and GMP, respectively, by a class of enzymes called cyclic nucleotide phosphodiesterases (PDEs). The PDE superfamily consists of 11 families differing in tissue distribution, regulation, and substrate affinity (e.g., cAMP versus cGMP) (Francis et al., 2011). Potent, selective inhibitors of nearly all family members are available (Bender and Beavo, 2006). Inhibition of PDEs would serve as a novel and potentially efficacious drug target to induce MB. As such, we studied inhibitors of PDE3, PDE4, and PDE5 for their ability to induce MB in the kidney and promote recovery from FA-induced AKI.
Materials and Methods
Reagents.
Cilostamide, trequinsin, (R)-(−)-rolipram, 4-(3-butoxy-4-methoxyphenyl)methyl-2-imidazolidone (Ro 20-1724), sildenafil, 8-Br-cAMP, and 8-Br-cGMP were purchased from Tocris Bioscience (Ellisville, MO). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Animal Care and Use.
Studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina and all efforts were made to minimize animal suffering.
Isolation and Culture of Proximal Tubules.
Female New Zealand white rabbits (1.5–2.0 kg) were purchased from Charles River Laboratories (Wilmington, MA). RPTCs were isolated using the iron oxide perfusion method previously described (Nowak and Schnellmann, 1995). For respirometry experiments, cells were plated on 100-mm culture-grade Petri dishes at 37°C in a 5% CO2/95% air environment. Dishes were continuously swirled on an orbital shaker at 80 rpm. Cell culture media consisted of a 1:1 mixture of Dulbecco’s modified Eagle’s essential medium and Ham’s F-12 (lacking glucose, phenol red, and sodium pyruvate; Invitrogen, Carlsbad, CA), supplemented with HEPES (15 mM), glutamine (2.5 mM), pyridoxine HCl (1 μM), sodium bicarbonate (15 mM), and lactate (6 mM). Hydrocortisone (50 nM), selenium (5 ng/ml), human transferrin (5 μg/ml), bovine insulin (10 nM), and l-ascorbic acid-2-phosphate (50 μM) were added daily to fresh culture media. After 3 days of culture, dedifferentiated RPTCs were trypsinized and replated on XF-96 polystyrene cell culture microplates (Seahorse Bioscience, North Billerica, MA) at a concentration of 18,000 cells per well. Cells were maintained at 37°C for an additional 2 days before experimentation (Beeson et al., 2010). For all other RPTC experiments, cells were plated and cultured in 35-mm dishes in the above-described media. Experiments were performed on the sixth day after plating when cells had formed a confluent monolayer. RPTCs were treated with various compounds for 24 hours unless otherwise noted.
Oxygen Consumption.
The oxygen consumption rate (OCR) of RPTCs was measured using the Seahorse Bioscience XF-96 Extracellular Flux Analyzer according to a previously described protocol (Beeson et al., 2010). Each assay plate was treated with vehicle control (dimethylsulfoxide <0.5%), and increasing concentrations of the compounds of interest. Basal OCR was measured before injection of carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (0.5 μM), allowing for the measurement of uncoupled OCR.
Testing of Compounds in C57BL/6 Mice.
Male C57BL/6 mice (aged 6–8 weeks) were obtained from the National Institutes of Health National Cancer Institute (Bethesda, MD). Mice were housed individually in a temperature-controlled room under a 12-hour light/dark cycle. Mice were randomly assigned to saline control, cilostamide (0.3 or 3 mg/kg), trequinsin (0.3 or 3 mg/kg), or sildenafil (0.3 or 3 mg/kg) treatment groups. Mice were given a single intraperitoneal injection of saline or compound at the above-described doses. Mice were euthanized by CO2 asphyxiation followed by cervical dislocation 24 hours after treatment. Kidneys were removed and preserved by flash-freezing in liquid nitrogen. Tissues were stored at −80°C for later analysis.
FA Animal Model.
Male C57BL/6 mice (aged 8–10 weeks) were given a single intraperitoneal injection of 250 mg/kg FA dissolved in 250 mM sodium bicarbonate or saline control based on previous literature (Doi et al., 2006). Mice were injected with sildenafil (0.3 mg/kg) or diluent every 24 hours beginning 1 day after FA injection. Mice were euthanized at 7 days via isoflurane asphyxiation and cervical dislocation. Kidneys were removed and preserved by flash-freezing.
Quantitative Real-Time Polymerase Chain Reaction.
Total RNA was extracted from RPTCs or renal cortex samples using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized via reverse transcription using the iScript Advanced cDNA synthesis kit (Bio-Rad, Hercules, CA) with 2 μg of RNA. Quantitative real-time polymerase chain reaction (qPCR) analysis was performed with cDNA using SsoAdvanced SYBR Green Supermix (Bio-Rad) at a concentration of 1× and primers at a concentration of 750 nM (Integrated DNA Technologies, Coralville, IA). mRNA expression of all genes was calculated using the 2-ΔΔCT method normalized to tubulin in RPTCs or β-actin in renal cortical tissue. Primer sequences for PGC-1α, NADH dehydrogenase 6 (ND6), NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 8 (NDUFβ8), and tubulin were described previously (Funk and Schnellmann, 2012). Primer sequences for NADH dehydrogenase 1 (ND1) and β-actin were as follows: ND1, sense 5′-TAGAACGCAAAATCTTAGGG-3′ and antisense 5′-TGCTAGTGTGAGTGATAGGG-3′; and β-actin, sense 5′- GGGATGTTTGCTCCAACCAA-3′ and antisense 5′-GCGCTTTTGACTCAAGGATTTAA-3′.
mtDNA Content.
Real-time PCR was used to determine the relative quantity of mtDNA in both RPTC and mouse renal cortical tissue samples. After treatment, DNA was extracted from cells or tissue using the DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA) and 5 ng of cellular DNA was used for qPCR. For RPTC samples, mitochondrial-encoded ND6 was used to measure mitochondrial copy number and was normalized to nuclear-encoded tubulin expression. For renal cortex, ND1 was used as the mitochondrial gene and expression was normalized to nuclear-encoded β-actin expression.
cAMP and cGMP Enzyme-Linked Immunosorbent Assay.
RPTCs in 35-mm dishes were treated with vehicle control (dimethylsulfoxide) or the compound of interest for 20 minutes. RPTCs were then harvested according to the manufacturer’s protocol and cAMP or cGMP levels were measured using a commercially available enzyme-linked immunosorbent assay kit (Cayman Chemical, Ann Arbor, MI).
Tissue ATP Levels.
ATP was isolated from renal cortical tissue via phenol-Tris-EDTA extraction as previously described (Chida et al., 2012). In brief, freshly prepared tissue was homogenized in 3.0 ml ice-cold Tris-EDTA saturated phenol. One milliliter of the homogenate was combined with 200 μl chloroform and 150 μl of deionized water and vortexed and centrifuged at 10,000g for 5 minutes at 4°C. An aliquot from the supernatant was diluted 200-fold in deionized water, and ATP levels were measured using a luciferin-luciferase–based ATP determination kit (Invitrogen).
Statistical Analysis.
Data are presented as the mean ± S.E.M. Single comparisons were performed using the t test. Multiple comparisons were subjected to one-way analysis of variance followed by the Newman–Keuls test, with P < 0.05 considered to be a statistically significant difference between means. RPTCs isolated from a single rabbit represented an individual experiment (n = 1) and were repeated until n ≥ 4 was obtained. Mouse studies were repeated until n ≥ 3 was obtained.
Results
PDE3 Inhibitors, but not PDE4 Inhibitors, Increase FCCP-Uncoupled OCR in RPTCs.
We treated RPTCs in XF-96 culture plates with the PDE3 inhibitors cilostamide or trequinsin, the PDE4 inhibitors (R)-(−)-rolipram or Ro 20-1724, or vehicle control for 24 hours. PDE3 hydrolyzes both cAMP and cGMP to their noncyclic forms, AMP and GMP, whereas PDE4 specifically hydrolyzes cAMP to AMP (Francis et al., 2011). FCCP-OCR increased in RPTCs compared with vehicle control after 24-hour exposure to cilostamide (25–100 nM) and trequinsin (30–100 nM) (Fig. 1, A and B), but no significant changes were observed in FCCP-OCR after treatment with (R)-(−)-rolipram (0.5–50 µM) or Ro 20-1724 (5–20 µM) (Fig. 1, C and D). These data suggest a functional selectivity for the MB response between PDE3 and PDE4 inhibition in RPTCs.
Fig. 1.
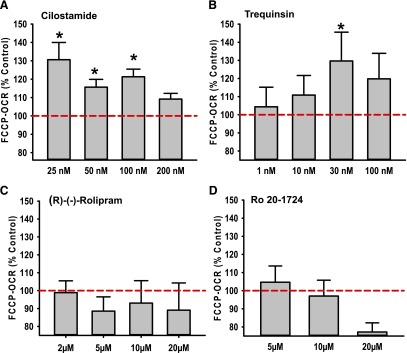
PDE3 inhibitors, but not PDE4 inhibitors, increase FCCP-induced uncoupled mitochondrial respiration in RPTCs. RPTCs were treated with cilostamide (A), trequinsin (B), (R)-(−)-rolipram (C), or Ro 20-1724 (D) for 24 hours. FCCP-uncoupled mitochondrial respiration was measured using the Seahorse XF-96 instrument. Data are presented as the mean ± S.E.M. (n ≥ 3). *P < 0.05 vs. vehicle control.
PDE3 Inhibitors Induce MB in RPTCs.
To validate that the increased FCCP-OCR observed in RPTCs after treatment with PDE3 inhibitors was due to MB, mRNA levels for PGC-1α, the mitochondrial-encoded complex I protein ND6, and the nuclear-encoded complex I protein NDUFβ8 were measured via qPCR. Gene expression was normalized to tubulin. PGC-1α levels increased versus control after treatment with cilostamide (1.8-fold) or trequinsin (2.5-fold) (Fig. 2). In addition, mRNA expression of mitochondrial-encoded ND6 and the nuclear-encoded NDUFβ8 mitochondrial proteins were increased versus control with cilostamide (1.5- and 2.2-fold, respectively) and trequinsin (1.8- and 2.4-fold, respectively). These data provide strong evidence that inhibition of PDE3 causes functional MB in RPTCs.
Fig. 2.

PDE3 inhibitors cilostamide or trequinsin induce mitochondrial protein gene expression in RPTCs. RPTCs were exposed to cilostamide (25 nM) or trequinsin (30 nM) for 24 hours and evaluated for changes in mRNA expression of PGC-1α, ND6, and NDUFβ8 relative to dimethylsulfoxide controls. Data are presented as the mean ± S.E.M. (n ≥ 4). *P < 0.05 vs. vehicle control.
Increased cGMP, but Not cAMP, Induces MB in RPTCs.
To examine the functional selectivity of PDE3 and PDE4 inhibitors under conditions that induce MB, RPTCs were treated with the PDE3 inhibitors cilostamide and trequinsin, the PDE4 inhibitor (R)-(−)-rolipram, or vehicle control for a period of 20 minutes. Sildenafil, a specific inhibitor of PDE5 (cGMP-specific PDE), was included as a control. Both cAMP and cGMP levels increased in response to cilostamide and trequinsin compared with vehicle control, whereas cAMP only increased in RPTCs treated with rolipram (Fig. 3, A and B). RPTCs treated with sildenafil resulted in increased cGMP, but not cAMP. These data agree with the classic mechanisms of PDE3 (hydrolyzes both cAMP and cGMP), PDE4 (hydrolyzes only cAMP), and PDE5 (hydrolyzes only cGMP) (Bender and Beavo, 2006). The inability of rolipram and other PDE4 inhibitors tested to induce MB suggests that cGMP may be the primary mediator of MB in RPTCs.
Fig. 3.
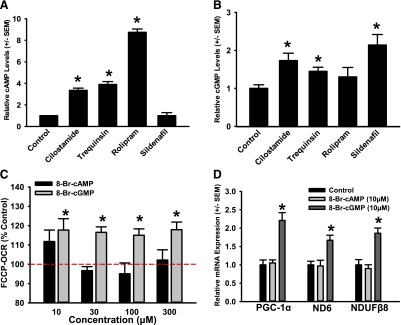
PDE inhibitor–induced increases in cGMP, but not cAMP, stimulate MB in RPTCs. cAMP (A) and cGMP (B) levels were measured in RPTCs by enzyme-linked immunosorbent assay 20 minutes after treatment with dimethylsulfoxide, cilostamide (25 nM), trequinsin (30 nM), rolipram (0.5 μM), or sildenafil (10 nM). (C) FCCP-uncoupled mitochondrial respiration was measured using the Seahorse XF-96 instrument after 24-hour treatment with 8-Br-cAMP or 8-Br-cGMP. (D) RPTCs were exposed to 8-Br-cAMP (10 μM) or 8-Br-cGMP (10 μM) for 24 hours and evaluated for changes in mRNA expression of PGC-1α, ND6, and NDUFβ8 relative to dimethylsulfoxide controls. Data are presented as the mean ± S.E.M. (n ≥ 3). *P < 0.05 vs. vehicle control.
To test this hypothesis, we treated RPTCs with the cell-permeable cyclic nucleotide analogs, 8-Br-cAMP and 8-Br-cGMP for a 24-hour period. RPTCs treated with 8-Br-cGMP (10–300 μM) showed an approximately 20% increase in FCCP-uncoupled OCR at all concentrations tested, whereas treatment with 8-Br-cAMP resulted in no change (Fig. 3C). To validate that this increase in FCCP-OCR is due to stimulation of MB, mRNA expression of PGC-1α, ND6, and NDUFB8 was measured by qPCR. RPTCs treated with 8-Br-cGMP had elevated mRNA levels of PGC-1α (2.2-fold), ND6 (1.7-fold), and NDUFB8 (1.9-fold). 8-Br-cAMP had no effect on mitochondrial gene expression (Fig. 3D). Furthermore, to test the ability of a PDE5 inhibitor to induce MB in vitro, RPTCs were treated with sildenafil for 24 hours (1 nM–1 μM) and FCCP-OCR was measured using the Seahorse XF96. RPTCs treated with sildenafil showed an approximately 20% increase in FCCP-uncoupled OCR versus controls (Fig. 4A) at 10 and 100 nM. To validate that the increase in respiration was due to MB, mRNA levels of PGC-1α, ND6, and NDUFB8 were measured and were found to increase 1.8-, 2.0-, and 1.5-fold, respectively (Fig. 4B).
Fig. 4.

The PDE5 inhibitor sildenafil stimulates MB in RPTCs. Sildenafil increases FCCP-uncoupled mitochondrial respiration at various doses (A) and mitochondrial gene expression at 10 nM (B) in RPTCs. mRNA expression of PGC-1α, ND6, and NDUFβ8 is presented as the mean ± S.E.M. of at least three biologic replicates. *P < 0.05 vs. vehicle control.
PDE3 Inhibitors Induce MB in Mouse Renal Cortex.
In kidneys of cilostamide-treated mice, PGC-1α was induced 2- and 2.7-fold at doses of 0.3 and 3 mg/kg, respectively. Trequinsin induced PGC-1α 2.7- and 2.8- fold in the kidney at doses of 0.3 and 3 mg/kg. mRNA expression of the nuclear-encoded mitochondrial genes NDUFB8 and ATPSβ both increased greater than 2-fold in kidneys of mice treated with either cilostamide or trequinsin at 0.3 or 3 mg/kg (Fig. 5, A and B). The mitochondrial-encoded mitochondrial genes ND1 and cytochrome c oxidase subunit I (COX1) increased in the kidneys of these mice. The mtDNA copy number was also increased in the kidneys of mice treated with cilostamide at 0.3 mg/kg, whereas mice treated with 3 mg/kg cilostamide had no effect (Fig. 5C). Trequinsin increased mtDNA copy number in the kidneys 1.6- and 2-fold at doses of 0.3 and 3 mg/kg, respectively (Fig. 5D). These data provide strong evidence that pharmacological inhibition of PDE3 induces MB in the kidney of naïve mice.
Fig. 5.
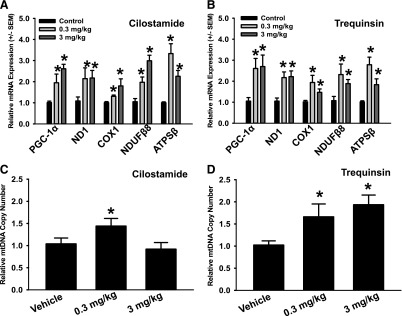
PDE3 inhibitors cilostamide and trequinsin induce mitochondrial gene expression and mtDNA copy number in mouse renal cortex. mRNA expression and mtDNA copy number were evaluated in the renal cortex of mice 24 hours after a single intraperitoneal injection of cilostamide (A and C) or trequinsin (B and D). Values indicate fold change relative to dimethylsulfoxide controls. Data are presented as the mean ± S.E.M. (n ≥ 4). *P < 0.05 vs. vehicle control.
Sildenafil Induces MB in Mouse Renal Cortex.
The selectivity of the MB response for cGMP in RPTCs indicates that inhibitors of cGMP-specific PDEs, such as PDE5, may in fact be a better therapeutic target and could eliminate off-target effects due to the accumulation of cAMP. PDE5 inhibitors also have a much more favorable safety protocol than PDE3 inhibitors, particularly for extended administration (Cruickshank, 1993).
To determine whether PDE5 inhibition is capable of inducing MB in the kidney in vivo, mice were given a single intraperitoneal injection of sildenafil (0.3 or 3 mg/kg) or saline control. Mice were euthanized and kidneys were harvested 24 hours after treatment. mRNA levels of PGC-1α, NDUFB8, ND1, ATPβ, and COX1 were measured by qPCR. All mitochondrial genes, except for COX1 and ATPSβ, were increased in mice treated with 3 mg/kg sildenafil versus saline-treated animals(Fig. 6A). mtDNA copy number was assessed by qPCR in kidneys of sildenafil-treated mice and was found to increase 1.6-fold in mice treated with 0.3 mg/kg sildenafil. No change in mtDNA copy number was observed in mice treated with 3 mg/kg sildenafil (Fig. 6B).
Fig. 6.

Sildenafil induces mitochondrial gene expression, mtDNA copy number, and ATP levels in mouse renal cortex. mRNA expression (A), mtDNA copy number (B), and ATP levels (C) were evaluated in the renal cortex of mice 24 hours after a single intraperitoneal injection of sildenafil. Values indicate fold change relative to dimethylsulfoxide controls. Data are presented as the mean ± S.E.M. (n ≥ 4). *P < 0.05 vs. vehicle control.
To assess whether sildenafil-induced MB increased mitochondrial function in the kidney cortex, we measured ATP levels. ATP levels increased 32% in mice treated with 0.3 mg/kg sildenafil compared with control mice (Fig. 6C). These data strongly support our hypothesis that PDE5 inhibitors induce MB and mitochondrial function in vitro and in vivo.
Sildenafil Promotes Recovery of MB and Renal Function after FA-Induced AKI.
To test the hypothesis that sildenafil-induced MB will accelerate recovery of mitochondrial and renal function after AKI, we induced AKI by injecting FA and then treated these mice with sildenafil or vehicle once daily starting at 24 hours after injury for 6 days. mRNA expression of COX1 and Tfam were reduced to 27 and 36% of control, respectively, in FA-treated mice receiving vehicle control at 6 days. Sildenafil-treated FA mice demonstrated a 1.6-fold increase in mRNA COX1 expression to 43% of control mice, and a 1.4-fold increase in Tfam expression to 50% of control (Fig. 7A). mtDNA copy number was reduced to 36% of animals receiving FA alone, and treatment with sildenafil caused an approximately 2-fold induction to 63% of control (Fig. 7B). These data demonstrate that sildenafil can induce MB in a model of AKI.
Fig. 7.
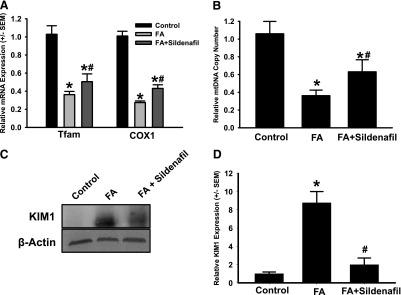
Sildenafil stimulates MB after FA-induced AKI. AKI was induced in C57BL/6 by a single intraperitoneal injection of FA. Mice received daily injections of sildenafil (0.3 mg/kg) or saline vehicle beginning 24 hours after FA. Mice were killed and kidneys were collected 7 days after FA administration. mRNA expression (A) and mtDNA copy number (B) were evaluated in the renal cortex. Immunoblotting was performed for renal cortical assessment of KIM-1 expression (C) and quantified via densitometry (D). Data are presented as the mean ± S.E.M. (n ≥ 3). *P < 0.05 versus vehicle control; #P < 0.05 vs. FA.
To examine whether MB promoted renal recovery, kidney injury molecule-1 (KIM-1), a specific marker of tubular injury, was measured in renal cortex. KIM-1 levels were markedly increased (approximately 6-fold) in FA-treated animals compared with control animals and treatment of FA mice with sildenafil restored KIM-1 expression to control levels (Fig. 7, C and D). These data demonstrate that sildenafil promotes renal recovery with its induction of mitochondrial gene expression and mtDNA copy number.
Discussion
Mitochondria are highly regulated organelles whose function is tightly linked to the metabolic demands and health of a cell (Brooks et al., 2009; Shaw and Winge, 2009; Funk and Schnellmann, 2012; Kubli and Gustafsson, 2012). Mitochondrial function is necessary for normal cell and tissue function, and is critical in energy-dependent repair processes. A wide array of disease states are characterized by mitochondrial dysfunction, including diabetes, neurodegenerative disease, traumatic brain injury, and acute organ injury (Lifshitz et al., 2004; Pundik et al., 2012; Cheng and Ristow, 2013; Hwang, 2013; Yan et al., 2013). I/R and drug/toxicant-induced renal injury demonstrate mitochondrial dysfunction and suppression of MB, and recovery of renal function is tightly linked to the restoration of mitochondrial number and function (Funk and Schnellmann, 2012). This suggests that development of therapies capable of inducing MB may have great potential in the treatment of a broad range of disease states.
Despite strong evidence supporting mitochondria as a therapeutic target, there are very few drugs/chemicals available that promote mitochondrial function or MB. Many of the agents that are available suffer from lack of specificity, low potency, or poor toxicity profiles. There is a clinical need to develop new pharmacological agents or identify existing therapeutics that induce MB. Because of the role of cyclic nucleotides as regulators of PGC-1α, in this study, we sought to determine the efficacy of various classes of PDE inhibitors at stimulating MB.
The cAMP/PKA/CREB signaling cascade is a well characterized regulator of PGC-1α expression and activity (Fernandez-Marcos and Auwerx, 2011). Increases in intracellular cAMP levels cause activation of PKA and subsequent phosphorylation and activation of CREB, an important transcriptional regulator of PGC-1α. Induction of cAMP levels in the cell occurs after activation of various G protein-coupled receptors. Our laboratory recently identified the β2-adrenergic agonist, formoterol, as a potent inducer of MB in the kidney and heart (Wills et al., 2012). β-agonism was previously shown to induce PGC-1α in skeletal muscle of treated mice (Miura et al., 2007). In addition, exercise-induced MB can be blocked by treatment with β-receptor antagonists, propranolol and ICI-118,551. cAMP levels in the cell are controlled both by the rate of synthesis and the rate of turnover by cyclic nucleotide PDEs. Therefore, inhibition of PDEs that hydrolyze cAMP may serve as a viable intervention to induce MB.
To test this hypothesis, we screened a panel of PDE3, PDE4, and PDE5 inhibitors using a phenotypic respirometric assay. FCCP-uncoupled OCR was used as a marker of increased energetic capacity and MB. Interestingly, PDE3 and PDE5 inhibitors increased FCCP-uncoupled OCR in RPTCs, whereas none of the PDE4 inhibitors tested caused an increase (Figs. 1 and 4). To further probe the functional selectivity of PDE3, PDE4, and PDE5 inhibition in promoting MB, cAMP and cGMP levels were measured in RPTCs after treatment with the PDE3 inhibitors cilostamide and trequinsin, the PDE4 inhibitor rolipram, or the PDE5 inhibitor sildenafil. PDE3 inhibition led to increases in levels of both cAMP and cGMP in RPTCs, whereas rolipram increased only cAMP levels and sildenafil increased only cGMP levels (Fig. 3). These data correspond with the classic substrate affinities of the various PDE family members: PDE3 hydrolyzes both cAMP and cGMP with nearly equal affinity, PDE4 specifically hydrolyzes cAMP, and PDE5 specifically hydrolyzes cGMP (Bender and Beavo, 2006; Francis et al., 2011). Finally, 8-Br-cGMP increased FCCP-uncoupled OCR in the respirometric assay and increased mRNA expression of mitochondrial genes after 24-hour treatment. 8-Br-cAMP had no effect on respiration of mitochondrial gene expression in RPTCs. This multipronged approach strongly supports our hypothesis that cGMP, rather than cAMP, is important for regulation of MB in renal tubules.
cGMP was previously demonstrated to induce MB through the eNOS/NO soluble guanylate cyclase/cGMP signaling cascade. Nisoli et al. (2004) showed that long-term administration of NO mimetics, guanylyl cyclase activators, or 8-Br-cGMP increased mRNA expression of mitochondrial genes, mtDNA copy number, mitochondrial respiration, and ATP levels in multiple cell lines. eNOS-deficient mice have a reduction in metabolic rate and accelerated weight gain, which has been correlated with reduced mitochondrial content and function (Nisoli et al., 2003).
Both the PDE3 inhibitors cilostamide or trequinsin (0.3–3 mg/kg) and the PDE5 inhibitor sildenafil (0.3–3 mg/kg) when administered to naïve mice induced renal cortical mRNA expression of PGC-1α, nuclear-encoded mitochondrial genes (NDUFB8 and ATPSβ), and mitochondrial-encoded mitochondrial genes (ND1 and COX1). mtDNA copy number was also increased in the renal cortex of these mice (Figs. 5 and 6). Sildenafil increased the number of functional mitochondria in the renal cortex as evidenced by a significant increase in tissue ATP levels (Fig. 6). These data confirm that PDE3 and PDE5 inhibitors are capable of inducing MB both in vitro in RPTCs and in vivo in mouse kidney.
Cyclic nucleotides including both cAMP and cGMP were shown to be activators of signaling pathways promoting MB in various model systems (Nisoli et al., 2003, 2004; Tadaishi et al., 2011; Muller et al., 2013). Acute ex vivo administration of the PDE5 inhibitor vardenafil to human skeletal muscle stimulated MB as evidenced by increases in mitochondrial gene expression and mtDNA copy number (De Toni et al., 2011). This is the first report of pharmacological induction of MB in vivo by inhibition of either PDE3 or PDE5, and could represent a novel use for these classes of compounds. Despite the evidence of their role in MB, these compounds have yet to be evaluated as potential therapies for mitochondrial damage and dysfunction.
Previous studies reported the ability of various classes of PDE inhibitors to protect against AKI. Pretreatment with rolipram, a specific PDE4 inhibitor, blunted I/R-induced renal dysfunction in rat kidney and reduced oxidative damage (Mammadov et al., 2012). Sildenafil was shown to be protective in cisplatin-induced AKI, whereas tadalafil, a long-acting PDE5 inhibitor, protected against early I/R injury in rats (Lee et al., 2009; Sohotnik et al., 2013). However, limitations of these studies have been the lack of a clear mechanism for the renoprotective effects and the use of pretreatment protocols. To address these issues, we examined the ability of sildenafil to promote recovery from FA-induced AKI by administering the drug 24 hours after induction of injury, and examined the effects of FA and sildenafil on both renal and mitochondrial function. Sildenafil promoted recovery mitochondrial gene expression (i.e., COX1 and Tfam) and mtDNA copy number. In addition, renal KIM-1 expression was reduced in sildenafil-treated mice, indicating an enhanced recovery from the renal injury. These results demonstrate that sildenafil accelerates recovery from AKI by activating MB pathways.
Our results indicate that PDE inhibitors that are capable of increasing tissue levels of cGMP, including sildenafil, are promising treatments for diseases characterized by mitochondrial dysfunction and suppression of MB, including AKI.
Abbreviations
- AKI
acute kidney injury
- ATPSβ
ATP synthase subunit β
- COX1
cytochrome c oxidase subunit 1
- CREB
cAMP-response element-binding protein
- DOI
1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane hydrochloride
- eNOS
endothelial nitric-oxide synthase
- FA
folic acid
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- I/R
ischemia reperfusion
- KIM-1
kidney injury molecule-1
- MB
mitochondrial biogenesis
- mtDNA
mitochondrial DNA
- ND1
NADH dehydrogenase 1
- ND6
NADH dehydrogenase 6
- NDUFβ8
NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 8
- NO
nitric oxide
- Nrf1
nuclear respiratory factor 1
- Nrf2
nuclear respiratory factor 2
- OCR
oxygen consumption rate
- PDE
phosphodiesterase
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator-1α
- PKA
protein kinase A
- qPCR
quantitative real-time polymerase chain reaction
- Ro 20-1724
4-(3-butoxy-4-methoxyphenyl)methyl-2-imidazolidone
- ROS
reactive oxygen species
- RPTC
renal proximal tubular cell
- SIRT1
silent mating type information regulation 2 homolog 1
- SRT1720
N-[2-[3-(piperazin-1-ylmethyl)imidazo[2,1-b][1,3]thiazol-6-yl]phenyl]quinoxaline-2-carboxamide
- Tfam
mitochondrial transcription factor A
Authorship Contributions
Participated in research design: Whitaker, Wills, Stallons, Schnellmann.
Conducted experiments: Whitaker, Stallons, Wills.
Performed data analysis: Whitaker.
Wrote or contributed to the writing of the manuscript: Whitaker, Schnellmann.
Footnotes
This study was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants R01-GM084147 (to R.G.S.) and P20-GM103542-02 (to SC COBRE in Oxidants, Redox Balance, and Stress Signaling)]; the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants F30-DK096964 (to R.M.W.) and F32-DK098053 (to L.J.S.)]; the National Institutes of Health National Heart, Lung, and Blood Institute [Grant T32-HL007260]; the National Institutes of Health National Center for Research Resources [Grant C06-RR015455]; and the Department of Veterans Affairs Biomedical Laboratory Research and Development Program [Grant BX000851]. This publication was supported, in part, by the South Carolina Clinical and Translational Research Institute, with an academic home at the Medical University of South Carolina, and funded by the National Institutes of Health National Center for Research Resources [Grant UL1-RR029882].
This work was previously presented at the following meeting: Whitaker RM, Wills LP, and Schnellmann RG (2012) Phosphodiesterase inhibitors stimulate mitochondrial biogenesis: a potential therapy for AKI. American Society of Nephrology 2012 Kidney Week; 2012 October 30–November 4; San Diego, CA.
References
- Andreux PA, Houtkooper RH, Auwerx J. (2013) Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12:465–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayeva M, Gheorghiade M, Ardehali H. (2013) Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 61:599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeson CC, Beeson GC, Schnellmann RG. (2010) A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem 404:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58:488–520 [DOI] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Cho SG, Dong Z. (2009) Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119:1275–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z and Ristow M (2013) Mitochondria and metabolic homeostasis. Antioxid Redox Signal 19:240–242. [DOI] [PubMed]
- Chida J, Yamane K, Takei T, Kido H. (2012) An efficient extraction method for quantitation of adenosine triphosphate in mammalian tissues and cells. Anal Chim Acta 727:8–12 [DOI] [PubMed] [Google Scholar]
- Cho SG, Du Q, Huang S, Dong Z. (2010) Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am J Physiol Renal Physiol 299:F199–F206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choumar A, Tarhuni A, Lettéron P, Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R, Pessayre D, et al. (2011) Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal 15:2837–2854 [DOI] [PubMed] [Google Scholar]
- Cooper MP. (2013) Interplay of mitochondrial biogenesis and oxidative stress in heart failure. Circulation 127:1932–1934 [DOI] [PubMed] [Google Scholar]
- Cruickshank JM (1993) Phosphodiesterase III inhibitors: long-term risks and short-term benefits. Cardiovasc Drugs Ther 7:655–660. [DOI] [PubMed]
- De Toni L, Strapazzon G, Gianesello L, Caretta N, Pilon C, Bruttocao A, Foresta C. (2011) Effects of type 5-phosphodiesterase inhibition on energy metabolism and mitochondrial biogenesis in human adipose tissue ex vivo. J Endocrinol Invest 34:738–741 [DOI] [PubMed] [Google Scholar]
- Doi K, Okamoto K, Negishi K, Suzuki Y, Nakao A, Fujita T, Toda A, Yokomizo T, Kita Y, Kihara Y, et al. (2006) Attenuation of folic acid-induced renal inflammatory injury in platelet-activating factor receptor-deficient mice. Am J Pathol 168:1413–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Auwerx J. (2011) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93:884S–890S [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Blount MA, Corbin JD. (2011) Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 91:651–690 [DOI] [PubMed] [Google Scholar]
- Funk JA, Odejinmi S, Schnellmann RG. (2010) SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther 333:593–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk JA, Schnellmann RG. (2012) Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302:F853–F864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AM, Unwin RJ. (2007) The not so 'mighty chondrion': emergence of renal diseases due to mitochondrial dysfunction. Nephron Physiol 105:1–10 [DOI] [PubMed] [Google Scholar]
- Hwang O (2013) Role of oxidative stress in Parkinson's disease. Exp Neurobiol 22:11–17. [DOI] [PMC free article] [PubMed]
- Jassem W, Fuggle SV, Rela M, Koo DD, Heaton ND. (2002) The role of mitochondria in ischemia/reperfusion injury. Transplantation 73:493–499 [DOI] [PubMed] [Google Scholar]
- Jassem W, Heaton ND. (2004) The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int 66:514–517 [DOI] [PubMed] [Google Scholar]
- Kang C, Li Ji L. (2012) Role of PGC-1α signaling in skeletal muscle health and disease. Ann N Y Acad Sci 1271:110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Gustafsson AB. (2012) Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 111:1208–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Jeong JY, Lim BJ, Chang YK, Lee SJ, Na KR, Shin YT, Choi DE. (2009) Sildenafil attenuates renal injury in an experimental model of rat cisplatin-induced nephrotoxicity. Toxicology 257:137–143 [DOI] [PubMed] [Google Scholar]
- Liang H, Ward WF. (2006) PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ 30:145–151 [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Sullivan PG, Hovda DA, Wieloch T, McIntosh TK. (2004) Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 4:705–713 [DOI] [PubMed] [Google Scholar]
- Mammadov E, Aridogan IA, Izol V, Acikalin A, Abat D, Tuli A, and Bayazit Y (2012) Protective effects of phosphodiesterase-4-specific inhibitor rolipram on acute ischemia-reperfusion injury in rat kidney. Urology 80:1390.e1391–1396. [DOI] [PubMed]
- Menzies KJ, Singh K, Saleem A, Hood DA. (2013) Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288:6968–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S, Kawanaka K, Kai Y, Tamura M, Goto M, Shiuchi T, Minokoshi Y, Ezaki O. (2007) An increase in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to exercise is mediated by beta-adrenergic receptor activation. Endocrinology 148:3441–3448 [DOI] [PubMed] [Google Scholar]
- Müller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, et al. (2013) p62 links β-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest 123:469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, et al. (2003) Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299:896–899 [DOI] [PubMed] [Google Scholar]
- Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, et al. (2004) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA 101:16507–16512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Schnellmann RG. (1995) Integrative effects of EGF on metabolism and proliferation in renal proximal tubular cells. Am J Physiol 269:C1317–C1325 [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24:78–90 [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839 [DOI] [PubMed] [Google Scholar]
- Pundik S, Xu K, Sundararajan S. (2012) Reperfusion brain injury: focus on cellular bioenergetics. Neurology 79(13, Suppl 1)S44–S51 [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Funk JA, Jayavelu T, Green PT, Schnellmann RG. (2010) 5-hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J Pharmacol Exp Ther 332:632–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. (2007) Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem 282:2355–2362 [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. (2008) Isoflavones promote mitochondrial biogenesis. J Pharmacol Exp Ther 325:536–543 [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638 [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23:459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JM, Winge DR, Conference on Mitochondrial Assembly and Dynamics in Health and Disease (2009) Shaping the mitochondrion: mitochondrial biogenesis, dynamics and dysfunction. EMBO Rep 10:1301–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohotnik R, Nativ O, Abbasi A, Awad H, Frajewicki V, Bishara B, Sukhotnik I, Armaly Z, Aronson D, Heyman SN, et al. (2013) Phosphodiesterase-5 inhibition attenuates early renal ischemia-reperfusion-induced acute kidney injury: assessment by quantitative measurement of urinary NGAL and KIM-1. Am J Physiol Renal Physiol 304:F1099–F1104 [DOI] [PubMed] [Google Scholar]
- Tadaishi M, Miura S, Kai Y, Kawasaki E, Koshinaka K, Kawanaka K, Nagata J, Oishi Y, Ezaki O. (2011) Effect of exercise intensity and AICAR on isoform-specific expressions of murine skeletal muscle PGC-1α mRNA: a role of β₂-adrenergic receptor activation. Am J Physiol Endocrinol Metab 300:E341–E349 [DOI] [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, et al. (2011) PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121:4003–4014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam MA, Weinberg JM. (2012) The tubule pathology of septic acute kidney injury: a neglected area of research comes of age. Kidney Int 81:338–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JM. (2011) Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol 22:431–436 [DOI] [PubMed] [Google Scholar]
- Wenz T. (2013) Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion 13:134–142 [DOI] [PubMed] [Google Scholar]
- Wills LP, Trager RE, Beeson GC, Lindsey CC, Peterson YK, Beeson CC, Schnellmann RG. (2012) The β2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J Pharmacol Exp Ther 342:106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124 [DOI] [PubMed] [Google Scholar]
- Yan MH, Wang X, andZhu X. (2013) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62:90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]