

Abstract
Duloxetine is a serotonin and norepinephrine reuptake inhibitor (SNRI) with central nervous system activity. Its analgesic efficacy in central pain is putatively related to its influence on descending inhibitory pain pathways. The analgesic efficacy of duloxetine has been demonstrated in four distinct chronic pain conditions. These include neuropathic pain associated with diabetic peripheral neuropathy, fibromyalgia, chronic low back pain, and osteoarthritis knee pain (OAKP). The purpose of this review is to examine the clinical efficacy and safety of duloxetine in the management of chronic OAKP. Three separate randomized, double-blind placebo-controlled trials have demonstrated that (1) a clinically meaningful decrease in pain severity occurs at about 4 weeks relative to placebo, (2) patients receiving duloxetine report better improvements in physical functioning relative to placebo, (3) duloxetine is safe and effective when used adjunctively with nonsteroidal anti-inflammatory drugs, and (4) that there are no new safety signals beyond what has been observed in other indications.
Keywords: Chronic pain management, drug interactions, duloxetine, osteoarthritis
Introduction
Osteoarthritis (OA) is recognized as one of the most prevalent chronic musculoskeletal diseases worldwide [World Health Organization, 2002]. The joints typically affected are located in the hands, knees, hips, and spine with varying degrees of joint deformity and swelling [Sarzi-Puttini et al. 2005]. Usually beginning when adults are in their 40s, it is estimated that 9.6% of men and 18% of women >60 years of age are affected with symptomatic OA. Since age is a significant risk factor in its development, it is predicted that OA will be the fourth leading cause of disability by 2020 [Sarzi-Puttini et al. 2005; Woolf and Pfleger, 2003]. In fact, the prevalence of knee OA has been reported to be 44% in those who are 80 years old or greater [Felson et al. 1987].
The yearly global economic burden of OA measured by direct and indirect costs is in the tens of billions of dollars annually [Chen et al. 2012].
Pain is recognized as one of the hallmark symptoms in knee OA and is the primary reason why patients seek medical attention [Creamer, 2000]. It is a significant determinant of functional impairment and disability, even more so than radiographic findings [Jinks et al. 2002; McAlindon et al. 1993]. Therefore, it is necessary to consider other factors which may be involved in the maintenance of pain when it cannot solely be explained by peripheral nociceptive factors. As such, alterations in the central nervous system (CNS) may be implicated and understanding the role of central sensitization in pain modulation is important in conditions such as chronic knee OA since its treatment requires an approach that differs from the treatment of pain in a peripheral context [Mease et al. 2011; Phillips and Clauw, 2013].
Osteoarthritis of the knee: pathophysiology and diagnosis
Knee OA has been characterized as an insidious disease related to structural changes in the joint over many years and decades. Progressive and irreversible articular damage results in a loss of the extracellular matrix of cartilage in addition to changes in subchondral bone. These degenerative changes result in cartilage loss, synovitis, subchondral cysts, bone marrow lesions, and osteophyte formation. It is also characterized by an attempt of the joint to regenerate tissue such as fibrocartilage [Iannone and Lapadula, 2003].
A clinical diagnosis may be made on the basis of three symptoms (persistent knee pain, short-lived morning stiffness, and reduced function) and identification of three signs on examination (crepitus, restricted movement, and bony enlargement) without an absolute requirement for imaging. The estimated probability of having radiographic knee OA increases with increasing number of positive features, to 99% when all six are present [Zhang et al. 2010].
Osteoarthritis knee pain
It was thought that pain was caused by cartilage damage; however, this view has since evolved given that cartilage is both an avascular and aneural tissue and therefore not capable of generating pain [Sofat et al. 2011]. In the earlier stages of the disease, patients typically report pain that is deep and aching which may worsen with joint use and improve with rest. In the later stages of the disease, patients may experience pain after minimal motion, and pain at rest can be severe enough to awaken the patient. Morning stiffness, lasting usually <30 minutes, is a common complaint. Stiffness of an affected joint is often described when the patient mobilizes after a period of rest. This phenomenon is referred to as articular gelling which resolves after several flexation and extension cycles [Sarzi-Puttini et al. 2005].
Radiographic studies have demonstrated that the amount of joint deformity does not reliably predict the amount of pain a patient may experience [Cubukcu et al. 2012]. Therefore, the experience of pain may involve the CNS and become more centralized [Sofat et al. 2011]. Through quantitative sensory testing, Finan and colleagues [Finan et al. 2013] elegantly demonstrated that patients with high levels of pain and an absence of moderate to severe radiographic findings show more centrally mediated pain processing.
Pharmacological treatment of osteoarthritis knee pain
Since 2000, numerous professional societies have published recommendations for the management of knee OA, including those developed by the European League Against Rheumatism (EULAR) [Jordan et al. 2003], the Osteoarthritis Research Society International (OARSI) [Zhang et al. 2008], the American Academy of Orthopaedic Surgeons (AAOS) [American Academy of Orthopaedic Surgeons, 2008], and the American College of Rheumatology (ACR) [Hochberg et al. 2012a].
These professional societies recommended the following steps in the pharmacological management of knee OA pain. If the healthcare provider chooses to initiate acetaminophen in the full dosage up to 4000 mg/day, the patient should be counseled to avoid all other products that contain acetaminophen, including over-the-counter cold remedies as well as combination products with opioid analgesics. If the patient does not have a satisfactory clinical response to full-dose acetaminophen, it is strongly recommended to use oral or topical nonsteroidal anti-inflammatory drugs (NSAIDs) or intra-articular corticosteroid injections. If they are ineffective or contraindicated, then it is conditionally recommended that tramadol, duloxetine, or intra-articular hyaluronan injections be used.
Pain processing, modulation and central sensitization
Both ascending nociceptive [Iyengar et al. 2004] and descending modulatory [Woolf, 2004] pathways are involved in pain perception (Figure 1). Ascending nociceptive pathways carry pain signals from the periphery to the brain [Scholz and Woolf, 2002]. More specifically, transduction occurs with the activation of peripheral nociceptors (C and Aδ-fibers) through stimulation of peripheral nerve endings which creates an electrical signal, a potential pain impulse, that is conducted to the spinal cord. The signal is then transmitted to central nerves in the spinal dorsal horn where peripheral nociceptors synapse with dorsal horn neurons. This is the first stage at which a pain signal can be modulated: either amplified or inhibited. Modulation of the signal is accomplished by neuronal, glial, and endocrine factors in the dorsal horn. Inhibition of the pain signal is referred to as gate control theory [Melzack and Wall, 1965]. This theory has been updated to reflect the notion that pain in itself is a multidimensional experience involving a distributed neural network, or ‘neuromatrix’ [Finan et al. 2013; Melzack, 1999]. Various neurotransmitters and neuromodulators are involved in the gating process. Both substance P and glutamate are involved in the amplification of pain signals whereas gamma aminobutyric acid (GABA), glycine, endocannabinoids, endorphins, monoamines, and neurosteroids are linked to the inhibition of pain signals. Through ascending pathways, the signal is then conducted further until it reaches the brain [Bolay and Moskowitz, 2002; Marchand, 2012].
Figure 1.
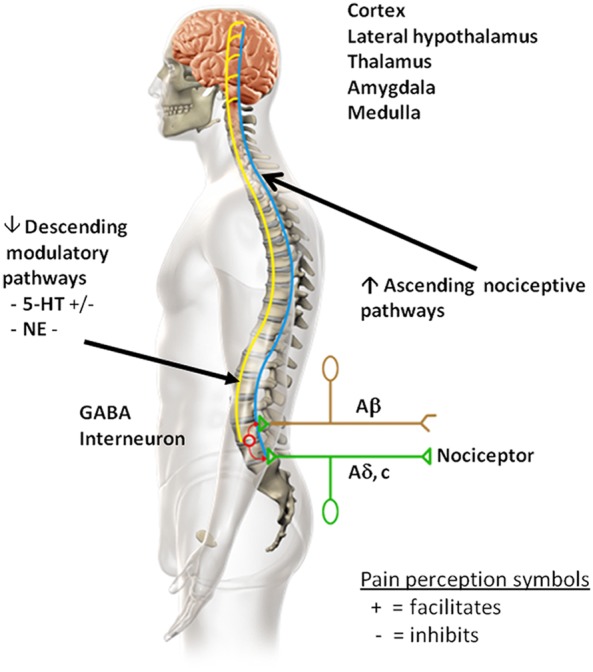
Ascending nociceptive pathways transmit/conduct noxious stimuli from the peripheral regions of the body to the brain. Descending modulatory pain pathways alter the processing of pain signals. Chronic pain, like that associated with knee osteoarthritis (OA), results in changes in the central nervous system, which likely reflect alterations in supraspinal modulation of nociception, and include increases in excitatory and decreases in inhibitory modulation pathways. In the descending modulatory pain system, the neurotransmitters 5-hydroxytryptamine (5-HT) and norepinephrine (NE) modulate pain signals. 5-HT both inhibits and facilitates the perception of pain. 5-HT inhibits pain via the descending inhibitory arm of the descending modulatory pathway and facilitates the perception of pain via the descending facilitatory arm of the descending modulatory pathway. NE inhibits the perception of pain via the descending inhibitory arm of the descending modulatory pathway. NE does not seem to be involved in the facilitatory aspect of pain perception in the descending modulatory pathway.
A painful sensory input is not perceived as painful until it reaches the brain. This is the stage at which the signal is processed by various cortical and subcortical regions responsible for the overall experience and interpretation of pain for an individual. The brain is not only implicated in the interpretation and experience of pain, but it can also modulate pain signals [Apkarian et al. 2005; Schweinhardt and Bushnell, 2010].
Pain signals are also modulated via descending pathways [Woolf, 2004]. These pathways travel down from the brain to subcortical nuclei within the mid-brain until they reach the dorsal horn of the spinal cord and can either amplify or attenuate pain signals. Two essential neurotransmitters involved in the attenuation of pain signals are serotonin (5-HT) and norepinephrine (NE). While 5-HT can both amplify and attenuate pain signals, NE only has an attenuation effect. In addition to 5-HT and NE, opioids and GABA are involved in the inhibition of pain signals whereas glutamate and aspartate are involved in their amplification [Benn and Woolf, 2004; Fields et al. 1991; Richardson, 1990]. Chronic pain, similar to pain associated with knee OA, results in changes in the CNS, which likely reflect alterations in supraspinal modulation of nociception, and include increases in excitatory and decreases in inhibitory modulation pathways [DeSantana and Sluka, 2008; Staud, 2011].
Duloxetine as an analgesic
Duloxetine is a serotonin norepinephrine reuptake inhibitor (SNRI) and it is hypothesized that potentiation of 5-HT and NE activity in the CNS results in pain inhibition [Woolf, 2004]. The analgesic properties of duloxetine have been demonstrated in several chronic pain conditions. These include neuropathic pain associated with diabetic peripheral neuropathy [Goldstein et al. 2005; Raskin et al. 2005; Wernicke et al. 2006], fibromyalgia [Arnold et al. 2004, 2005; Chappell et al. 2009a; Russell et al. 2008], and chronic low back pain [Skljarevski et al. 2009, 2010a, 2010b].
Duloxetine has also been evaluated in three separate double-blind, randomized, placebo-controlled trials in chronic osteoarthritis knee pain (OAKP). These trials will herein be referred to as OA-1 [Chappell et al. 2009c], OA-2 [Chappell et al. 2011], and OA-3 [Frakes et al. 2011]. The recommended dose for OAKP is 60 mg once daily. Some patients may benefit from dosages above the recommended 60 mg once daily up to a maximum dose of 120 mg per day, although the higher dose has been associated with a higher rate of adverse reactions. A lower starting dose of 30 mg may be considered for tolerability reasons in some patients, with a target dose of 60 mg/day within 1–2 weeks [Eli Lilly Canada Inc., 2012].
Duloxetine in OAKP
A concerted effort is underway to ensure that clinical trial research in pain is conducted according to the best available standards. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) aims to systematically address the challenges related to pain research and has proposed recommendations related to clinical trial methodology and design [Dworkin et al. 2010], the inclusion of appropriate core outcome measures and domains [Dworkin et al. 2005; Turk et al. 2003], and the clinical interpretation of statistical results [Dworkin et al. 2008, 2009].
With the purpose of drawing clinically relevant information, a review of duloxetine’s analgesic efficacy in OAKP will be presented within the context of IMMPACT recommendations.
Trial methodology
OA-1 [Chappell et al. 2009c] was a 13-week, randomized, double-blind, placebo-controlled trial. The trial design was divided into three study periods. Study period I was a 1-week screening phase in order to determine patient eligibility. In study period II, the 13-week treatment phase, eligible patients were randomly assigned 1:1 to receive either duloxetine 60 mg/day or placebo. During this period, patients started duloxetine at 30 mg/day during the first week and were then titrated to 60 mg/day. At week 7, patients receiving duloxetine were randomly re-assigned at a 1:1 ratio to receive either 60 mg/day or 120 mg/day for the remainder of the trial. At the end of the treatment period, week 13, patients underwent a 2-week taper phase.
The design for OA-2 [Chappell et al. 2011] is almost identical to that for OA-1 [Chappell et al. 2009c]. The primary difference is related to the dose escalation of patients at week 7. Here, patients had their dose increased to 120 mg/day based on their response. Response was defined as having less than a 30% pain reduction from baseline using the Brief Pain Inventory (BPI) average pain score [Cleeland and Ryan, 1994].
The aim of OA-3 [Frakes et al. 2011] was to evaluate duloxetine’s efficacy in OAKP when added to patients already optimized on NSAID therapy. Patients were optimized on NSAIDs over a 2-week period. Those still experiencing at least a 4/10 on average weekly pain severity during the previous week were included into the study and were randomly assigned to 10 weeks of duloxetine or placebo treatment. However, the primary efficacy end point was at week 8. Efficacy ratings may be influenced by the impending end to a study; therefore both patients and investigators were unaware of the sham end point at week 10.
Similar to the other two studies, patients were started on duloxetine 30 mg/day for the first week of blinded therapy and were then titrated to 60 mg/day. To reflect what may occur in clinical practice, patients were re-evaluated at week 3 to determine response to treatment. Patients with a mean average pain severity score of at least 4/10 had their dose augmented to 120 mg/day of blinded treatment for the remainder of the trial.
Study patients
IMMPACT has identified several important characteristics which should be considered in subject selection. These include information on demographic and related characteristics, diagnosis, disease and pain duration, pain intensity, medical and psychiatric comorbidities, concomitant and rescue medications, and response to previous medications [Dworkin et al. 2010].
At a minimum, trials should report key demographic variables which include age, sex, and race/ethnicity. All three trials examining the effect of duloxetine on OAKP included age, sex and race [Chappell et al. 2009c, 2011; Frakes et al. 2011] (Table 1). IMMPACT has recommended that pain trials utilize already-existing validated diagnostic criteria in patient selection such as those from the ACR. ACR clinical and radiographic criteria for knee OA were used as the diagnostic inclusionary criteria for the three studies [Altman et al. 1986].
Table 1.
Baseline patient characteristics.
| Characteristics | OA-1 [Chappell et al. 2009] |
OA-2 [Chappell et al. 2011] |
OA-3 [Frakes et al. 2011] |
|||
|---|---|---|---|---|---|---|
| Duloxetine 60–120 mg/day (N = 111) | Placebo (N = 120) | Duloxetine 60–120 mg/day (N = 128) | Placebo (N = 128) | Duloxetine 60–120 mg/day (N = 264) | Placebo (N = 260) | |
| Age, mean (SD), years | 62.1 (9.6) | 62.5 (9.3) | 63.2 (8.8) | 61.9 (9.2) | 61.6 (9.2) | 60.3 (9.2) |
| Ethnicity, n (%) Caucasian | 94 (84.7) | 100 (83.3) | 126 (98.4) | 124 (96.9) | 218 (82.6) | 206 (79.5) |
| Female gender, n (%) | 70 (63.1) | 81 (67.5) | 89 (69.5) | 107 (83.6)* | 152 (57.6) | 147 (56.5) |
| Duration of osteoarthritis since diagnosis, mean (SD), years | 6.9 (8.4) | 7.1 (7.2) | 6.2 (5.9) | 5.6 (6.2) | 9.8 (8.9)‡ | 9.2 (8.9)‡ |
| Mean duration of pain, mean (SD), years | 9.0 (8.7) | 9.3 (8.3) | 8.1 (7.6) | 6.7 (6.6) | 9.8 (8.9) | 9.2 (8.9) |
| Weekly average pain severity score, mean (SD) | 6.1 (1.3) | 6.2 (1.3) | 6.0 (1.2) | 6.1 (1.3) | 6.27 (1.41) | 6.36 (1.41) |
| NSAID use, yes, n (%) | 58 (52.3) | 59 (49.2) | 47 (36.7) | 53 (41.4) | 100 | 100 |
Significantly different from placebo (p = 0.012).
SD, standard deviation; NSAID, nonsteroidal anti-inflammatory drug.
Duration of osteoarthritis pain, years mean.
The duration of the disease as well as the duration of the pain can also influence the outcome of a trial. Patients must be in a chronic phase of their illness; however, they should not necessarily be in a treatment refractory stage either. Pain which has persisted at least 3 months can be considered as chronic [Dworkin et al. 2010]. Inclusion criteria for the three duloxetine trials required that patients be at least 40 years of age and have OAKP for a minimum of 14 days of each month for 3 months. The duration of pain ranged from 6.7 to 9.8 years across all three trials [Chappell et al. 2009c, 2011; Frakes et al. 2011] (Table 1) indicating that patients included in the duloxetine trials were well within the chronic phase of their illness.
Homogeneity of pain intensity at baseline is a very important variable. To minimize floor effects (patients with very little pain at baseline), IMMPACT has stated that randomized controlled trials (RCTs) include patients who have a moderate pain severity of least 4 on a 0–10 numeric rating scale [Dworkin et al. 2010]. This cutoff was used in the three RCTs investigating duloxetine, and the range in pain severity at baseline was remarkably consistent across groups and trials and ranged from 6.0/10 to 6.4/10 [Chappell et al. 2009c, 2011; Frakes et al. 2011] (Table 1).
Overall, patients participating in these three trials were fairly consistent across trials and representative of patients suffering from chronic OA pain.
Other medical and psychiatric comorbidities may influence the outcome and interpretation of efficacy and safety results in RCTs, and IMMPACT recommends that these be exclusionary criteria [Dworkin et al. 2010]. All three duloxetine trials excluded patients with potentially confounding painful conditions and psychiatric illnesses [Chappell et al. 2009c, 2011; Frakes et al. 2011]. Patients with major depressive disorder (MDD), a previous diagnosis of psychosis, bipolar disorder, or schizoaffective disorder were also excluded from studies OA-1 and OA-2 [Chappell et al. 2009c, 2011; Frakes et al. 2011].
Patients receiving concomitant analgesic medications prior to study entry is a noteworthy potentially confounding variable [Dworkin et al. 2010]. Therefore, RCTs must adjust for this reality in their design. In trials which permit the use of concomitant analgesics, IMMPACT recommends that these patients remain on stable doses of the medication(s) and that rescue medication be available for pain unrelated to the condition under investigation (e.g. pain related to a dental procedure) and that it only be used temporarily [Dworkin et al. 2010]. NSAIDs were allowed to be continued in all three duloxetine trials. In OA-1 [Chappell et al. 2009c] and OA-2 [Chappell et al. 2011], patients entering the trial were permitted to continue taking their NSAID/acetaminophen if they were taking therapeutic doses for at least 14 days per month for more than 3 months and were not permitted to increase their dose during the study. In order to ensure that groups were balanced for NSAID/acetaminophen use, patients were stratified at randomization. The third trial, OA-3, required that all patients be optimized on NSAIDs over the course of 2 weeks prior to study entry [Frakes et al. 2011].
Results from OA-1 [Chappell et al. 2009c] revealed that both the placebo (49.2%) and duloxetine (52.3%) groups were balanced at baseline for NSAID/acetaminophen use. However, a treatment-by-subgroup analysis between NSAID users and nonusers revealed that the nonuser group appeared to have a better response to both placebo and duloxetine relative to the user group. The authors posit that NSAID users may represent a subset of patients who may be more difficult to treat. This finding was not observed in OA-2 [Chappell et al. 2011]. Here, 41.4% of patients in the placebo group and 36.7% of patients in the duloxetine group were receiving NSAIDs at trial entry [Chappell et al. 2011].
Overall, the three trials examining the efficacy of duloxetine in OAKP [Chappell et al. 2009c, 2011; Frakes et al. 2011] adhered to key IMMPACT recommendations for clinical trial designs related to pain studies [Dworkin et al. 2010].
Core outcome measure and domains
In addition to pain intensity, IMMPACT has recommended five other core domains which should be considered in clinical trials of analgesia efficacy. These include (1) physical functioning, (2) emotional functioning, (3) participant ratings of improvement and satisfaction with treatment, (4) symptoms and adverse events, and (5) participant disposition (e.g. adherence to the treatment regimen and reasons for premature withdrawal from the trial) [Dworkin et al. 2005; Turk et al. 2003]. IMMPACT has also proposed reliable and validated measures which address these core domains [Dworkin et al. 2005] and recommendations regarding the translation of statistical results into clinically relevant terms [Dworkin et al. 2008, 2009]. The following section on the efficacy and safety of duloxetine will be presented within the context of these IMMPACT recommendations.
Effect of duloxetine on pain intensity
The majority of RCTs evaluating analgesic efficacy utilize reductions in pain intensity as their primary outcome measure. Visual analog scales, numerical rating scales, as well as verbal rating scales have all been demonstrated as being valid and reliable measures of pain [Dworkin et al. 2005].
All three duloxetine trials utilized changes in pain intensity as their primary efficacy outcome variable. OA-1 [Chappell et al. 2009c] and OA-3 [Frakes et al. 2011] measured changes in pain intensity by the weekly mean of the 24-h average pain scores. This numerical rating scale is based on an 11-point Likert scale (an ordinal scale with 0 = ‘no pain’ and 10 = ‘worst pain imaginable’). OA-2 [Chappell et al. 2011] utilized the average pain score item from the BPI [Cleeland and Ryan, 1994]. The BPI is a validated self-reported tool which assesses pain severity (BPI-S) in addition to its interference on daily functions (BPI-I). The BPI-S subscale is composed of four questions which ask the subject to rate their pain on a 0 to 10 Likert scale (with 0 = no pain to 10 = pain as bad as you can imagine). They are asked about their (1) worst pain in the past 24 h, (2) least pain in the last 24 h, (3) their average pain in the past 24 h, and (4) their pain right now [Cleeland and Ryan, 1994].
Results from OA-1 [Chappell et al. 2009c] revealed that the duloxetine 60/120 mg/day group had statistically significant reductions in average pain scores relative to placebo beginning at week 1 and at each weekly time point thereafter. The mean change from baseline to end-point in the 24-h average pain score also showed a statistically significant difference in favor of duloxetine 60/120 mg/day (duloxetine = −2.92; placebo = −2.08). A similar finding was observed in OA-2 [Chappell et al. 2011]. Reductions in BPI average pain severity at each time point (weeks 4, 7, and 13) were statistically significantly different in favor of duloxetine 60/120 mg/day versus placebo. The mean change from baseline to end point at week 13 was −2.72 for duloxetine 60/120 mg/day and −1.88 for placebo. A similar pattern emerges in OA-3 where duloxetine or placebo is added to patients optimized on NSAID therapy [Frakes et al. 2011]. Here, there is a statistically significant separation between groups beginning at week 1 which is continued through to week 8. The mean change from baseline to end point on the average pain rating was −2.46 for duloxetine 60/120 mg/day and −1.55 for placebo.
From a clinical stand point, what do these magnitudes of change on an ordinal scale that ranges from 0 to 10 mean? IMMPACT recommends that a decrease of two points from baseline to end point is clinically meaningful for patients and represents a important decrease in chronic pain intensity [Dworkin et al. 2008]. Although statistical significance separated duloxetine from placebo as early as week 1 (OA-1 and OA-3), a statistical significance does not necessarily equate to clinical significance. These results across the three trials suggest that the impact of duloxetine on a clinically meaningful reduction in pain occurs at about 4 weeks and is maintained for the remainder of the trial periods. Placebo also achieved a clinically meaningful change in OA-1 [Chappell et al. 2009c].
According to IMMPACT recommendations on the interpretation of group differences, it is suggested that responder analyses be used to elucidate the question of whether or not the magnitude of differences between groups is clinically meaningful despite statistical significance [Dworkin et al. 2008]. IMMPACT provisionally suggests that a 30% change from baseline to end point on the primary outcome variable is considered to be a moderate improvement in pain severity whereas a 50% improvement is reflective of a substantial improvement. A comparison of responder rates between an active medication and placebo provides clinically meaningful information regarding the magnitude of improvement between groups.
Responder analyses were conducted in all three duloxetine trials. OA-1 [Chappell et al. 2009c] reported a statistically significant 30% improvement in 59% of duloxetine patients and 45% of placebo patients. A similar statistically significant pattern emerged for the 50% response rate; 47% for duloxetine patients and 29% for placebo. These results suggest that there was a clinically meaningful difference in the reduction of pain severity for patients treated with duloxetine relative to placebo in OA-1.
Similar to OA-1, statistically significant differences in 30% responder rates for OA-2 were observed; 65% for duloxetine and 44% for placebo [Chappell et al. 2011]. However, differences in 50% responder rates failed to show a statistically significant difference; 44% for duloxetine and 32% for placebo. In OA-3, both 30% and 50% responder rates were statistically significant in favor of duloxetine. More specifically, 54% of patients in the duloxetine group and 34% in the placebo group achieved a 30% improvement. Furthermore, 36% of duloxetine patients and 16% of placebo patients achieved a 50% response [Frakes et al. 2011]. With the exception of 50% improvement in OA-2, these results suggest that patients receiving duloxetine 60/120 mg/day show moderate to substantial improvements in their pain severity.
Effect of duloxetine on physical functioning
The assessment of physical functioning in each of the three duloxetine trials used the BPI-I [Cleeland and Ryan, 1994] and the physical functioning subscale of the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC-pf) [Bellamy et al. 1988].
As mentioned previously, the BPI is a validated self-reported tool which assesses pain severity (BPI-S) in addition to its interference on daily functions (BPI-I). The BPI-I subscale uses a 0 (does not interfere) to 10 (completely interferes) Likert scale and is composed of seven items. Patients are asked to rate the amount of interference their pain has had in the last 24 h on (1) general activity, (2) mood, (3) walking ability, (4) normal work, (5) relations with other people, (6) sleep, and (7) enjoyment of life [Cleeland, 1994].
The general activity item was the only item which was consistently statistically significantly different from placebo in each of the three trials. The mean change from baseline to end point for duloxetine ranged from −2.16 in OA-2 [Chappell et al. 2011] to −2.72 in OA-1 [Chappell et al. 2009c]. The mean change from baseline to end point for placebo ranged from −1.56 in OA-3 [Frakes et al. 2011] to −2.01 in OA-1 [Chappell et al. 2009c]. In considering each trial individually, patients treated with duloxetine in OA-3 had statistically significantly greater improvements in each of the seven items from the BPI-I compared to placebo [Frakes et al. 2011]. Duloxetine patients in OA-1 [Chappell et al. 2009c] had significant improvements relative to placebo on four of the seven items. In addition to general activity, these included walking ability, sleep, and enjoyment of life. Finally, the only two items to show a statistically significant difference between duloxetine and placebo in OA-2 were general activity and normal work [Chappell et al. 2011].
The WOMAC assess pain, stiffness, and physical function in patients with OA of the knee or hip. It consists of 24 questions: five on pain, two on stiffness, and 17 on physical function, each answered using a 5-point scale ranging from 0 (none) to 4 (extreme) [Bellamy et al. 1988]. A composite score of these 17 items is then calculated (range 0−68) and lower scores reflect better functioning. Statistically significant differences between duloxetine patients and placebo were observed in all three OA trials [Chappell et al. 2009c, 2011; Frakes et al. 2011]. Decreases in WOMAC-pf change scores from baseline to end point ranged from −12.69 [Chappell et al. 2011] to −21.10 [Frakes et al. 2011] for duloxetine and from −9.43 [Chappell et al. 2011] to −13.81 [Frakes et al. 2011] for placebo.
In considering results from the BPI-I and the WOMAC-pf, duloxetine patients demonstrated greater improvements in physical functioning relative to placebo patients.
Effect of duloxetine on emotional functioning
IMMPACT recommends [Dworkin et al. 2005] that emotional symptoms be assessed with an instrument such as the Beck Depression Inventory-II (BDI-II) [Beck et al. 1988]. However, both OA-1 and OA-2 [Chappell et al. 2009c, 2011] excluded patients if they had current MDD or any other psychiatric diagnosis. For instance, patients in OA-1 had scores <6 on the BDI-II at baseline indicating minimal depressive symptoms well within the normal range at study entry [Chappell et al. 2009c]. This floor effect would preclude observing significant decreases in depressive symptoms over the course of the trials. Nevertheless, depressive as well as anxiety symptoms were evaluated using path analysis in both OA-1 [Chappell et al. 2009c] and OA-2 [Chappell et al. 2011] in order to determine the contribution of emotional symptoms (depression and anxiety) to the overall analgesic effect of duloxetine. This analysis revealed that changes from baseline to end point in depression and anxiety had very little influence on the analgesic effect of duloxetine on changes in pain severity [Chappell et al. 2009c, 2011].
Patient global impression of change
Other secondary measures including the Patient Global Impression of Improvement (PGI-I) [Guy, 1976] were included in all three studies. The PGI-I is a one-item, seven-point scale which evaluates a patient’s impression of their overall change from randomization to the end of the trial. The scale ranges from 1 (very much better) to the midpoint of 4 (no change) to 7 (very much worse). While there was no significant improvement on the PGI-I between the duloxetine patients and the placebo group in OA-2 [Chappell et al. 2011], significant differences in both OA-1 and OA-3 were observed. The duloxetine patients in OA-1 reported that their improvement was 2.38 whereas the placebo group reported their improvement to be 2.91 (2 = much better and 3 = a little better) [Chappell et al. 2009c]. OA-3 reports that 53% of duloxetine patients reported feeling at least much better compared with 32% of placebo patients [Frakes et al. 2011]. Overall, duloxetine patients report better global improvements relative to placebo.
Health outcomes
Health-related outcomes were assessed in OA-1 and OA-2 with the 36-Item Short-Form Health Status Survey (SF-36) [Ware et al. 1993] and the EuroQol: 5 Dimensions Questionnaire (EQ-5D) [Kind, 1996]. The SF-36 is composed of eight items which measure changes related to (1) bodily pain, (2) general health, (3) mental health, (4) physical functioning, (5) role: emotional, (6) role: physical, (7) social functioning, and (8) vitality. Results for the SF-36 in OA-1 revealed significant differences between the duloxetine and placebo groups on three of the eight items. These were bodily pain, mental health and vitality [Chappell et al. 2009c]. Results in OA-2 showed significant differences in bodily pain, physical functioning, and role: physical [Chappell et al. 2011]. Both the US and UK indices for the EQ-5D were significantly improved in favor of the duloxetine patients in OA-1 [Chappell et al. 2009c].
Safety
The safety of duloxetine has been well characterized in clinical trials in more than 32,000 patients across all indications. Since its first approval in 2004, over 53 million patients have been treated with duloxetine worldwide, accounting for over 19 million patient-years of therapy. Duloxetine has been shown to be generally safe and well tolerated with no new safety concerns identified in the OA population. An analysis of safety data from 52 completed RCTs of duloxetine identified that the proportion of patients experiencing treatment-emergent adverse events was lowest in studies of OAKP versus other indications [Brunton et al. 2010]. The incidence of adverse events occurring at a rate of ≥3% in the three OA trials is summarized in Table 2. The most commonly experienced adverse events in OA knee patients include nausea, constipation, dry mouth, diarrhea, fatigue, dizziness, somnolence and insomnia [Chappell et al. 2009c, 2011; Frakes et al. 2011]. Nausea is typically mild–moderate and usually resolves within 8 days [Brunton et al. 2010]. Two strategies can be utilized in order to mitigate nausea. Patients can start at 30 mg/day or they can take the 60 mg dose with food in order to improve tolerability [Whitmyer et al. 2007].
Table 2.
Adverse events.
| Characteristics | OA-1 [Chappell et al. 2009] |
OA-2 [Chappell et al. 2011] |
OA-3 [Frakes et al. 2011] |
|||
|---|---|---|---|---|---|---|
| Duloxetine 60–120 mg/day (N = 111) | Placebo (N = 120) | Duloxetine 60–120 mg/day (N = 128) | Placebo (N = 128) | Duloxetine 60–120 mg/day (N = 264) | Placebo (N = 260) | |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Discontinuation due to adverse event | 15(13.5) | 7(5.8) | 24(18.8) | 7(5.5) | 40 (15.2) | 23 (8.8) |
| Nausea | 7(6.3) | 2(1.7) | 13(10.2)* | 3(2.3) | 41(15.5)*** | 12(4.6) |
| Fatigue | 7(6.3)* | 1(0.8) | 2(2.3) | 1(0.8) | 18(6.8)** | 4(1.5) |
| Constipation | 4(3.6) | 0 | 10(7.8)* | 2(1.6) | 23(8.7)** | 8(3.1) |
| Somnolence | 5(4.5) | 1(0.8) | 5(3.9) | 3(2.3) | 14(5.3) | 7(2.7) |
| Dizziness | 4(3.6) | 2(1.7) | 6(4.7) | 2(1.6) | 17(6.4) | 7(2.7) |
| Diarrhea | 5(4.5) | 3(2.5) | 6(4.7) | 3(2.3) | 18(6.8) | 10(3.8) |
| Abdominal pain | 1(0.9) | 2(1.7) | 6(4.7) | 1(0.8) | – | – |
| Insomnia | 3(2.7) | 1(0.8) | 6(4.7) | 3(2.3) | 13(4.9)* | 3(1.2) |
| Hyperhidrosis | 1(0.9) | 1(0.8) | 7(5.5)* | 0 | 10(3.8)* | 1(0.4) |
| Dry mouth | 2(1.8) | 2(1.7) | 6(4.7) | 1(0.8) | 25(9.5)* | 7(2.7) |
| Decreased appetite | 3(2.7) | 1(0.8) | 1(0.8) | 0 | 15(5.7)*** | 1(0.4) |
| Headache | 3(2.7) | 1(0.8) | 4(3.1) | 5(3.9) | 16(6.1) | 10(3.8) |
| Decreased libido | 4(3.6) | 0 | 1(0.8) | 0 | – | – |
| Hypertension | 4(3.6) | 1(0.8) | 0 | 1(0.8) | – | – |
| Vomiting | - | - | 1(0.8) | 0 | 11(4.2) | 3(1.2) |
| Dysgeusia | 4(3.1) | 1(0.8) | – | – | ||
| Asthenia | 2(1.8) | 0 | 4(3.1) | 0 | – | – |
Incidence of treatment-emergent adverse events that occurred in ≥3% of patients treated with duloxetine and with an incidence greater than placebo.
p ≤ 0.05.
p ≤ 0.01.
p ≤ 0.001.
The prescribing information for duloxetine contains warnings about certain rare but potentially clinically important adverse events. Cases of elevated liver enzymes, hepatitis, jaundice, and hepatic failure have been reported. Duloxetine should not be prescribed to patients with hepatic impairment or to patients with substantial alcohol use. As part of class labeling for all serotonin reuptake inhibitors, the duloxetine label includes precautions related to potential increases in suicidality in adolescents and young adults, the possibility of increased risk of bone fracture and potential increase risk of bleeding events due to serotonergic effects on platelet aggregation [Eli Lilly Canada Inc., 2012].
Drug interactions
Duloxetine is extensively metabolized, predominantly by CYP1A2 and also by CYP2D6, and its metabolites are not pharmacologically active. Duloxetine is also a moderate inhibitor of CYP2D6. Duloxetine should not be used concomitantly with potent inhibitors of CYP1A2 and with other drugs that are primarily metabolized by CYP2D6 (Table 3) [Eli Lilly Canada Inc., 2012].
Table 3.
Drug interactions.
| Substrates of CYP2D6 | Potential for increased concentrations of these agents or reduced efficacy of prodrugs that require conversion to their active metabolite, e.g. TCAs, tramadol (↑ tramadol concentration and ↓ conversion to M1), codeine (↓ conversion to morphine), tamoxifen(↓ conversion to endoxifen) |
| Potent inhibitors of CYP1A2 | Potential for increased duloxetine concentrations, e.g. fluvoxamine (↓ clearance of approx. 77%), ciprofloxacin |
| Potent inhibitors of CYP2D6 | Potential for higher concentrations of duloxetine, e.g. fluoxetine, paroxetine, bupropion |
| Seronergic agents | Potential for serotonin syndrome, e.g. MAOIs (absolute contraindication), caution is advised with triptans, serotonin reuptake inhibitors, tramadol, tapentadol, methadone, pentazocine or St. John’s Wort |
| Drugs affecting platelet function or bleeding risk | Potential to potentiate the anticoagulant effects caused by serotonin reuptake inhibition of duloxetine, e.g. caution is advised with warfarin, NSAIDs, ASA, and other anticoagulants |
ASA, acetylsalicylic acid; MAOI, monoamine oxidase inhibitor; NSAID, nonsteroidal anti-inflammatory drug; TCA, tricyclic antidepressant.
Discussion
The importance of understanding pain mechanisms is crucial in the selection of appropriate analgesic treatments [Martel-Pelletier et al. 2012; Mease et al. 2011]. Duloxetine is an SNRI with CNS activity and its analgesic efficacy is putatively thought to involve its effect on descending inhibitory pain pathways [Woolf, 2004].
The efficacy of duloxetine as an analgesic for OAKP was evaluated in three randomized, double-blind, placebo-controlled trials. With respect to its effect on reducing pain intensity, IMMPACT recommends that a decrease of two points from baseline to end point is clinically meaningful for patients and represents an important decrease [Dworkin et al. 2008]. Results from the three trials suggest that impact of duloxetine on a clinically meaningful reduction in pain occurs at about 4 weeks and is maintained for the remainder of the trial periods [Chappell et al. 2009c, 2011; Frakes et al. 2011]. Furthermore, 30% and 50% responder analyses [Dworkin et al. 2008] suggest that a greater proportion of patients treated with duloxetine have moderate to substantial improvement in their pain intensity when compared with placebo [Chappell et al. 2009c, 2011; Frakes et al. 2011].
Similar results are reported in an independent 16-week trial evaluating duloxetine 60 mg/day and placebo in OAKP in older patients; ≥65 years [bou-Raya et al. 2012]. Here, OARSI 2004 clinical response criteria were used as the primary efficacy measure [Pham et al. 2004]. Response was defined as having at least a 50% reduction in pain scores or in physical function scores and had at least a 20 mm reduction on a visual analog pain scale (range 0–100 mm). The authors report that the duloxetine group had a significantly greater reduction in pain and physical function (WOMAC function scores) relative to placebo. They also report that the duloxetine group had a significantly greater reduction in paracetamol use at 16 weeks. Adverse events were also consistent with the three trials reported above. Duloxetine patients had significantly more constipation, nausea, hyperhidrosis, cough, myalgia, arthralgia, and palpitation.
A pooled analysis [Hochberg et al. 2012b] of OA-1 [Chappell et al. 2009c] and OA-2 [Chappell et al. 2011] revealed that duloxetine patients were 33% more likely to have a clinically meaningful response to treatment than placebo patients and that the number needed to treat (NNT) = 6. A clinically meaningful response is based on criteria developed by the Outcome Measures in Rheumatoid Arthritis Clinical Trials (OMERACT) and the OARSI criteria [Pham et al. 2004]. Also, more duloxetine than placebo patients reported >30% improvement in pain from baseline to end point with an NNT = 5 and improvements >50% occurred more often in the duloxetine group with an NNT = 7. The authors conclude that duloxetine has a clinically meaningful effect on both pain and function. Also, duloxetine patients were more likely than placebo patients to experience a treatment-emergent adverse event and the NNH was 8 [Hochberg et al. 2012b].
A second recently published pooled analysis [Micca et al. 2013] of OA-1 [Chappell et al. 2009c] and OA-2 [Chappell et al. 2011] examining potential differences between older (≥65 years) and younger patients (40–65 years) revealed that there was no statistically significant difference between groups on OAKP when treated with duloxetine. Both groups did show statistically significant improvement over placebo. In addition, increasing the dose of duloxetine to 120 mg/day did not confer any additional benefit in either age category.
At the present time, there are no long-term studies evaluating duloxetine in OAKP. However, duloxetine has been evaluated in a 1-year extension trial of fibromyalgia [Chappell et al. 2009b]. Effectiveness was maintained over the course of the year and the safety of duloxetine was consistent with that observed in other indications.
Acknowledgments
The authors would like to thank Ms Monica Kirk for her assistance with this article.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: JPB has received research grants from Abbott, Amgen, Bristol-Myers Squibb, Eli Lilly, Merck, Novartis, Pfizer, Roche, Sanofi-Aventis, Servier, Takeda, and Warner Chilcott. JPB has received consulting fees or other remuneration from Amgen, Eli Lilly, Merck, Novartis, Sanofi-Aventis, and Warner Chilcott, and has served on the speaker’s bureau for Amgen, Eli Lilly, and Novartis. LJB is an employee in Research and Development at Eli Lilly Canada Inc.
Contributor Information
Jacques P. Brown, CHU de Québec (CHUL) Research Centre, Rheumatology and Bone Diseases Research Group, S-763, 2705 Laurier Boulevard, Quebec City, QC, G1V 4G2, Canada
Luc J. Boulay, Eli Lilly, Canada
References
- Altman R., Asch E., Bloch D., Bole G., Borenstein D., Brandt K., et al. (1986) Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum 29: 1039–1049 [DOI] [PubMed] [Google Scholar]
- American Academy of Orthopaedic Surgeons (2008) American Academy of Orthopaedic Surgeons clinical practice guideline on the treatment of osteoarthritis of the knee (non-arthroplasty). Rosemont, IL: American Academy of Orthopaedic [Google Scholar]
- Apkarian A., Bushnell M., Treede R., Zubieta J. (2005) Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain 9: 463–484 [DOI] [PubMed] [Google Scholar]
- Arnold L., Lu Y., Crofford L., Wohlreich M., Detke M., Iyengar S., et al. (2004) HMBO - A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 50: 2974–2984 [DOI] [PubMed] [Google Scholar]
- Arnold L., Rosen A., Pritchett Y., D’Souza D., Goldstein D., Iyengar S., et al. (2005) HMCA - A randomized, double-blind, placebo-controlled trial of duloxetine in the treatment of women with fibromyalgia with or without major depressive disorder. Pain 119: 5–15 [DOI] [PubMed] [Google Scholar]
- Beck A., Epstein N., Brown G., Steer R. (1988) An inventory for measuring clinical anxiety: psychometric properties. J Consult Clin Psychol 56: 893–897 [DOI] [PubMed] [Google Scholar]
- Bellamy N., Buchanan W., Goldsmith C., Campbell J., Stitt L. (1988) Validation study of WOMAC: a health status instrument for measuring clinically important patient relevant outcomes to antirheumatic drug therapy in patients with osteoarthritis of the hip or knee. J Rheumatol 15: 1833–1840 [PubMed] [Google Scholar]
- Benn S., Woolf C. (2004) Adult neuron survival strategies–slamming on the brakes. Nat Rev Neurosci 5: 686–700 [DOI] [PubMed] [Google Scholar]
- Bolay H., Moskowitz M. (2002) Mechanisms of pain modulation in chronic syndromes. Neurology 59: S2–S7 [DOI] [PubMed] [Google Scholar]
- bou-Raya S., bou-Raya A., Helmii M. (2012) Duloxetine for the management of pain in older adults with knee osteoarthritis: randomised placebo-controlled trial. Age Ageing 41: 646–652 [DOI] [PubMed] [Google Scholar]
- Brunton S., Wang F., Edwards S., Crucitti A., Ossanna M., Walker D., et al. (2010) Profile of adverse events with duloxetine treatment: a pooled analysis of placebo-controlled studies. Drug Saf 33: 393–407 [DOI] [PubMed] [Google Scholar]
- Chappell A., Bradley L., Wiltse C., Detke M., D’Souza D., Spaeth M. (2009a) A six-month double-blind, placebo-controlled, randomized clinical trial of duloxetine for the treatment of fibromyalgia. Int J Gen Med 1: 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell A., Desaiah D., Liu-Seifert H., Zhang S., Skljarevski V., Belenkov Y., et al. (2011) A double-blind, randomized, placebo-controlled study of the efficacy and safety of duloxetine for the treatment of chronic pain due to osteoarthritis of the knee. Pain Pract 11: 33–41 [DOI] [PubMed] [Google Scholar]
- Chappell A., Littlejohn G., Kajdasz D., Scheinberg M., D’Souza D., Moldofsky H. (2009b) A 1-year safety and efficacy study of duloxetine in patients with fibromyalgia. Clin J Pain 25: 365–375 [DOI] [PubMed] [Google Scholar]
- Chappell A., Ossanna M., Liu-Seifert H., Iyengar S., Skljarevski V., Li L., et al. (2009c) Duloxetine, a centrally acting analgesic, in the treatment of patients with osteoarthritis knee pain: a 13-week, randomized, placebo-controlled trial. Pain 146: 253–260 [DOI] [PubMed] [Google Scholar]
- Chen A., Gupte C., Akhtar K., Smith P., Cobb J. (2012) The global economic cost of osteoarthritis: how the UK compares. Arthritis 2012:698709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeland C., Ryan K. (1994) Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore 23: 129–138 [PubMed] [Google Scholar]
- Creamer P. (2000) Osteoarthritis pain and its treatment. Curr Opin Rheumatol 12: 450–455 [DOI] [PubMed] [Google Scholar]
- Cubukcu D., Sarsan A., Alkan H. (2012) Relationships between pain, function and radiographic findings in osteoarthritis of the knee: a cross-sectional study. Arthritis 2012: 984060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantana J., Sluka K. (2008) Central mechanisms in the maintenance of chronic widespread noninflammatory muscle pain. Curr Pain Headache Rep 12: 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin R., Turk D., Farrar J., Haythornthwaite J., Jensen M., Katz N., et al. (2005) Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain 113: 9–19 [DOI] [PubMed] [Google Scholar]
- Dworkin R., Turk D., McDermott M., Peirce-Sandner S., Burke L., Cowan P., et al. (2009) Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. Pain 146: 238–244 [DOI] [PubMed] [Google Scholar]
- Dworkin R., Turk D., Peirce-Sandner S., Baron R., Bellamy N., Burke L., et al. (2010) Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. Pain 149: 177–193 [DOI] [PubMed] [Google Scholar]
- Dworkin R., Turk D., Wyrwich K., Beaton D., Cleeland C., Farrar J., et al. (2008) Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain 9: 105–121 [DOI] [PubMed] [Google Scholar]
- Eli Lilly Canada Inc (2012) Product Monograph: Cymbalta duloxetine. [Google Scholar]
- Felson D., Naimark A., Anderson J., Kazis L., Castelli W., Meenan R. (1987) The prevalence of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum 30: 914–918 [DOI] [PubMed] [Google Scholar]
- Fields H., Heinricher M., Mason P. (1991) Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci 14: 219–245 [DOI] [PubMed] [Google Scholar]
- Finan P., Buenaver L., Bounds S., Hussain S., Park R., Haque U., et al. (2013) Discordance between pain and radiographic severity in knee osteoarthritis: findings from quantitative sensory testing of central sensitization. Arthritis Rheum 65: 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes E., Risser R., Ball T., Hochberg M., Wohlreich M. (2011) Duloxetine added to oral nonsteroidal anti-inflammatory drugs for treatment of knee pain due to osteoarthritis: results of a randomized, double-blind, placebo-controlled trial. Curr Med Res Opin 27: 2361–2372 [DOI] [PubMed] [Google Scholar]
- Goldstein D., Lu Y., Detke M., Lee T., Iyengar S. (2005) Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain 116: 109–118 [DOI] [PubMed] [Google Scholar]
- Guy W. (1976) ECDEU assessment manual for psychopharmacology, revised. Rockville, MD: US Department of Health, Education, and Welfare Publication (ADM). National Institute of Mental Health [Google Scholar]
- Hochberg M., Altman R., April K., Benkhalti M., Guyatt G., McGowan J., et al. (2012a) American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 64: 465–474 [DOI] [PubMed] [Google Scholar]
- Hochberg M., Wohlreich M., Gaynor P., Hanna S., Risser R. (2012b) Clinically relevant outcomes based on analysis of pooled data from 2 trials of duloxetine in patients with knee osteoarthritis. J Rheumatol 39: 352–358 [DOI] [PubMed] [Google Scholar]
- Iannone F., Lapadula G. (2003) The pathophysiology of osteoarthritis. Aging Clin Exp Res 15: 364–372 [DOI] [PubMed] [Google Scholar]
- Iyengar S., Webster A., Hemrick-Luecke S., Xu J., Simmons R. (2004) Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther 311: 576–584 [DOI] [PubMed] [Google Scholar]
- Jinks C., Jordan K., Croft P. (2002) Measuring the population impact of knee pain and disability with the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC). Pain 100: 55–64 [DOI] [PubMed] [Google Scholar]
- Jordan K., Arden N., Doherty M., Bannwarth B., Bijlsma J., Dieppe P., et al. (2003) EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: Report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 62: 1145–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind P. (1996) The EuroQoL instrument: an index of health-related quality of life. In: Spilker B. (ed.), Quality of Life and Pharmacoeconomics in Clinical Trials. Philadelphia, PA: Lippincott-Raven Publishers, pp. 191–201 [Google Scholar]
- Marchand S. (2012) The Phenomenon of Pain. Seattle, WA: IASP Press [Google Scholar]
- Martel-Pelletier J., Wildi L., Pelletier J. (2012) Future therapeutics for osteoarthritis. Bone 51: 297–311 [DOI] [PubMed] [Google Scholar]
- McAlindon T., Cooper C., Kirwan J., Dieppe P. (1993) Determinants of disability in osteoarthritis of the knee. Ann Rheum Dis 52: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease P., Hanna S., Frakes E., Altman R. (2011) Pain mechanisms in osteoarthritis: understanding the role of central pain and current approaches to its treatment. J Rheumatol 38: 1546–1551 [DOI] [PubMed] [Google Scholar]
- Melzack R. (1999) From the gate to the neuromatrix. Pain Suppl 6: S121–S126 [DOI] [PubMed] [Google Scholar]
- Melzack R., Wall P. (1965) Pain mechanisms: a new theory. Science 150: 971–979 [DOI] [PubMed] [Google Scholar]
- Micca J., Ruff D., Ahl J., Wohlreich M. (2013) Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 14: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T., van der Heijde D., Altman R., Anderson J., Bellamy N., Hochberg M., et al. (2004) OMERACT-OARSI initiative: Osteoarthritis Research Society International set of responder criteria for osteoarthritis clinical trials revisited. Osteoarthritis Cartilage 12: 389–399 [DOI] [PubMed] [Google Scholar]
- Phillips K., Clauw D. (2013) Central pain mechanisms in the rheumatic diseases: future directions. Arthritis Rheum 65: 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin J., Pritchett Y., Wang F., D’Souza D., Waninger A., Iyengar S., et al. (2005) A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med 6: 346–356 [DOI] [PubMed] [Google Scholar]
- Richardson B. (1990) Serotonin and nociception. Ann N Y Acad Sci 600: 511–519 [DOI] [PubMed] [Google Scholar]
- Russell I., Mease P., Smith T., Kajdasz D., Wohlreich M., Detke M., et al. (2008) HMCJ - Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled, fixed-dose trial. Pain 136: 432–444 [DOI] [PubMed] [Google Scholar]
- Sarzi-Puttini P., Cimmino M., Scarpa R., Caporali R., Parazzini F., Zaninelli A., et al. (2005) Osteoarthritis: an overview of the disease and its treatment strategies. Semin Arthritis Rheum 35: 1–10 [DOI] [PubMed] [Google Scholar]
- Scholz J., Woolf C. (2002) Can we conquer pain? Nat Neurosci 5(Suppl.): 1062–1067 [DOI] [PubMed] [Google Scholar]
- Schweinhardt P., Bushnell M. (2010) Pain imaging in health and disease - how far have we come? J Clin Invest 120: 3788–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skljarevski V., Desaiah D., Liu-Seifert H., Zhang Q., Chappell A., Detke M., et al. (2010a) Efficacy and safety of duloxetine in patients with chronic low back pain. Spine (Phila Pa 1976) 35: E578–E585 [DOI] [PubMed] [Google Scholar]
- Skljarevski V., Ossanna M., Liu-Seifert H., Zhang Q., Chappell A., Iyengar S., et al. (2009) A double-blind, randomized trial of duloxetine versus placebo in the management of chronic low back pain. Eur J Neurol 16: 1041–1048 [DOI] [PubMed] [Google Scholar]
- Skljarevski V., Zhang S., Desaiah D., Alaka K., Palacios S., Miazgowski T., et al. (2010b) Duloxetine versus placebo in patients with chronic low back pain: a 12-week, fixed-dose, randomized, double-blind trial. J Pain 11: 1282–1290 [DOI] [PubMed] [Google Scholar]
- Sofat N., Ejindu V., Kiely P. (2011) What makes osteoarthritis painful? The evidence for local and central pain processing. Rheumatology (Oxford) 50: 2157–2165 [DOI] [PubMed] [Google Scholar]
- Staud R. (2011) Evidence for shared pain mechanisms in osteoarthritis, low back pain, and fibromyalgia. Curr Rheumatol Rep 13: 513–520 [DOI] [PubMed] [Google Scholar]
- Turk D., Dworkin R., Allen R., Bellamy N., Brandenburg N., Carr D., et al. (2003) Core outcome domains for chronic pain clinical trials: IMMPACT recommendations. Pain 106: 337–345 [DOI] [PubMed] [Google Scholar]
- Ware J., Snow K., Kosinski M., Gandek B. (1993) SF-36 Health Survey Manual and Interpretation Guide. Boston, MA: The Health Institute, New England Medical Center [Google Scholar]
- Wernicke J., Pritchett Y., D’Souza D., Waninger A., Tran P., Iyengar S., et al. (2006) A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology 67: 1411–1420 [DOI] [PubMed] [Google Scholar]
- Whitmyer V., Dunner D., Kornstein S., Meyers A., Mallinckrodt C., Wohlreich M., et al. (2007) A comparison of initial duloxetine dosing strategies in patients with major depressive disorder. J Clin Psychiatry 68: 1921–1930 [DOI] [PubMed] [Google Scholar]
- Woolf A., Pfleger B. (2003) Burden of major musculoskeletal conditions. Bull World Health Organ 81: 646–656 [PMC free article] [PubMed] [Google Scholar]
- Woolf C. (2004) Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med 140: 441–451 [DOI] [PubMed] [Google Scholar]
- World Health Organization (2002) World Health Report 2002. Reducing Risks, Promoting Healthy Life. Geneva: WHO [Google Scholar]
- Zhang W., Doherty M., Peat G., Bierma-Zeinstra M., Arden N., Bresnihan B., et al. (2010) EULAR evidence-based recommendations for the diagnosis of knee osteoarthritis. Ann Rheum Dis 69: 483–489 [DOI] [PubMed] [Google Scholar]
- Zhang W., Moskowitz R., Nuki G., Abramson S., Altman R., Arden N., et al. (2008) OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage 16: 137–162 [DOI] [PubMed] [Google Scholar]