

Several proteasome inhibitors have been identified, including peptide boronates (Bortezomib, its bidendate analog BU-321, and MG-262), epoxiketones (Carfilzomib, also called Kyprolis, which has recently been approved for treatment of multiple myeloma (MM) patients by the FDA), PR047, and Epoxomicin, peptide aldehydes (MG-132 and PSI), and β-lactones (NPI-0052, also called Salinosporamide A and lactacystin).2–5 Several proteasome inhibitors that are currently in clinical trials including MLN9708, CEP-18770, Carfilzomib, PR-047, and NPI-0052, a β-lactone.6 In addition, allosteric chemical inhibitors that target the proteasome outside of the active site have been developed, including Chloroquine and its analog 5-amino-8-hydroxyquinoline (5AHQ).7 The inhibitory potency and target selectivity towards the 20S proteasome of boronic acid-based inhibitors are quite remarkable (in the nM range). This is presumably due to the fact that an empty p-orbital on a boron atom is positioned to accept the oxygen lone pair of the amino terminal threonine residue of the 20S proteasome to form a stable tetrahedral intermediate.8 To date, Bortezomib and Carfilzomib are the only proteasome inhibitors approved for treatment of MM. Despite its remarkable effect in myeloma therapy, significant toxicities, such as peripheral neuropathy and thrombocytopenia, have restricted the intensity of Bortezomib dosing and its clinical efficacy has been hampered by the emergence of resistance. There is an urgent need for new approaches to selectively target the delivery of effective anticancer drugs such as Bortezomib specifically to bone tissue, where myeloma cells reside, to ameliorate and/or prevent the toxic side effects arising from systemic distribution of these drugs and to make dose escalation possible in order to improve therapeutic outcome.
Almost all patients with MM have an early excessive bone resorption leading to lytic lesions and sometimes to hypercalcemia.9,10 Interactions between myeloma cells and bone cells and the extracellular matrix proteins within the bone microenvironment underlie the progression of multiple myeloma and are mediated through cell surface receptors.11 These interactions trigger a self-amplifying cascade of events that result in the secretion of cytokines and growth factors that promote the growth and proliferation of myeloma cells, increase bone resorption and enhance drug resistance by inducing antiapoptotic pathways.11–13 Bisphosphonates, which are degradation-resistant analogs of inorganic pyrophosphate, exhibit exceptional affinity for bone and have traditionally been the mainstay therapy for the treatment of skeletal related events associated with myeloma.13 However, there may be a greater role for the use of bisphosphonates than has previously been considered. Recent reports suggest that they may act as antitumor agents14, able to delay disease progression and prolong survival in multiple myeloma15, and in solid tumors such as breast16 and prostate cancers.17,18 These observations, together with the efficacy of proteasome inhibitors in myeloma treatment and recent reports that suggest that proteasome inhibitors may stimulate new bone formation,19,20 strongly argue for the use of proteasome inhibitor-bisphosphonate conjugates to target these potent drugs selectively to bone tissue in order to reduce off-target adverse effects and improve treatment outcomes. The important message is that by targeting proteasome inhibitors to bone, it may be possible to accentuate the beneficial bone anabolic effects of these compounds, kill myeloma cells more effectively, and prevent or reduce toxic side effects associated with the current mode of administration.
In order to evaluate the therapeutic potential of bone-targeted proteasome inhibitors, we used the procedures outlined in Schemes 1–3 to synthesize a series of boronated proteasome inhibitor-bisphosphonate conjugates: PS-341-BP-1, PS-341-BP-2 and MG-262-BP (Figure 1) containing the proteasome inhibitors PS-341 and MG-262 as the active components. To synthesize PS-341-BP-1 (Scheme 1), 2,5-dimethylpyrazine was converted to pyrazine-2,5-dicarboxylic acid (2) according to the literature procedure.21 Hydrogen chloride gas was bubbled through a solution of compound 2 in methanol to form the di-ester 3 which was treated with 1.0M sodium hydroxide to afford the mono-ester 4. The key boronic acid dipeptide pinanediol ester (5) intermediate, which was synthesized in six steps using the literature procedure,22,23 was coupled to compound 4 to produce 6. The methyl ester 6 was hydrolyzed to the acid 7 which was then conjugated to the bisphosphonate alendronate followed by pinanediol deprotection to afford PS-341-BP-1. The synthesis of PS-341-BP-2 was achieved using the procedure outlined in Scheme 2. The key intermediate pinanediol leucine boronated trifluoroacetate salt 8 was synthesized using the published procedure,22,23 and was coupled to N-Boc-tyrosine followed by deprotection of the Boc group to produce compound 9. Next, compound 9 was coupled to 2-pyrazine carboxylic acid to give 10 which was then coupled to ethyl 4-bromobutanoate to yield the corresponding ether 11. After hydrolysis of the ethyl ester, the carboxylic acid was converted to the N-hydroxysuccinimide ester and then coupled to alendronate to give the pinanediol boronate ester which was deprotected to afford PS-341-BP-2. Next, the synthesis of MG-262-BP was achieved using the procedure outlined in Scheme 3. 4-(Methoxycarbonyl)benzyl 1H-imidazole-1-carboxylate 12 was reacted with the boronated tripeptide 13 to give the carbamate intermediate 14. After hydrolysis of the methyl ester and activation of the carboxylic acid followed by reaction with alendronate, compound 15 was obtained. After deprotection of the boronate, MG-262-BP was obtained. Details of the experimental procedures are provided in the supplementary data.
Scheme 1.
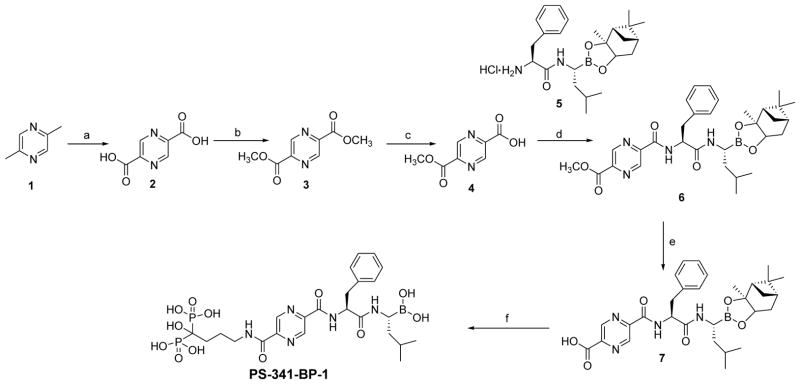
Synthesis of PS-341-BP-1
Key: (a) SeO2, H2O, pyridine, 68%; (b) MeOH, HCl gas, 94%; (c) MeOH, NaOH, 73%; (d) 5, TBTU, DIEA, DMF, 60%; (e) LiOH, THF/MeOH/H2O, 47%; (f) (i) NHS, DCC, DME; (ii) alendronate, DIEA, 2-propanol/H2O; (iii) (2-methylpropyl)boronic acid, MeOH/Hexane/2M HCl
Scheme 3.
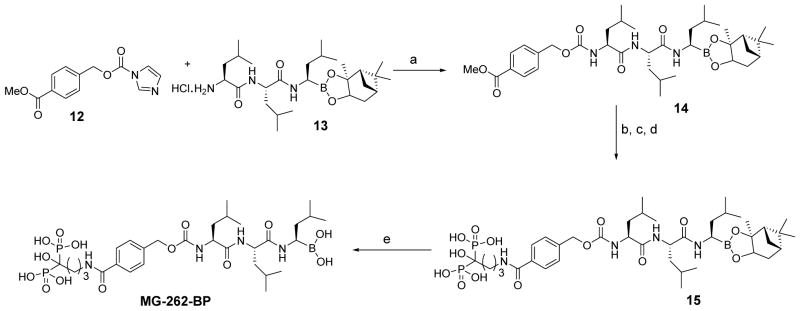
Synthesis of MG-262-BP
Key: (a) DMAP, CH2Cl2; (b) LiOH, THF/MeOH/H2O; (c) NHS, DCC, DMF; (d) alendronate, DIEA, H2O/2-propanol; (e) (2-methylpropyl)boronic acid, MeOH/hexane/2M HCl
Figure 1.
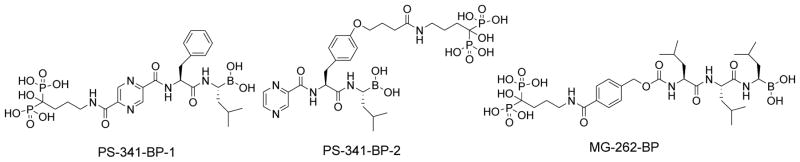
Chemical structures of bone-targeted proteasome inhibitors
Scheme 2.
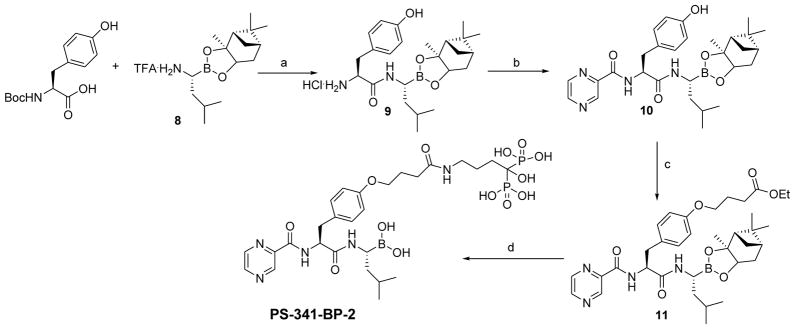
Synthesis of PS-341-BP-2
Key: (a) TBTU, DIEA, DMF, followed by deprotection with HCl in dioxane; (b) 2-pyrazine carboxylic acid, TBTU, DIEA, DMF, 40%; (c) ethyl 4-bromobutyrate, K2CO3, DMF; (d) (i) LiOH, MeOH/H2O/THF; (ii) NHS, DCC, DME; (iii) alendronate, DIEA, H2O/2-propanol; (iv) (2-methylpropyl)boronic acid, MeOH/hexane/2M HCl
The sulforhodamine B assay (SRB assay) was used to evaluate the antiproliferative and cytotoxic effects of the bone-targeted proteasome inhibitors PS-341-BP-1, PS-341-BP-2 and MG-262-BP using the mouse 5TGM1 and human RPMI 8226 cell lines in vitro as described.24,25 RPMI 8226 and 5TGM1 cells were purchased from the American Type Culture Collection. The cell lines were seeded in triplicate at 3,000 cells per well in 96-well plates in RPMI containing 10% fetal bovine serum. After 24 h, the cells were treated with various drug concentrations. Colorimetric analyses were done after 72 h of growth in the presence of drug. The IC50 represents the mean of at least 3 independent experiments using triplicate points in each experiment. The results of the SRB assay revealed that all three BP conjugates inhibited cell viability and proliferation, after 72 h of treatment, in a dose-dependent manner (Figure 2), with a half maximal inhibitory concentration (IC50) ranging from 4.89 – 9.39 nM compared with 6.78 nM for PS-341 in 5TGM1 and 10.18 – 13.76 nM compared with 9.50 nM for PS-341 in RPMI 8226 (Table 1). The choice of a suitable linker for the bisphosphonate is of fundamental importance to the success of these bone-targeted pro-drug conjugates, as linkers that are too stable cannot release the drug whereas linkers that are too labile result in premature release of the drug. To ensure ready release of the proteasome inhibitors in the tumor microenvironment, we plan to investigate a series of BP-drug pharmacophores containing more labile linkers such as carbamate, carbonate, and ester moieties to determine the best linker for targeting these drugs to bone. The use of bone-targeted nanoparticles loaded with these drugs will be an alternative approach to deliver them to the bone microenvironment, which we are currently investigating.
Figure 2.

Cell viability studies of proteasome inhibitors against RPMI 8226 (A) and 5TGM1 (B)
Effect of proteasome inhibitor on myeloma cell proliferation. Proliferation assay of RPMI8226 (A) and 5TGM1 (B) myeloma cells were treated with different concentrations (1–18 nM) of proteasome inhibitors for 72h. Results are mean of three independent experiments.
Table 1.
Cell viability was measured with SRB assay reagent after compound exposure for 48 h.
| Cell line | PS-341 (nM) | PS341-BP-1 (nM) | PS341-BP-2 (nM) | MG-262 (nM) | MG-262 BP (nM) |
|---|---|---|---|---|---|
| 5TGM1 | 6.78+0.57 | 7.59+ 0.68 | 9.39+ 0.78 | 9.18+0.54 | 4.89+ 0.12 |
| RPMI8226 | 9.50+ 0.48 | 10.18+ 0.87 | 11.51+ 0.92 | 13.76+ 0.89 | 12.47+ 0.87 |
* IC50 values were determined in each of the cell lines from a graph of at least six drug concentrations that span the entire curve of growth inhibition as means of 3 independent experiments ±SD.
In summary, we have synthesized a series of proteasome inhibitor-bisphosphonate conjugates that are potent inhibitors of cell viability and proliferation in vitro in 5TGM1 and RPMI 8226 myeloma cell lines. This study could serve as a basis to provide promising bone-targeted drugs for effective treatment of multiple myeloma. We plan to address this in future studies.
Supplementary Material
Acknowledgments
This work was supported by grants KO1 CA113468 and P30 CA054174 (to JKA) from the National Cancer Institute, and by grants from the Kerr, and William & Ella Owens Research Foundations (JKA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Agyin JK, Santhamma B, Nair HB, Roy SS, Tekmal RR. Breast Cancer Res. 2009;11:R74. doi: 10.1186/bcr2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traenckner EB, Wilk S, Baeuerle PA. EMBO J. 1994;13:5433. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Proc Natl Acad Sci U S A. 1999;96:10403. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mc Cormack T, Baumeister W, Grenier L, Moomaw C, Plamondon L, Pramanik B, Slaughter C, Soucy F, Stein R, Zuhl F, Dick L. J Biol Chem. 1997;272:26103. doi: 10.1074/jbc.272.42.26103. [DOI] [PubMed] [Google Scholar]
- 5.Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL, Rock KL. J Biol Chem. 1997;272:13437. doi: 10.1074/jbc.272.20.13437. [DOI] [PubMed] [Google Scholar]
- 6.Dick LR, Fleming PE. Drug Discov Today. 2010;15:243. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Wood TE, Sprangers R, Jansen G, Franke NE, Mao X, Wang X, Zhang Y, Verbrugge SE, Adomat H, Li ZH, Trudel S, Chen C, Religa TL, Jamal N, Messner H, Cloos J, Rose DR, Navon A, Guns E, Batey RA, Kay LE, Schimmer AD. J Natl Cancer Inst. 2010;102:1069. doi: 10.1093/jnci/djq198. [DOI] [PubMed] [Google Scholar]
- 8.Myung J, Kim KB, Crews CM. Med Res Rev. 2001;21:245. doi: 10.1002/med.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barille S, Akhoundi C, Collette M, Mellerin MP, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Blood. 1997;90:1649. [PubMed] [Google Scholar]
- 10.Roodman GD. J Cell Biochem. 2010;109:283. doi: 10.1002/jcb.22403. [DOI] [PubMed] [Google Scholar]
- 11.Corso A, Ferretti E, Lazzarino M. Hematology. 2005;10:215. doi: 10.1080/10245330500094714. [DOI] [PubMed] [Google Scholar]
- 12.Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Lancet. 2009;374:324. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- 13.Modi ND, Lentzsch S. Leukemia. 2012;26:589. doi: 10.1038/leu.2011.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costa L, Harper P, Coleman RE, Lipton A. Crit Rev Oncol Hematol. 2011;77(Suppl 1):S31. doi: 10.1016/S1040-8428(11)70006-3. [DOI] [PubMed] [Google Scholar]
- 15.Morgan GJ, Davies FE, Gregory WM, Cocks K, Bell SE, Szubert AJ, Navarro-Coy N, Drayson MT, Owen RG, Feyler S, Ashcroft AJ, Ross F, Byrne J, Roddie H, Rudin C, Cook G, Jackson GH, Child JA National Cancer Research Institute Haematological Oncology Clinical Study, G. . Lancet. 2010;376:1989. [Google Scholar]
- 16.Gnant M, Mlineritsch B, Schippinger W, Luschin-Ebengreuth G, Postlberger S, Menzel C, Jakesz R, Seifert M, Hubalek M, Bjelic-Radisic V, Samonigg H, Tausch C, Eidtmann H, Steger G, Kwasny W, Dubsky P, Fridrik M, Fitzal F, Stierer M, Rucklinger E, Greil R, Marth C. N Engl J Med. 2009;360:679. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 17.Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, Miller K, Sieber P, Karsh L, Damiao R, Tammela TL, Egerdie B, Van Poppel H, Chin J, Morote J, Gomez-Veiga F, Borkowski T, Ye Z, Kupic A, Dansey R, Goessl C. Lancet. 2012;379:39. doi: 10.1016/S0140-6736(11)61226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coleman R, Gnant M, Morgan G, Clezardin P. J Natl Cancer Inst. 2012;104:1059. doi: 10.1093/jnci/djs263. [DOI] [PubMed] [Google Scholar]
- 19.Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. Am J Hematol. 2009;84:6. doi: 10.1002/ajh.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zangari M, Esseltine D, Lee CK, Barlogie B, Elice F, Burns MJ, Kang SH, Yaccoby S, Najarian K, Richardson P, Sonneveld P, Tricot G. Br J Haematol. 2005;131:71. doi: 10.1111/j.1365-2141.2005.05733.x. [DOI] [PubMed] [Google Scholar]
- 21.Schut WJ, Mager HIX, Berends W. Recueil Des Travaux Chimiques Des Pays-Bas-Journal of the Royal Netherlands Chemical Society. 1961;80:391. [Google Scholar]
- 22.Shenvi AB, Kettner CA. 4,499,082 Patent, U S, Ed USA. 1985
- 23.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Bioorg Med Chem Lett. 1998;8:333. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 24.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. Cancer Res. 2008;68:8881. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 26.Uludag H. Curr Pharm Des. 2002;8:1929. doi: 10.2174/1381612023393585. [DOI] [PubMed] [Google Scholar]
- 27.Pierce WM, Jr, Waite LC. Proc Soc Exp Biol Med. 1987;186:96. doi: 10.3181/00379727-186-42590a. [DOI] [PubMed] [Google Scholar]
- 28.Gil L, Han Y, Opas EE, Rodan GA, Ruel R, Seedor JG, Tyler PC, Young RN. Bioorg Med Chem. 1999;7:901. doi: 10.1016/s0968-0896(99)00045-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.