

Abstract
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene cause late-onset Parkinson's disease (PD). Emerging evidence suggests a role for LRRK2 in the endocytic pathway. Here, we show that LRRK2 is released in extracellular microvesicles (i.e. exosomes) from cells that natively express LRRK2. LRRK2 localizes to collecting duct epithelial cells in the kidney that actively secrete exosomes into urine. Purified urinary exosomes contain LRRK2 protein that is both dimerized and phosphorylated. We provide a quantitative proteomic profile of 1673 proteins in urinary exosomes and find that known LRRK2 interactors including 14-3-3 are some of the most abundant exosome proteins. Disruption of the 14-3-3 LRRK2 interaction with a 14-3-3 inhibitor or through acute LRRK2 kinase inhibition potently blocks LRRK2 release in exosomes, but familial mutations in LRRK2 had no effect on secretion. LRRK2 levels were overall comparable but highly variable in urinary exosomes derived from PD cases and age-matched controls, although very high LRRK2 levels were detected in some PD affected cases. We further characterized LRRK2 exosome release in neurons and macrophages in culture, and found that LRRK2-positive exosomes circulate in cerebral spinal fluid (CSF). Together, these results define a pathway for LRRK2 extracellular release, clarify one function of the LRRK2 14-3-3 interaction and provide a foundation for utilization of LRRK2 as a biomarker in clinical trials.
INTRODUCTION
Missense mutations in the Leucine-Rich Repeat Kinase 2 (LRRK2) gene cause late-onset Parkinson's disease (PD) in both autosomal-dominant disease transmitting families (1,2) and sporadic late-onset disease populations (3–5). In addition to linkage to PD through genome-wide association studies (6), LRRK2 genetic variants impose susceptibility risks to inflammation-linked diseases that include Crohn's disease and mycobacterium infection (7,8). The LRRK2 gene encodes a protein with a unique multi-domain composition, including functional GTPase and protein kinase domains, and missense mutations that cause PD alter these enzymatic activities (9,10). Understanding the function of LRRK2 may provide insight into pathogenic mechanisms as well as uncover particular targets for disease-modifying therapeutics.
LRRK2 expression in mammals is widely distributed in many cell types, but particularly enriched in the kidney and in activated macrophages of the innate immune system (11–13). In the brain, LRRK2 expression is relatively modest and includes medium spiny neurons in the striatum that form striosomes (14). On a subcellular level, LRRK2 associates with a number of vesicle types and with intraluminal vesicles within multivesicular bodies (MVBs) (15–18). Assignment of LRRK2 function within the endocytic pathway has been suggested (15–25), with recent evidence for action in retrograde vesicle trafficking from endosomes to the trans-Golgi network (26), and for modifying chaperone-mediated autophagy (27). Over expression of the late-endosome GTPase Rab7 and related Rab7 isoforms rescue LRRK2-mediated phenotypes in neurons (26), while RNAi knockdown of ArfGAP1, a protein associated with vesicle biogenesis, also rescues LRRK2 phenotypes in neurons (24). Thus, compelling experimental evidence suggests that interaction in the endocytic pathway is at the heart of LRRK2 function in health and disease, yet it is not known how LRRK2 interacts in this pathway and whether there are other aspects to LRRK2 interaction with vesicles that have not been considered.
Elevated LRRK2 kinase activity is linked to PD susceptibility since the most common pathogenic LRRK2 mutation G2019S increases kinase activity ∼3-fold in most assays (9,10,28,29), and overall LRRK2 subcellular localization has been described as exquisitely sensitive to acute LRRK2 kinase inhibition (30). LRRK2 interaction with 14-3-3 proteins appears to be one of the main modulators of LRRK2 subcellular localization (30), consistent with the notion that 14-3-3 isoforms principally function by modulating cell localization of binding partners. However, there are no known physiological roles described for the 14-3-3 LRRK2 interaction or impact that 14-3-3-mediated localization may have on LRRK2 functionality, particularly within the endocytic pathway.
Here, we find that LRRK2 is secreted from intra-luminal vesicles from MVBs (i.e. exosomes) from a variety of cells where LRRK2 is natively expressed, including cells in the kidney, brain and immune system. LRRK2 can be readily detected through purification of exosomes from urine or cerebral spinal fluid (CSF) in clinical populations, or through exosomes from cell culture media. We find that a major role for the 14-3-3 LRRK2 interaction may be the regulation of LRRK2 association with late endosomes and uptake into MVBs with a subsequent extracellular release of LRRK2 protein via exosome secretion. These studies elucidate a new component to LRRK2 functionality in the endocytic pathway and open the door for LRRK2-targeted biomarker and clinical trial studies through characterization of a convenient source for LRRK2 protein (e.g. urine and CSF). Future studies may show that direct LRRK2 action extends to cells that lack autonomous LRRK2 expression through the process of exosome uptake and incorporation of active LRRK2 protein.
RESULTS
Identification of LRRK2 in exosomes
LRRK2 action in the endocytic pathway may include aspects of lysosomal and autophagy-mediated degradation, receptor recycling, and vesicle budding. Evidence of LRRK2 association with MVBs, the obligate source of exosomes, was observed previously (16). Our past studies demonstrated that the highest expression of endogenous LRRK2 in mice is in the kidney (13), and the kidneys are known to secrete high quantities of exosomes into urine. To test whether LRRK2 can be secreted in urinary exosomes, we isolated a 100K × g pellet (P100) into lysis buffer together with an equivalent-volume concentrated supernatant from healthy human subjects and measured the LRRK2 protein content via western blot (Fig. 1A). LRRK2 was present at an approximate concentration of 1 pg/ml of urine, and all of the LRRK2 protein localized to the P100 (exosome) fraction. Exosomes are organelles that maintain the orientation of the proteins from the parental cells, so that extracellular proteins or extracellular protein domains remain extra-exosomal, whereas cell cytosolic proteins are encapsulated within the exosome (31). A trypsin-sensitivity assay demonstrated that the addition of triton-X 100 detergent to break the vesicles was required for proteolytic digestion of LRRK2, suggesting that LRRK2 is localized within exosomes (Fig. 1B). The LRRK2 binding partner 14-3-3 was also sensitive to trypsin degradation in the presence of a detergent. In contrast, CD9 is known to be present in a large trans-luminal protein complex within the plasma membrane that was resistant to trypsin digestion whether or not the detergent was added.
Figure 1.

Localization of LRRK2 to urinary exosomes. (A) Human urine was processed into equivalent volumes of supernatant S100 (exosome depleted), P100 (exosome enriched) or the P100 wash buffer. Recombinant LRRK2 is Δ970-LRRK2 (Invitrogen). (B) Ten micrograms of purified urinary exosomes, treated with or without 200 ng trypsin, with or without 1% Triton X-100, for 5 min at 37°C. n.s. is non-specific band. (C) P100 urinary exosomes separated across a density gradient and probed for expression of the indicated protein. (D) Representative cryo- EM image, scale bar is 50 nm. (E) LRRK2-fluorescent imaging in urinary exosomes visualized by STED microscopy, scale bar is 500 nm. (F) Fluorescent co-labeling of LRRK2 (green) together with TSG101 or CD9. Arrows indicate positive co-localization, scale bar is 2 μm. (G) Blue NativePAGE of LRRK2 protein from purified urinary exosomes, treated with or without 1% Triton X-100. (H) Comparison of relative LRRK2 expression and phospho-serine 935 in 10 µg purified urinary exosomes with 10 µg of protein lysate from human brain (frontal cortex), or 1 ng of recombinant LRRK2 isolated from HEK-293 T cells.
Next, the P100 fraction was separated on an iodixanol density gradient and LRRK2 was found to float in the same fraction as the canonical exosome proteins Alix and TSG101 (Fig. 1C), suggesting that LRRK2 specifically resides within exosomes. CD9 is a transmembrane protein that is abundant in plasma membrane-derived vesicles that traffic to MVBs (32,33). We found that CD9 diffusely spreads across the high-density fractions, with the highest concentration in the exosome-containing fraction. Under native (non-crosslinked or dehydrated) conditions, the exosomes were perfectly spherical with intra-exosome puncta composing a dense vesicle core, as visualized using cryo-electron microscopy (EM, Fig. 1D). We localized LRRK2 immunoreactivity to isolated exosomes using a super-resolution technique (stimulated emission depletion microscopy) and found structures also of ∼100 nm in size with LRRK2 protein primarily concentrated in the exosome core (Fig. 1E). CD9 has been previously used in fluorescent and immunoaffinity approaches to purify exosomes, but we found complete exclusion of LRRK2 immunoreactivity in CD9-positive exosomes via immunofluorescence in dilute exosome preparations (Fig. 1F). In contrast, LRRK2 partially co-localized to TSG101-enriched exosomes, although examples of TSG101-positive exosomes with weak or no detectable LRRK2 expression could also be observed. As CD9 is a ubiquitously expressed plasma membrane protein, this could indicate that the intracellular source of LRRK2-positive exosomes could be distinct from plasma membrane-derived exosomes.
Both LRRK2 dimerization and phosphorylation are activities that have been linked to LRRK2 kinase activity (34,35). To assess LRRK2 dimerization, urinary exosomes were resuspended in buffer containing the non-ionic detergent triton X-100 and lysates analyzed by native-PAGE (Fig. 1G). Urinary exosome LRRK2 migration was identical to that of endogenous and kinase-active LRRK2 isolated from cell cytosols (34). Low amounts of LRRK2 in preparations without detergent treatment were likely due to exosome breakage during processing, but show that the detergent did not alter the migration pattern of LRRK2 in native gels. To measure LRRK2 phosphorylation, 10 µg of total protein from exosomes was compared with 10 µg of total protein isolated from low-post-mortem interval human brain cerebral cortex (brain samples described previously for LRRK2 expression (36)), together with 1 ng of recombinant LRRK2 protein derived from HEK-293T cells (Fig. 1H). The recombinant protein is known to be heavily phosphorylated at serine 935 (pS935) (10). pS935 levels were comparable between recombinant kinase-active LRRK2 and urinary exosome-derived LRRK2. In comparison, much lower levels of total and pS935 LRRK2 were detected in human brain.
These results demonstrate LRRK2 can be readily measured from urinary exosomes, and that LRRK2 protein in exosomes derives from the cell cytosol, either captured in vesicles that fuse with MVBs, or packaged during the inward internalization of vesicles fusing with MVBs. Although LRRK2 protein concentration is appreciable in urinary exosomes, the overall levels are not sufficient to directly assess LRRK2 kinase activity with any described kinase activity assay.
Localization of LRRK2 to kidney luminal tubule cells and characterization of urinary exosome proteomes
The sources of urinary exosomes are not fully understood, although the kidney is thought to be the major contributor (37). Staining for LRRK2 in healthy rat kidneys revealed intense LRRK2 expression in virtually all segments of the outer and inner collecting duct epithelium, with reactive cells directly abutting luminal spaces (Fig. 2A). In contrast, rat LRRK2 KO kidneys are non-reactive under identical staining conditions, albeit with minor nonspecific reactivity observed in a subset of nuclei (Fig. 2B). Since the collecting duct epithelium cells specifically are hypothesized to be contributors toward the urinary exosome pool, these results explain in part the source of LRRK2 in urinary exosomes.
Figure 2.

Localization of LRRK2 in the kidney and proteomic characterization of urinary exosomes. (A) Coronal rat kidney sections from WT or (B) KO animals, stained with LRRK2 antibody(c41-2, Epitomics) and counterstained with H&E. Scale bars for low-magnification images (left panels) are 200 μm, and 25 μm for high magnification (right panels). (C) Quantitative TMT-MudPIT results for the most abundant exosome proteins in comparison to known LRRK2 protein interactors. Error bars are SD generated by spectral counts from six differential isobaric tags. (D) Urinary exosome proteins identified by the tissue-specific gene expression and regulation database.
It is possible that LRRK2 protein interactors in exosomes may chaperone and control LRRK2 vesicular association. Although several urinary exosome proteomic studies have been performed, these have been qualitative assessments that have not capitalized on recent and dramatic improvements in the proteomic technology. To quantitatively measure the exosome proteome, urinary exosomes were pooled from six healthy controls, and the resultant exosomes were split into six equal fractions that were labeled with tandem isobaric mass tags and analyzed by long-column multi-dimensional protein identification (TMT-MudPIT). Five two hundred and sixty-eight peptides were identified and quantifiable that corresponds to 1673 protein identifications (Supplementary Material, Database 1). Of the 1673 proteins, 965 mapped to unique GeneGO objects with assigned function, and the GO process ‘cellular component organization’ was significantly enriched, owing to the high concentration of components of the actin cytoskeleton and known vesicle components [false discovery rate (FDR)-adjusted P = 5.2 × 10−41]. A number of proteins associated with neurodegenerative diseases were identified in exosomes besides LRRK2, such as DJ-1, ApoE and Nerprelysin. LRRK2-interacting proteins, including 14-3-3ɛ (Supplementary Material, Fig. S1), HSP70/90, ezrin/moesin/ radixin, and Rab7, were all identified at levels comparable with the most abundant exosome proteins (Fig. 2C). Despite previous studies that indicate some of these interactors as potential kinase substrates, we were unable to show that LRRK2 could phosphorylate any proteins from exosome lysates (Supplementary Material, Fig. S2).
Among the urinary exosome proteins detected, several highly expressed proteins were identified that are considered exclusive or highly enriched in organs other than the kidney (38). These organs include prostate, stomach, pancreas, liver and colon (Fig. 2D), where LRRK2 expression of differing levels can also be detected (13). Notably, we did not obtain any evidence that brain specific proteins were present in urinary exosomes. Thus, it is possible that LRRK2 protein in urinary exosomes originate from these organs, in addition to the kidney.
14-3-3 Binding to LRRK2 controls LRRK2 exosome release
We and others have found that LRRK2 may be tightly bound to heat-shock proteins and 14-3-3 chaperones that may control LRRK2 solubility and oligomerization (10,30,39,40). We sought to test whether interactions with these proteins may control LRRK2 extracellular secretion. First, we determined that HEK-293T cells transfected with LRRK2 actively secrete exosomes into cell culture media (Fig. 3A). While knockdown of all 14-3-3 isoforms in HEK-293T cells is difficult to accomplish, a short-peptide inhibitor known as difopein has been developed in HEK-293T cells that effectively acts as a pan 14-3-3 inhibitor by blocking 14-3-3 dimerization (41). Transfection of difopein in LRRK2-expressing HEK-293T cells resulted in a very efficient ablation of LRRK2 binding to 14-3-3 proteins, as observed through immunoprecipitation assays using a pan-14-3-3 antibody (Fig. 3B). In cells expressing both LRRK2 and difopein, LRRK2 could no longer be detected in resultant exosome fractions, yet cytosolic levels of LRRK2 and 14-3-3 (pan) remained unaltered. Likewise, difopein treatment did not have any significant effects on total exosome release, indicating 14-3-3 proteins are dispensable for exosome biogenesis and processing.
Figure 3.

LRRK2 exosome release is regulated by 14-3-3 (A) Representative cryo-EM image of exosomes purified from HEK-293 T cells expressing LRRK2 protein, scale bar is 100 nm. (B) HEK-293 T cells expressing LRRK2 protein were co-transfected with eGFP, scrambled difopein (a control for difopein, see (41)) or difopein. LRRK2 was immunoprecipitated and exosomes collected from culture media. (C, D) Dose-response curves measuring p1503 autophosphorylation (Alpha Screen signal) in in vitro kinase assays at indicated drug concentrations. IC50 concentrations were calculated through non-linear regression. (E) HEK-293T cells expressing LRRK2 were treated with the indicated drug (1 µm) or equivalent DMSO concentrations (0.01%) for 36 h. Cells were harvested into total cell lysates, LRRK2 immunoprecipitated and exosomes collected.
Acute LRRK2 kinase inhibition via small molecules causes a reduction in 14-3-3 binding to LRRK2 (30). To test whether acute kinase inhibition-mediated loss of 14-3-3 binding would also disrupt LRRK2 release in exosomes, we first characterized the two most potent and specific LRRK2 kinase inhibitors described. HG-10-102 (42), that is known to be a selective LRRK2 inhibitor, and the widely utilized L2in1 compound (35), were first defined for potency in a kinase inhibition assay measuring LRRK2 autophosphorylation in vitro (Fig. 3C,D). When applied to HEK-293T cells at 1 µm concentration, these two compounds had comparable effects in reducing 14-3-3 (pan) binding to LRRK2 and reducing LRRK2 release in exosomes (Fig. 3E). Unexpectedly, treatment with L2in1, a known inhibitor of ERK5, and possibly Aurora A and CHK2, also blocked overall exosome release in HEK-293T cells, as determined by lower levels of TSG101 (Fig. 3E) and other markers evaluated such as Alix and CD9. Over expression of 14-3-3ɛ, the most abundant 14-3-3 isoform identified in urinary exosomes (Supplementary Material, Fig. and Database 1), restored 14-3-3 binding to LRRK2 and exosome release (Fig. 3E).
The cytosolic distribution of LRRK2 may be important for LRRK2 packaging into exosomes. Using immunofluorescence localization in HEK-293T cells over-expressing LRRK2 protein, we observe a diffuse yet punctate cytoplasmic localization of LRRK2 Figure 4A). In cells co-expressing the small peptide difopein, LRRK2 redistributes to concentrated perinuclear structures (Fig. 4C), whereas exposure to LRRK2 kinase inhibitors renders LRRK2 to skein-like structures (Fig. 4E,F), consistent with previous reports evaluating L2in1 exposures (30). We found that 14-3-3ɛ over expression rescued the normal localization of LRRK2 (Fig. 4G,H). These results demonstrate how 14-3-3 binding to LRRK2 alters subcellular localization, where diffuse cytoplasmic distribution correlates to extracellular secretion.
Figure 4.
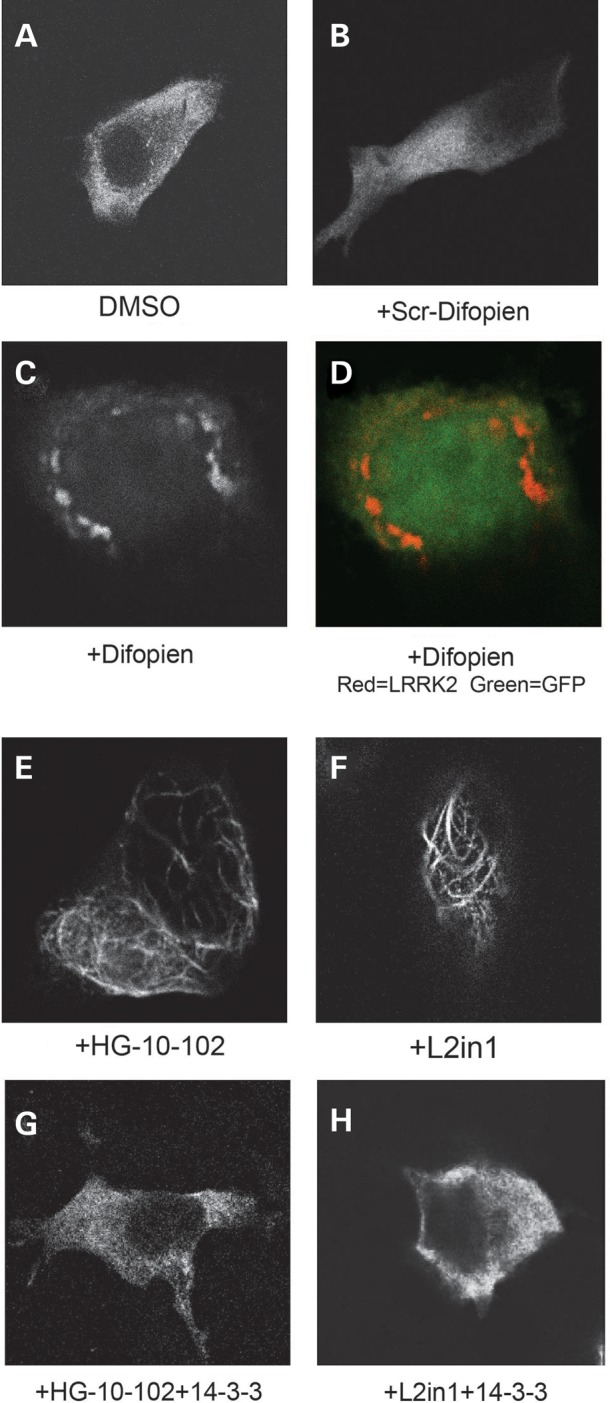
LRRK2 cytoplasmic localization is regulated by 14-3-3 (A–H) Representative confocal images of HEK-293T cells expressing mKate2-tagged LRRK2 (N-terminal tag), with cells treated with the indicated drug and/or co-transfected with the indicated construct. Drugs were used at 1 µm concentration for 36 h.
Because LRRK2 cytoplasmic distribution appears to critically mediate extracellular secretion, we next co-localized LRRK2 with the exosome marker TSG101 in HEK-293T cells that constitutively secrete exosomes. While TSG101 had a discrete vesicular-like distribution, LRRK2 was more evenly expressed across the cytosol with rarer examples of discrete vesicle-sized puncta, clearly distinguishing LRRK2 from a canonical vesicular protein (Fig. 5A). Some previous evidence suggests that the G2019S mutation may alter this cytoplasmic distribution to LRRK2-positive intracellular puncta, so we tested whether this mutation affects LRRK2 secretion. Neither G2019S nor a kinase-inactivating mutation altered overall LRRK2 secretion (Fig. 5B). These results are also consistent with past data that these alterations in LRRK2 do not affect binding to 14-3-3 proteins (39).
Figure 5.

Lack of effect of LRRK2 mutations and lack of localization with α-synuclein in exosomes (A) Immunofluorescence co-localization of mKate2-LRRK2 (red signal) with the MVB marker TSG101 using confocal immunofluorescence. A representative cell is shown. (B) LRRK2-myc, with the indicated mutation, was transiently transfected into HEK-293T cells and exosomes and cytosol lysates collected 48 h after transfection. (C) Purified exosomes from HEK-293 T cells transfected with mKate2-LRRK2 (red signal) and α-synuclein (blue signal). No overlapping blue/red signal could be observed. All data are representative of at least three independent experiments, scale bars represent 10 µm.
The other protein linked to autosomal-dominant PD, α-synuclein, can also be secreted in exosomes (43). To determine whether LRRK2 may co-exist in the same exosomes as α-synuclein, we transiently co-expressed both proteins in HEK-293T cells, isolated exosomes, and analyzed the composition of the structures by immunofluorescence. No examples of co-localized exosomes were observed, suggesting that α-synuclein and LRRK2 may derive from exosomes of divergent origin. (Fig. 5C). α-Synuclein is also a binding partner for 14-3-3 isoforms, although the α-synuclein complex in exosomes has not been described.
Quantification of LRRK2 in clinical exosome samples
We collected a cohort of late-onset PD without a family history of disease (n = 20) and age-matched control samples (n = 15) from the Movement Disorder Clinic at the University of Alabama at Birmingham, following informed consent and institutional approval, and measured LRRK2 and 14-3-3(pan) expression relative to the canonical exosome marker TSG101. Age between the PD and control cohort was similar at 62.8 ± 9.4 and 62.5 ± 13.1, respectively, and the modified Hoehn and Yahr score of the PD group was 2.2 ± 0.38.
We found that both LRRK2 and 14-3-3(pan) had unexpectedly high variability in both controls and PD, as LRRK2 levels varied in the PD affected group by over two orders of magnitude, in contrast to TSG101 that showed lower variability (Fig. 6A). After exclusion of outliers from the series (identified with at least one order of magnitude above or below the population average), there were no significant differences in the expression of LRRK2 or 14-3-3 in PD versus controls (P > 0.1, Fig. 6B and C), nor did LRRK2 expression correlate well with 14-3-3 (pan) expression (Fig. 6D). There were no effects of age or gender on LRRK2 or 14-3-3 expression in urinary exosomes. High levels of LRRK2 in exosomes, more than 50-fold above the cohort average, were detected in one PD-affected subject (Fig. 6E). This subject had normal quantities of exosomes in urine and had typical late-onset PD, and we did not identify a clinical cause for these extreme levels of LRRK2 (Fig. 6F). To determine whether LRRK2 expression in urine changes over time, we collected urine from healthy control subjects over the course of a week at different times of the day and found that LRRK2 levels did not significantly vary within this period of time (Fig. 6F shows one such subject, one-way ANOVA P > 0.5). These data demonstrate the relative ease of collection and measurement of LRRK2 in clinical samples, but the data emphasize the importance of baseline measures of LRRK2 expression due to extreme inter-sample variability.
Figure 6.

Characterization of exosomal LRRK2 in clinical populations. (A) Exosomes were isolated from urine of PD and control patients, and lysates were analyzed by western blot in quadruplicate independent runs. The ‘pool’ value is a sample comprised of 10% of each exosome lysate of the 20 PD and 15 control individuals mixed together, allowing comparison of samples analyzed on different gels. (B–D) Plots showing relative LRRK2 expression normalized to TSG101 expression; data points of each subject are the mean of four independent runs, with removal of subject outliers. Error bars are SEM, and the horizontal line is the cohort mean. (E) Titration of an outlier PD affected subject, in comparison to the cohort geometric mean (GM) sample that has average LRRK2 and TSG101 expression. (F) Clinical summary of the PD affected case analyzed in (E). (G) Urinary exosomes collected from a neurologically normal individual over the course of 1 week, and analyzed for LRRK2 and TSG101 expression.
LRRK2 exosomes secreted by macrophages
As opposed to constitutive exosome secretion in active neurons and HEK-293T cells, other cells, such as macrophages, have been described with strong acute regulation of exosome release (44). We and others have found that activated macrophages of the periphery and brain express high levels of LRRK2 protein that can be induced with stimulation (11,12,45). Upon lipopolysaccharide (LPS) stimulation of RAW 264.7 macrophage cell line that express endogenous LRRK2 protein, we find that the exosome-associated protein TSG101 nearly evacuates from the cell cytosol, and abundant exosomes were released that contained high levels of LRRK2 expression (Fig. 7A). Thus, past-assessments of the magnitude of LRRK2 induction in macrophages would be underestimates if extracellular LRRK2 were not measured. To confirm and expand on these results, we isolated into culture primary macrophages from the mouse peritoneum of LRRK2 transgenic mice that express mouse WT-LRRK2 or G2019S-LRRK2 from BAC transgenes. Stimulation of these cells with LPS resulted in abundant LRRK2 secretion, but again the G2019S-mutation had no effect on the total quantity of LRRK2 released (Fig. 7B and C).
Figure 7.

LRRK2 exosome secretion by macrophages (A) Macrophage cell line raw 264.7 cells were treated with or without LPS (100 ng/ml) and exosomes and cell cytosols collected 24 h later. (B) Thioglycollate collected primary macrophages were isolated from WT or G2019S-BAC mice and exosomes and cytosol lysates collected 24 h post-LPS transduction. (C) Quantification of exosomal LRRK2 from (B). (D, E) Confocal images of thioglycollate collected peripheral primary macrophages isolated from LRRK2 KO or LRRK2 BAC-transgenic mice were stained for LRRK2 protein (antibody c41-2) or TSG101. LPS (100 ng/ml) was used for 24 h prior to fixing cells. Representative images are shown; results were typical of three independent experiments. Scale bars are 10 μm for all panels.
Due to the high levels of LRRK2 expressed in primary macrophages, immunofluorescence becomes possible. While LPS had a striking effect on overall macrophage morphology from a polarized cell into an amoeboid body, a more modest perinuclear accumulation of LRRK2 and the remaining TSG101 could be observed (Fig. 7D). We did not notice an enhanced co-localization between LRRK2 and TSG101 after stimulation, suggesting that LRRK2 vesicular association is probably independent of stimulation, but overt release is critically modified by stimulation. Macrophage-derived exosomes provide an example of how LRRK2 exosome release can be physiologically regulated.
LRRK2 exosomes in CSF and secretion by neurons
In the brain, serial sectioning confirmed that the most intense LRRK2 staining was in the striatum, and many of the LRRK2-positive cells were grouped into patches resembling striosomes juxtaposed against the lateral ventricles (Fig. 8A). We rationalized that LRRK2 may be secreted into the CSF from these neurons and other LRRK2-positive cells in the brain. Two samples of CSF were prepared from control subjects and exosomes verified using cryo-EM (Fig. 8B). LRRK2 expression was determined at a concentration of ∼10 pg/ml of CSF in these samples (Fig. 8C). To test whether pathogenic LRRK2 mutations affect neuronal LRRK2 exosome secretion, viral transduction of cultured neurons resulted in high levels of LRRK2 in exosomes, but these levels did not vary due to the presence of pathogenic mutations (Fig. 8D,E). Within neurons, LRRK2 occasionally localized with TSG101-positive vesicles, particularly in dendrites (Fig. 8F). As neurons have been previously described as major sources of exosomes in the brain (46, 47), it seems likely that the source of LRRK2-positive exosomes in the CSF derives from neurons expressing LRRK2.
Figure 8.

LRRK2 exosome secretion by neurons and in the CSF. (A) Representative coronal section through the striatum of either WT or LRRK2 KO mice. Arrows indicate LRRK2 positive cells in striosome-like patches present in the striatum and juxtaposed to lateral ventricles. Scale bar is 0.2 mm. (B) Representative cryo-EM image of exosomes isolated from human CSF. Scale bar is 50 nm. (C) Western blot quantification of LRRK2 in purified CSF exosomes (Δ970-rLRRK2, Invitrogen). CSF loaded onto the lane represents exosomes from the equivalent of 1 ml of CSF. (D,E) Primary cortical neurons transduced with LRRK2 adenovirus were lysed 48 h post-transduction and exosomes purified. Quantification of relative G2019S and WT LRRK2 in exosomes secreted by neurons, normalized to cytosolic LRRK2 levels. (F) Primary neurons cultured for 10 days in vitro, transduced with low concentrations (∼0.1 multiplicity of infection) of the WT-LRRK2 virus at 5 days in vitro, were stained for LRRK2 protein (antibody N241) or TSG101. Arrow heads show co-labeled vesicles, dendrites were identified by intense MAP2 labeling (see Supplementary Material, Figs S3 and S4). White scale bars are 5 μm.
DISCUSSION
Our results define a pathway for LRRK2 secretion in exosomes (Fig. 9). Exosomes may be recruited to MVBs through plasma membrane-derived vesicles enriched in transmembrane plasma proteins such as CD9 in a clathrin-dependent manner, or through the direct uptake of late-endosomal vesicles into MVBs (31). LRRK2 cytoplasmic association with late-endosome vesicles is dependent on 14-3-3 binding. As opposed to canonical vesicular proteins, 14-3-3 bound LRRK2 is relatively diffuse and soluble, and this is critical for packaging into MVBs. During the process of vesicle uptake into MVBs, LRRK2 is likely captured inward into invaginating late-endosomal vesicles in the cytosol (Fig. 7). Cells can then release LRRK2-containing exosomes after MVB docking to the plasma membrane in either a tightly controlled manner, such as in macrophages, or a constitutive manner, such as in HEK-293T cells, or cells may traffic LRRK2-positive MVBs to other parts of the cell, such as along dendrites of a neuron, for possible release at a physical proximity different than the originating vesicle, and to a cell that does not normally express LRRK2 protein.
Figure 9.

Proposed model for LRRK2 exosomal release.
LRRK2 is a protein kinase of interest both as a potential disease-modifying target and as a window to pathogenic mechanisms important for disease susceptibilities. Strong genetic links to PD and possibly Crohn's disease, as well as some evidence for genetic susceptibility to mycobacterium infection, suggest LRRK2 action in several cell types. Since pathogenic LRRK2 mutations in PD are autosomal dominant and may increase kinase activity, LRRK2 kinase inhibitors are under development for clinical trials. One problem with past clinical trials, such as a recent trial in PD (PRECEPT) with mixed-lineage kinase inhibitors (48), is that confirmation of kinase inhibition in subjects was not achieved. Our findings here show that peripheral LRRK2 is readily accessible in urinary exosomes and also can be purified from CSF. Based on our results, we predict that the overall LRRK2 protein secreted into exosomes will diminish in subjects treated with LRRK2 kinase inhibitors. Because of the strong inter-individual variability noted in LRRK2 in urinary exosomes, it would be essential to compare changes to baseline measurements. Future studies are needed to address LRRK2 levels in CSF from PD cases and controls.
As a biomarker for PD, LRRK2 expression in urinary exosomes was unexpectedly variable in clinical populations, making it difficult to assess possible LRRK2 changes between PD cases and controls. Future studies with more samples may address some aspects of power, but it seems unlikely that the LRRK2 measurement in urine alone would provide a valuable diagnostic tool. Urinary and CSF exosomes as general sources for disease biomarkers are being widely considered, and we have developed a quantitative proteomics method for exosome characterization. Exosomes as biologically relevant structures are only recently understood, and in neurodegenerative disease exosomes have been speculated as propagating toxic species of Aβ in Alzheimer's disease (49), α-synuclein in PD (43,50,51), SOD-1 in ALS (52) and prions in CJD (53, 54). Here, we show that LRRK2 joins this club and opens the door for transient LRRK2 function in cells that do not normally express LRRK2 protein. This action may have important effects in disease initiation or propagation and further studies exploring LRRK2 extracellular trafficking are warranted.
MATERIALS AND METHODS
See Supplemental Materials and Methods for extended protocols.
Clinical samples and exosome isolation
All studies were approved by the Institutional Review Board at the University of Alabama at Birmingham. Urine specimens were collected from the Movement Disorder Clinic at the University of Alabama at Birmingham, and CSF and brain tissue samples were obtained from the NICHD Brain and Tissue bank at the University of Maryland. Cell culture supernatants and clinical samples were subjected to a differential centrifugation protocol for exosome isolation, in some cases this was followed by a density gradient separation.
Animals, cell culture and plasmids
Male WT and LRRK2 KO rats were obtained from Sigma and used at 10–12 weeks of age. Primary neurons and macrophages were prepared from C57BL/6J WT or LRRK2-BAC mice (JAX strain 012467) as previously described (11, 24). All procedures were approved by the relevant Institutional Committees. HEK-293T cells and macrophage Raw264.7 cells were maintained in exosome-free media composed of DMEM with 10% fetal-bovine serum. Cells were transfected as previously described with WT-LRRK2, G2019S-LRRK2, D1994A kinase dead LRRK2, 14-3-3ɛ, difopein, scrambled-difopein or eGFP, in the pcDNA 3.1 backbone (55).
LRRK2 inhibitors and antibodies
The small molecule inhibitor HG-10-102 was synthesized in-house according to the method previously described (42), and L2in1 compound was a kind gift from Dr Dario Alessi. Antibodies are listed in Supplemental Materials and Methods.
Immunohistochemistry, immunofluorescence, Cryo-EM and super resolution imaging
Dissociated cells were cultured on glass coverslips and stained for LRRK2 and other targets as previously described (34). In all experiments, cells from LRRK2 KO animals were used to confirm LRRK2 staining. Isolated exosome preparations were fixed with paraformaldehyde, permeabilized with triton X-100 and stained using standard immunofluorescence protocols. For cryo-EM, exosomes were resuspended in water, applied to a film, plunge frozen and imaged. STED microscopy of fluorescently labeled exosomes was performed on a custom-built super-resolution microscope.
Exosome proteomics
Sonicated exosome pellets were processed into trifluoroethanol, reduced, alkylated, digested with trypsin and tandem mass tag (TMT) labeling reagents (Thermo-Pierce) were added to the samples. Isobaric-labeled peptides from each sample were combined, purified and loaded onto a tandem long-flow SCX/C18 column, and an MuDPIT analysis was performed. Peptides were resolved on a high-resolution mass spectrometer, with results filtered to a 2% peptide FDR using concatenated (forward and reverse db's) approaches, with the requirement of a minimum of two quantifiable peptides per reported UniProt object. Raw data are available upon request.
Western blotting
Proteins were resuspended in Laemmli buffer and eletrophoresed on either 7.5% or 4–20% gradient TGX gels (BioRad) and proteins transferred onto PVDF membranes. Blue-Native PAGE gels were processed as previously described (10). Blots were exposed using an Luminata HRP Substrate system (Millipore) and the signal intensity recorded using an Alpha-Innotech Fluorchem HD system or autoradiography film.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest Statement:None declared.
FUNDING
Funding was provided by NIH R01NS064934, U18NS082132, The Michael J. Fox Foundation for Parkinson's Disease Research, and the American Parkinson's Disease Association. Brain samples and CSF were provided by the NICHD Brain Bank at the University of Maryland.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge the expert technical assistance of Ms Nour Sukar.
REFERENCES
- 1.Paisan-Ruiz C., Jain S., Evans E.W., Gilks W.P., Simon J., van der Brug M., Lopez de Munain A., Aparicio S., Gil A.M., Khan N., et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 2.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B., et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Gilks W.P., Abou-Sleiman P.M., Gandhi S., Jain S., Singleton A., Lees A.J., Shaw K., Bhatia K.P., Bonifati V., Quinn N.P., et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 4.Lesage S., Durr A., Tazir M., Lohmann E., Leutenegger A.L., Janin S., Pollak P., Brice A. French Parkinson's Disease Genetics Study, G. LRRK2 G2019s as a cause of Parkinson's disease in North African Arabs. N. Engl. J. Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 5.Ozelius L.J., Senthil G., Saunders-Pullman R., Ohmann E., Deligtisch A., Tagliati M., Hunt A.L., Klein C., Henick B., Hailpern S.M., et al. LRRK2 G2019s as a cause of Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 6.Sharma M., Ioannidis J.P., Aasly J.O., Annesi G., Brice A., Van Broeckhoven C., Bertram L., Bozi M., Crosiers D., Clarke C., et al. Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology. 2012;79:659–667. doi: 10.1212/WNL.0b013e318264e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umeno J., Asano K., Matsushita T., Matsumoto T., Kiyohara Y., Iida M., Nakamura Y., Kamatani N., Kubo M. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn's disease and ulcerative colitis. Inflamm. Bowel Dis. 2011;17:2407–2415. doi: 10.1002/ibd.21651. [DOI] [PubMed] [Google Scholar]
- 8.Zhang F.R., Huang W., Chen S.M., Sun L.D., Liu H., Li Y., Cui Y., Yan X.X., Yang H.T., Yang R.D., et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. [DOI] [PubMed] [Google Scholar]
- 9.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West A.B., Moore D.J., Choi C., Andrabi S.A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K.L., Dawson V.L., et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 11.Moehle M.S., Webber P.J., Tse T., Sukar N., Standaert D.G., DeSilva T.M., Cowell R.M., West A.B. LRRK2 Inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thevenet J., Pescini Gobert R., Hooft van Huijsduijnen R., Wiessner C., Sagot Y.J. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One. 2011;6:e21519. doi: 10.1371/journal.pone.0021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biskup S., Moore D.J., Rea A., Lorenz-Deperieux B., Coombes C.E., Dawson V.L., Dawson T.M., West A.B. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 2007;8:102. doi: 10.1186/1471-2202-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandemakers W., Snellinx A., O'Neill M.J., de Strooper B. LRRK2 Expression is enriched in the striosomal compartment of mouse striatum. Neurobiol. Dis. 2012;48:582–593. doi: 10.1016/j.nbd.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Alegre-Abarrategui J., Christian H., Lufino M.M., Mutihac R., Venda L.L., Ansorge O., Wade-Martins R. LRRK2 Regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J., et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 17.Dodson M.W., Zhang T., Jiang C., Chen S., Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012;21:1350–1363. doi: 10.1093/hmg/ddr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatano T., Kubo S., Imai S., Maeda M., Ishikawa K., Mizuno Y., Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet. 2007;16:678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- 19.Higashi S., Moore D.J., Yamamoto R., Minegishi M., Sato K., Togo T., Katsuse O., Uchikado H., Furukawa Y., Hino H., et al. Abnormal localization of leucine-rich repeat kinase 2 to the endosomal–lysosomal compartment in Lewy body disease. J. Neuropathol. Exp. Neurol. 2009;68:994–1005. doi: 10.1097/NEN.0b013e3181b44ed8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z., Hamamichi S., Lee B.D., Yang D., Ray A., Caldwell G.A., Caldwell K.A., Dawson T.M., Smith W.W., Dawson V.L. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson's disease models. Hum. Mol. Genet. 2011;20:3933–3942. doi: 10.1093/hmg/ddr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matta S., Van Kolen K., da Cunha R., van den Bogaart G., Mandemakers W., Miskiewicz K., De Bock P.J., Morais V.A., Vilain S., Haddad D., et al. LRRK2 Controls an endoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 22.Piccoli G., Condliffe S.B., Bauer M., Giesert F., Boldt K., De Astis S., Meixner A., Sarioglu H., Vogt-Weisenhorn D.M., Wurst W., et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin N., Jeong H., Kwon J., Heo H.Y., Kwon J.J., Yun H.J., Kim C.H., Han B.S., Tong Y., Shen J., et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Stafa K., Trancikova A., Webber P.J., Glauser L., West A.B., Moore D.J. GTPase activity and neuronal toxicity of Parkinson's disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012;8:e1002526. doi: 10.1371/journal.pgen.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J., III, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl Acad. Sci. USA. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macleod D.A., Rhinn H., Kuwahara T., Zolin A., Di Paolo G., Maccabe B.D., Marder K.S., Honig L.S., Clark L.N., Small S.A., et al. RAB7L1 Interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orenstein S.J., Kuo S.H., Tasset I., Arias E., Koga H., Fernandez-Carasa I., Cortes E., Honig L.S., Dauer W., Consiglio A., et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Jaleel M., Nichols R.J., Deak M., Campbell D.G., Gillardon F., Knebel A., Alessi D.R. LRRK2 Phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem. J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dzamko N., Deak M., Hentati F., Reith A.D., Prescott A.R., Alessi D.R., Nichols R.J. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thery C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 32.Boucheix C., Benoit P., Frachet P., Billard M., Worthington R.E., Gagnon J., Uzan G. Molecular cloning of the CD9 antigen. A new family of cell surface proteins. J. Biol. Chem. 1991;266:117–122. [PubMed] [Google Scholar]
- 33.Thery C., Boussac M., Veron P., Ricciardi-Castagnoli P., Raposo G., Garin J., Amigorena S. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001;166:7309–7318. doi: 10.4049/jimmunol.166.12.7309. [DOI] [PubMed] [Google Scholar]
- 34.Sen S., Webber P.J., West A.B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 2009;284:36346–36356. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng X., Dzamko N., Prescott A., Davies P., Liu Q., Yang Q., Lee J.D., Patricelli M.P., Nomanbhoy T.K., Alessi D.R., et al. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat. Chem. Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies P., Hinkle K.M., Sukar N.N., Sepulveda B., Mesias R., Serrano G., Alessi D.R., Beach T.G., Benson D.L., White Iii C.L., et al. Comprehensive characterization and optimization of leucine rich repeat kinase 2 (LRRK2) monoclonal antibodies. Biochem. J. 2013;453:101–113. doi: 10.1042/BJ20121742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pisitkun T., Shen R.F., Knepper M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X., Yu X., Zack D.J., Zhu H., Qian J. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics. 2008;9:271. doi: 10.1186/1471-2105-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nichols R.J., Dzamko N., Morrice N.A., Campbell D.G., Deak M., Ordureau A., Macartney T., Tong Y., Shen J., Prescott A.R., et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L., Xie C., Greggio E., Parisiadou L., Shim H., Sun L., Chandran J., Lin X., Lai C., Yang W.J., et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J. Neurosci. 2008;28:3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masters S.C., Fu H. 14-3-3 Proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 2001;276:45193–45200. doi: 10.1074/jbc.M105971200. [DOI] [PubMed] [Google Scholar]
- 42.Choi H.G., Zhang J., Deng X., Hatcher J.M., Patricelli M.P., Zhao Z., Alessi D.R., Gray N.S. Brain penetrant LRRK2 inhibitor. ACS Med. Chem. Lett. 2012;3:658–662. doi: 10.1021/ml300123a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danzer K.M., Kranich L.R., Ruf W.P., Cagsal-Getkin O., Winslow A.R., Zhu L., Vanderburg C.R., McLean P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gauley J., Pisetsky D.S. The release of microparticles by RAW 264.7 macrophage cells stimulated with TLR ligands. J. Leukoc. Biol. 2010;87:1115–1123. doi: 10.1189/jlb.0709465. [DOI] [PubMed] [Google Scholar]
- 45.Dzamko N., Inesta-Vaquera F., Zhang J., Xie C., Cai H., Arthur S., Tan L., Choi H., Gray N., Cohen P. The IkappaB kinase family phosphorylates the Parkinson's disease kinase LRRK2 at Ser935 and Ser910 during toll-like receptor signaling. PLoS One. 2012;7:e39132. doi: 10.1371/journal.pone.0039132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faure J., Lachenal G., Court M., Hirrlinger J., Chatellard-Causse C., Blot B., Grange J., Schoehn G., Goldberg Y., Boyer V., et al. Exosomes are released by cultured cortical neurones. Mol. Cell Neurosci. 2006;31:642–648. doi: 10.1016/j.mcn.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Lachenal G., Pernet-Gallay K., Chivet M., Hemming F.J., Belly A., Bodon G., Blot B., Haase G., Goldberg Y., Sadoul R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell Neurosci. 2011;46:409–418. doi: 10.1016/j.mcn.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Parkinson Study Group, P.I. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- 49.Rajendran L., Honsho M., Zahn T.R., Keller P., Geiger K.D., Verkade P., Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc. Natl Acad. Sci. USA. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez-Erviti L., Seow Y., Schapira A.H., Gardiner C., Sargent I.L., Wood M.J., Cooper J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emmanouilidou E., Melachroinou K., Roumeliotis T., Garbis S.D., Ntzouni M., Margaritis L.H., Stefanis L., Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomes C., Keller S., Altevogt P., Costa J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007;428:43–46. doi: 10.1016/j.neulet.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 53.Bellingham S.A., Coleman B.M., Hill A.F. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 2012;40:10937–10949. doi: 10.1093/nar/gks832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fevrier B., Vilette D., Archer F., Loew D., Faigle W., Vidal M., Laude H., Raposo G. Cells release prions in association with exosomes. Proc. Natl Acad. Sci. U.S.A. 2004;101:9683–9688. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Webber P.J., Smith A.D., Sen S., Renfrow M.B., Mobley J.A., West A.B. Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J. Mol. Biol. 2011;412:94–110. doi: 10.1016/j.jmb.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.