

Abstract
The isochorismate and salicylate synthases are members of the MST family of enzymes. The isochorismate synthases establish an equilibrium for the conversion chorismate to isochorismate and the reverse reaction. The salicylate synthases convert chorismate to salicylate with an isochorismate intermediate; therefore, the salicylate synthases perform isochorismate synthase and isochorismate-pyruvate lyase activities sequentially. While the active site residues are highly conserved, there are two sites that show trends for lyase-activity and lyase-deficiency. Using steady state kinetics and HPLC progress curves, we tested the “interchange” hypothesis that interconversion of the amino acids at these sites would promote lyase activity in the isochorismate synthases and remove lyase activity from the salicylate synthases. An alternative, “permute” hypothesis, that chorismate-utilizing enzymes are designed to permute the substrate into a variety of products and tampering with the active site may lead to identification of adventitious activities, is tested by more sensitive NMR time course experiments. The latter hypothesis held true. The variant enzymes predominantly catalyzed chorismate mutase-prephenate dehydratase activities, sequentially generating prephenate and phenylpyruvate, augmenting previously debated (mutase) or undocumented (dehydratase) adventitious activities.
Keywords: Isochorismate synthase, salicylate synthase, siderophore biosynthesis, enzyme engineering
Introduction
The redesign of enzyme active sites has been successful to interconvert substrate specificities in structurally homologous and functionally related enzymes. In some examples, the interconversion is as simple as a single amino acid substitution, such as the generation of a malate dehydrogenase from lactate dehydrogenase (1). In other cases the interconversion of substrate specificity requires more complex changes including modification to the active site and surface loop changes, such as the generation of a trypsin serine protease with the substrate specificity of chymotrypsin (2–4). Enzymes dependent on the cofactor pyridoxal 5′-phosphate, including ornithine decarboxylase (5) and amino acid aminotransferases (6–8), have been redesigned to have new activities when changes in the active site lead to a promiscuous addition of the proton to different locations on the substrate-cofactor covalent intermediate. Enzymatic activities have been interconverted in the N-acetylneuraminate lyase subfamily of (β,α) -barrel enzymes with a single point mutation (9). More recently, design of enzymes with augmented adventitious or promiscuous activities by directed evolution, looking for an increase of a desired activity after cycles of random mutagenesis, has been successful (10, 11).
The MST family of enzymes are structurally homologous, Mg2+-dependent, chorismate-utilizing enzymes involved in menaquinone, siderophore and tryptophan biosynthesis (12–14). This work will focus on two enzymes that isomerize the chorismate ring to form isochorismate for siderophore biosynthesis (Figure 1A): PchA, the isochorismate synthase from Pseudomonas aeruginosa, and Irp9, the salicylate synthase from Yersinia enterocolitica. These enzymes are required to make the salicylate cap of the structurally similar siderophores pyochelin and yersiniabactin (Figure 2). Both enzymes generate isochorismate from chorismate, but Irp9 is then capable of generating salicylate from isochorismate. An interesting question may be posed: what are the features of Irp9 that makes this enzyme bifunctional, whereas PchA is not? This question can be extrapolated more broadly to include other enzymes of this same family in similar biosynthetic pathways, including the salicylate synthase for mycobactin biosynthesis (MbtI) and the isochorismate synthases from E. coli (EntC of enterobactin biosynthesis and MenF of menaquinone biosynthesis)
Figure 1. Enzymatic activities of PchA and Irp9 and their variants.

A. Schematic of the isochorismate synthase, isochorismate-pyruvate lyase, salicylate synthase, chorismate mutase and prephenate dehydratase activities. B. The physiological functions of the isochorismate synthases and salicylate synthases of the MST family discussed herein.
Figure 2. Biosynthetic Products for the isochorismate and salicylate synthases.

The compounds in the left column are all generated in a pathway that includes an isochorismate synthase (PchA for the siderophore pyochelin, EntC for the siderophore enterobactin, and MenF for the electron carrier menaquinone). The compounds in the right column are both generated in siderophore biosynthetic pathways that include a salicylate synthase (Irp9 for yersiniabactin, and MbtI for mycobactin). The portion derived from chorismate is shown in bold.
The formation of isochorismate, common to all five enzymes, has been hypothesized to be catalyzed by general acid-general base chemistry, in which a lysine (K221 in PchA) is the general base for the activation of water to attack the C2 position and a glutamic acid (E269 in PchA) is the general acid for elimination of the C4-OH. This proposed mechanism is supported by mutational analyses in MenF (13), Irp9 (15) and MbtI (14). When the general base lysine was replaced with alanine or the general acid glutamic acid was replaced with alanine or glutamine, the enzymes showed a loss of isochorismate synthase activity. However, Ziebart and Toney (16) have shown that putative general base lysine to glutamine variants in EntC and MbtI retained some activity. Ferrer and colleagues (17) have suggested that the general base is a glutamic acid (E313 in PchA), but more recently Meneely et. al report a model based on pH/pD rate profiles and mutagenesis that is most consistent with the lysine general base and the glutamic acid general acid (18).
For MbtI, NMR studies have shown that the isochorismate-pyruvate lyase activity observed in the salicylate synthase enzymes is a pericyclic reaction (14). The proton transferred is derived from the chorismate ring and not from solvent (14), similar to that seen for the stand-alone isochorismate-pyruvate lyase (19). A comparison of the active site residues between enzymes that perform one activity (isochorismate synthase) such as PchA, EntC and MenF (Figure 1B) and enzymes that perform two activities (isochorismate synthase and isochorismate-pyruvate lyase) such as Irp9 and MbtI show these residues are conserved except for two (Figure 3) (20). One of the differing residues is an alanine in the isochorismate synthase enzymes that is a threonine in the salicylate synthase enzymes (cyan in Figure 3A). In the salicylate synthases, the backbone carbonyl of the threonine forms a hydrogen bond with the substrate hydroxyl-group and was hypothesized to be involved in orientation of the substrate for the lyase activity (13, 21). Replacement mutagenesis (A→T) has not proven enlightening, instead generating enzymes with no addition of lyase activity but also reduced or abrogated synthase activity (13, 21). Lyase-deficient and lyase-proficient enzymes differ in one of the two residues that chelate the Mg+2 ion through a water molecule (aspartic acid or asparagine in the isochorismate synthase enzymes but a glutamic acid in the salicylate synthase enzymes (yellow in Figure 3A) (20)). The longer sidechain of the lyase-proficient salicylate synthases was hypothesized to shift the magnesium ion to a position permitting the conversion of isochorismate into salicylate (20). This amino acid difference has been ignored previously due to the distance from the site of chemistry.
Figure 3. Active site residues of the salicylate and isochorismate synthases.

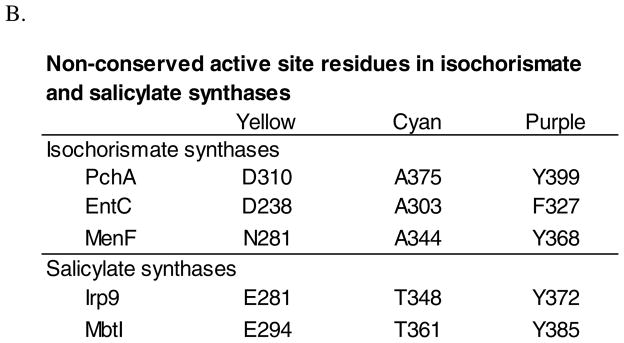
A. Irp9 (PDB: 2FN1) active site residues are shown in grey with the exception of the non-conserved residues within the isochorismate synthase and salicylate synthase enzymes. Salicylate is tan and pyruvate is brown. The magnesium ions (green) and coordinating waters (red) are shown as spheres. Amino acids are labeled with Irp9 (PchA) numbering. The highlighted residues, E281 (yellow), T348 (cyan) and Y372 (purple) are the non-conserved residues. The chart in part B indicates that for two of these residues (E281 and T348) there is a trend between lyase-deficient (the isochorismate synthases) and lyase-active (the salicylate synthases) enzymes. The final change (F327 in EntC) is a conservative change in only one of the five enzymes. This figure was generated with PyMOL (49).
Here, we initially test the “interchange” hypothesis: the activities of the isochorismate synthase PchA and the salicylate synthase Irp9 can be interconverted by mutational exchange of one or both of the nonconserved active site residues that show a trend relative to presence or absence of lyase activity. This is a simplistic view of enzymes, which are known to be dynamic and capable of compensating for mutational change. Structurally homologous MST enzymes have been classified as “chorismate-utilizing”(22) and can perform a variety of chemistries, including the isomerization investigated here, but also replacement of the C4-OH with an amine or removal of the C4-OH and addition of an amine at C-2. Adventitious chorismate mutase activity (not physiologically important and detected at low catalytic levels) has been debated in the salicylate synthases enzymes(14, 16). Since the MST enzymes seem evolutionarily designed to permute chorismate, the more complex, “permute” hypothesis would be that the enzyme catalyzes reactions based on a binding site designed for arrangement of chorismate, and therefore interconversion of the nonconserved residues may lead to other catalytic events, expected to be much lower in catalytic efficiency than the physiological reaction.1 Our results show that the exchange of activities (isochorismate versus salicylate synthase) is not achieved by the straightforward interchange path. Indeed, the variant enzymes develop chorismate mutase and prephenate dehydratase activities in agreement with the permute hypothesis, selectively catalyzing these activities relative to the isochorismate synthase and isochorismate-pyruvate lyase activities.
Materials and Methods
Preparation of overexpression plasmids
The pchA gene was cloned as previously described (18). The PchA variants were produced using QuikChange (Agilent) with wildtype pchA plasmid as the template and the following primers: D310E (5′-GAC AGC GCC AAG GAG AGG CAC GAA CAC-3′) and A375T (5′-GCA TCC GAC CCC CAC GGT GGG CGG CTA CC-3′) and complementary reverse primers for each variant. Each variant plasmid contained only the desired mutation.
The irp9 gene was amplified from Y. enterocolitica 33114 genomic DNA (ATCC) by polymerase chain reaction by use of Master Mix (Eppendorf) with 1.5 mM magnesium acetate. The forward primer (5′-GGA ATT CCA TAT GAA AAT CAG TGA ATT TCT ACA C-3′) includes an NdeI site (underlined) and the reverse primer (5′-AAA ACT CGA GCA CCA TTA AAT AGG GCG CAA TG-3′) contains a XhoI site (underlined). The amplified 1302 base pair fragment was digested with NdeI and XhoI and ligated into the pET29b plasmid (Novagen) digested with the same enzymes. The resultant plasmid encodes the irp9 gene with a C-terminal histidine tag and one mutation from that found in the published structure (PDB: 2FN1) that changes proline 38 to a glutamic acid. The homologous protein, YbtS, in Yersinia pestis also contains a glutamic acid at this position. The Irp9 variants were produced with QuikChange (Agilent) using wildtype irp9 plasmid as the template, the forward primers used were: E281D (5′-TGC TGC ACG ATA GTA AAG ATG TGC TTG AGC ATA TCC) and T348A (5′-CGT GCT GTT TCC GTC GAT TGC CGC CTC AGG-3′); in conjunction with complementary reverse primers. The irp9 variant plasmids contained only the desired mutation relative to the wildtype plasmid.
Protein overexpression and purification
Wildtype and variant PchA was purified as previously described (18) with a yield of 16–74 mg/L of culture for the variants. The P. aeruginosa isochorismate-pyruvate lyase PchB used in the coupled assay with PchA to convert isochorismate to salicylate was purified as previously described (23).
BL21 Star (DE3) pLysS E. coli containing the Irp9 wildtype or variant expression plasmid were grown in LB medium containing 50 μg/mL kanamycin at 37 °C with shaking (225 rpm) until an OD600 of ~1.0 was reached. The culture temperature was reduced to 18 °C and the cells were harvested by centrifugation (6,000×g, 10 min, 4 °C) after 20 h. The cell pellet was resuspended in 15 mL of 25 mM Tris-HCl pH 8.5, 500 mM NaCl, 5 mM imidazole (buffer A) per liter of culture. Cells were disrupted by use of a French pressure cell (35,000 psi), and cellular debris was removed by centrifugation (12,000×g, 30 min, 4 °C). The supernatant was applied to a chelating Sepharose fast-flow column (Amersham Biosciences) charged with nickel chloride and pre-equilibrated in buffer A. Irp9 protein eluted at 300 mM imidazole in a step gradient of 30 mM, 45 mM, 150 mM and 300 mM imidazole in buffer A. The pooled fractions were applied to a Superdex 200 size-exclusion column (Amersham Biosciences) equilibrated with 20 mM Tris-HCl pH 8, 100 mM NaCl. The fractions containing Irp9 were pooled and concentrated by use of an Amicon stirred cell with a YM-30 membrane to 125 – 333 μM as determined by absorbance at 280 nm (ε = 53,400 M−1cm−1) and stored at −80 °C. Wildtype Irp9 had a yield of 60 mg/L culture and the Irp9 variants had yields of 4–46 mg/L of culture.
Preparation of isochorismate and chorismate
Isochorismate was isolated from Klebsiella pneumoniae 62–1 harboring the entC plasmid pKS3-02 (24) with only minor changes, as described previously (25). Chorismate was isolated from K. pneumoniae 62–1 as previously described (26) with minor modifications (18).
Isochorismate synthase activity assays
Enzyme was added to the standard assay buffer containing 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10% glycerol and incubated with a 20-fold excess of PchB. The reaction was initiated by the addition of chorismate (0–300 μM). The accumulation of salicylate was measured by fluorescence with an excitation wavelength of 310 nm and an emission wavelength of 430 nm at 25 °C using a Cary Eclipse fluorescence spectrophotometer (Varian) and an electrothermal single cell holder for temperature control (0.1 °C). For the D310E-PchA variant assay, the enzyme concentration was 0.1 μM, and for all of the other PchA and Irp9 variants the enzyme concentration was 1 μM.
Isochorismate-pyruvate lyase activity assays
All isochorismate-pyruvate lyase assays were performed using a TgK Scientific SF-61DX2 stopped-flow apparatus equipped with a photomultiplier detector and a xenon lamp with the monochromator set at 310 nm, and a 360 nm cutoff filter operated at 25 °C. Equal volumes of enzyme and substrate in the standard reaction buffer were injected into a 22 μl cell and initial velocities of salicylate production were collected for 30 seconds during which the reaction was linear. Pre-injection enzyme and substrate concentrations were twice the final enzyme/substrate concentration in the cell (post mixing). Unless otherwise specified, all concentrations reported are final post-mixing concentrations. Enzyme concentrations used for the assays with PchA variants was 10 μM, for WT-Irp9 and E281D-Irp9 assays the enzyme concentration was 0.05 μM and for the T348A-Irp9 and E281D/T348A-Irp9 variants the enzyme concentration was 0.1 μM with isochorismate concentrations of 0 – 400 μM.
Salicylate synthase activity assays
Salicylate production with wildtype PchA, PchA variants, T348A-Irp9 and E281D/T348A-Irp9 was measured using 10 μM enzyme and the reaction was initiated with 1 mM chorismate. For reactions with wildtype Irp9 and E281D-Irp9, salicylate production was measured using 100 nM enzyme and initiated by the addition of chorismate (0 – 25 μM). Initial velocities were determined at 25 °C in the standard assay buffer by measuring the accumulation of salicylate by fluorescence with an excitation wavelength of 310 nm and an emission wavelength of 430 nm using a Cary Eclipse fluorescence spectrometer (Varian) with temperature controller.
Chorismate mutase activity assays
Chorismate mutase activity for wildtype and variant PchA and Irp9 was measured in 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10% glycerol or with 50 mM Tris-HCl pH 7.5, 1 mM EDTA, 10% glycerol at 25 °C according to the method optimized by Gaille et. al (27). The enzyme concentration was 25 μM and the reaction was initiated by the addition of chorismate (0 – 10 mM). The reaction was terminated at the various time points by the addition of 20 μl 1 M HCl to 40 μl of reaction mixture and the reaction product, prephenate, was converted to phenylpyruvate upon incubation at 37 °C for 20 min. The phenylpyruvate concentration was determined at 320 nm under basic conditions with the addition of 140 μl of 2.5 M NaOH (ε320 = 17,500 M−1cm−1) using a Cary 50 Bio spectrophotometer (Varian).
Salicylate production progress curves
Salicylate production by PchA and Irp9 variants was monitored over time at 25 °C using a TgK stopped-flow apparatus. Enzyme in the standard reaction buffer was mixed in the stopped-flow apparatus in a 1:1 ratio with reaction buffer containing chorismate such that the final concentration of enzyme was 10 μM and of chorismate was 1 μM. The fluorescence was measured with a 360 nm cutoff filter upon excitation at 310 nm and converted to salicylate concentration using a standard curve.
Nuclear magnetic resonance data
NMR time course reactions were obtained with 1 mM chorismate in 50 mM Tris-HCl pD 7.9, 10 mM MgCl2, using a protocol akin to that performed for the uncatalyzed reaction (28, 29) and for EntC (30). Enzyme (25 μM final concentration) was added to start the reaction and 1H-NMR spectra were collected at 25 °C with an Avance AV-III 500 with a dual carbon/proton (CPDUL) cryoprobe, every 5 min for the first 2 hours and every 30 min for the remaining 16 hours. The reaction products were analyzed using MestReNova (Mestrelab Research). NMR spectra peaks were assigned according to those of standards in 50 mM Tris-HCl pD 7.9, 10 mM MgCl2 in a range from 8 – 5.5 ppm. Chorismate δ 6.60 (s, 1H), 6.37 (dt, J = 10.1, 1.8 Hz, 1H), 6.01 (dd, J = 10.0, 2.7 Hz, 1H); isochorismate δ 6.86 (d, J = 5.6 Hz, 1H), 6.38 (dd, J = 9.6, 5.6 Hz, 1H), 6.24 (dd, J = 9.6, 4.3 Hz, 1H); salicylate δ 7.83 (dd, J = 7.8, 1.8 Hz, 1H), 7.49 – 7.44 (m, 1H), 7.00 – 6.94 (m, 2H); phenylpyruvate δ 7.45–7.40 (m, J = 7.4 2H), 7.39 – 7.33 (m, 1H), 7.33 – 7.26 (m, 2H);. Prephenate peaks were determined from a chorismate NMR time course by the addition of new peaks at δ 6.03 (dd, J = 10.3, 1.7 Hz, 2H), 5.94 (dd, J = 10.4, 3.2 Hz, 2H). No 4-hydroxybenzoate or isoprephenate (other decomposition products of chorismate and isochorismate) were detected under our experimental conditions, which differed from previous work by buffer, pH and temperature (28, 29).
Data analysis
Steady state kinetic data were fit to the Michaelis-Menten equation by the nonlinear regression function of KaleidaGraph (Synergy Software). At subsaturating concentrations of substrate, kinetic constants were determined using Henderson and Dixon plots (31). kobs values reported in Table 4 were determined by curve fitting using KaleidaGraph (Synergy Software). Simulation was conducted using KinTek Global Kinetic Explorer (32, 33) for the NMR time course experiments, following the manufacturer’s instructions.
Table 4.
kobs determined from NMR time course reactions
| k obs (min−1) | ||||||
|---|---|---|---|---|---|---|
| Irp9 | PchA | uncatalyzed | ||||
| WT | E281D/T348A | WT | D310E | D310E/A375T | ||
| Chorismate disappearance | 0.24 | 0.070 | 0.27 | 0.10 | 0.0018 | 0.0011 |
| Isochorismate formation | n.d. | n.d. | 0.29 | 0.12 | 0.0017 | n.d. |
| Salicylate formation | 0.23 | 0.056 | 0.0013 | 0.0040 | 0.00075 | n.d. |
| Prephenate formation | * | * | 0.00029 | 0.010 | 0.00027 | 0.00081 |
| Prephenate disappearance | * | * | n.d. | n.d. | n.d. | n.d. |
| Phenylpyruvate formation | 0.063 | 0.048 | 0.00027 | 0.050 | 0.0018 | 0.00000021 |
n.d. = not detected;
rates not reliably determined
Results
PchA and Irp9 steady state activity assays
PchA is an isochorismate synthase (Figure 1A) (18, 34). Isochorismate synthase activity was determined using a coupled assay in which isochorismate produced by PchA is converted to the fluorescent salicylate by the isochorismate-pyruvate lyase, PchB (18). Chorismate is provided as the substrate, and PchB is used in 20-fold molar excess such that any isochorismate produced is immediately converted into salicylate. Irp9 is a salicylate synthase (15, 35), in which isochorismate synthase and isochorismate-pyruvate lyase activities are performed sequentially. Isochorismate synthase activity cannot be determined for Irp9 independently because the isochorismate is immediately converted to salicylate in the same active site. Salicylate synthase activity is detected using chorismate as the substrate and measuring salicylate fluorescence. Isochorismate-pyruvate lyase activity in both PchA and Irp9 is measured with isochorismate as the substrate, detecting salicylate fluorescence. Chorismate mutase activity has previously been detected in the absence of magnesium for the salicylate synthase enzyme MbtI (14). More recently, chorismate mutase activity was not detected in MbtI or EntC, and the previous mutase activity was hypothesized to be the result of a co-purified chorismate mutase contaminant (16). Chorismate mutase activity is measured using chorismate as a substrate. The prephenate product is converted to phenylpyruvate at low pH for detection by absorbance.
PchA and Irp9 wildtype kinetic parameters
Wildtype PchA isochorismate synthase and isochorismate-pyruvate lyase kinetic parameters recorded here have been reported previously and are listed in the tables for easy comparison (Tables 1 and 2) (18). Wildtype Irp9 salicylate synthase activity was determined to be 42,000 M−1s−1 (Table 3), whereas isochorismate-pyruvate lyase activity was 159,000 M−1s−1 (Table 2). The salicylate synthase kinetic parameters for wildtype Irp9 are as expected based on those previously reported (35).
Table 1. Isochorismate Synthase activity for PchA of non-conserved amino acids.
The substrate is chorismate which is converted by PchA to isochorismate. The isochorismate-pyruvate lyase from P. aeruginosa (PchB) is added in excess to convert isochorismate to salicylate which is measure by fluorescence (excitation at 310 nm, emission at 430 nm).
| Isochorismate synthase activity | ||||
|---|---|---|---|---|
| kcat (×10−3 s−1) | Km (μM) | kcat/Km (M−1s−1) | Mutational effect (mutant/WT) | |
| PchA | ||||
| WTa | 50.4 ± 0.2 | 0.9 ± 0.1 | 57,000 ± 7,000 | |
| D310E | 22 ± 1 | 0.6 ± 0.1 | 41,000 ± 9,000 | within error |
| A375T | 9.3 ± 0.3 | 150 ± 20 | 62 ± 4 | 0.001 |
| D310E/A375T | 4.0 ± 0.3 | 119 ± 5 | 34 ± 4 | 0.0006 |
Wildtype PchA data from Meneely, Luo, Dhar and Lamb, reference 18.
Table 2. Isochorismate-Pyruvate Lyase activity for PchA and Irp9 variants of nonconserved amino acids.
The substrate is isochorismate. Salicylate production is measured by fluorescence (excitation at 310 nm, emission >360 nm using stopped-flow spectrometer).
| Isochorismate-pyruvate lyase activity | ||||
|---|---|---|---|---|
| kcat (×10−3 s−1) | Km (μM) | kcat/Km (M−1s−1) | Mutational effect (mutant/WT) | |
| PchA | ||||
| WTa | 0.70 ± 0.01 | 42 ± 2 | 17 ± 1 | |
| D310E | 0.69 ± 0.00 | 13.7 ± 0.1 | 50.4 ± 0.2 | 3 |
| A375T | 0.44 ± 0.00 | 12 ± 0 | 36.6 ± 0.3 | 2 |
| D310E/A375T | 0.38 ± 0.00 | 11 ± 2 | 37 ± 6 | 2 |
|
| ||||
| Irp9 | ||||
| WT | 92 ± 2 | 0.58 ± 0.01 | 159,000 ± 6,000 | |
| E281D | 50.4 ± 0.3 | 1.7 ± 0.2 | 31,000 ± 3,000 | 0.2 |
| T348A | 5.5 ± 0.2 | 0.59 ± 0.05 | 9,300 ± 500 | 0.06 |
| E281D/T348A | 34.3 ± 0.0 | 47 ± 1 | 740 ± 10 | 0.005 |
Wildtype PchA data from Meneely, Luo, Dhar and Lamb, reference 18.
Table 3. Salicylate Synthase activity for Irp9 variants of non-conserved amino acids.
The substrate is chorismate which is converted to salicylate with isochorismate as an intermediate. Salicylate production is measured by fluorescence (excitation at 310 nm, emission at 430 nm).
| Salicylate synthase activity | ||||
|---|---|---|---|---|
| kcat (×10−3 s−1) | Km (μM) | kcat/Km (M−1s−1) | Mutational effect (mutant/WT) | |
| Irp9 | ||||
| WT | 84 ± 7 | 1.98 ± 0.05 | 42,000 ± 2,000 | |
| E281D | 8 ± 1 | 6.0 ± 0.1 | 1,300 ± 200 | 0.03 |
A375T-PchA and T347A-Irp9 variants (H-bonds to substrate hydroxyl-group)
One of the differing residues between lyase-active and lyase-deficient enzymes is an alanine in the isochorismate synthases and a threonine in the salicylate synthases (cyan in Figure 3A). These residues are hypothesized to be involved in substrate orientation by forming a hydrogen bond using the backbone carbonyl (13, 21). A variant of PchA to change the native alanine to threonine (A375T-PchA) results in a loss of the physiological isochorismate synthase catalytic efficiency by three orders of magnitude (Table 1), and a 2-fold gain in isochorismate-pyruvate lyase catalytic efficiency (Table 2). A corresponding mutation in EntC (21) resulted in reduction of isochorismate synthetase catalytic efficiency by two orders of magnitude; a corresponding variant of MenF (13) had no detectable activity. In these previous studies, none of the A→T variants in the isochorismate synthases gained lyase activity to produce salicylate (13, 21). The converse T→A variant in the salicylate synthase Irp9 results in a reduction in salicylate synthase catalytic efficiency by 5 orders of magnitude: 42,000 M−1s−1 in WT-Irp9 to undetectable levels in T348A-Irp9. However, if the reaction is allowed to progress for a more extended period of time, salicylate formation can be detected. This result will be described in more detail in the section on progress curves. Isochorismate-pyruvate lyase catalytic efficiency in the T348A-Irp9 is reduced by 17-fold (Table 2).
D310E-PchA and E281D-Irp9 variants (change in Mg2+ chelator length)
A second active site residue that shows a trend between the isochorismate synthase and salicylate synthase enzymes is one of the two residues involved in chelating the Mg+2 ion through a water molecule. This residue is an aspartic acid or asparagine in the isochorismate synthase enzymes but a glutamic acid in the salicylate synthase enzymes (yellow in Figure 3A) (20). A PchA variant that changes the amino acid from the isochorismate synthase native sidechain to the salicylate synthase amino acid (D310E-PchA) has a kcat/Km value for the isochorismate synthase activity that is within error of the wildtype enzyme (Table 1). Using isochorismate as the substrate, the isochorismate-pyruvate lyase activity of the D310E-PchA variant was determined to have a kcat/Km value that is 3-fold greater than wildtype (Table 2). Previous work on wildtype PchA did not detect salicylate production (34). Therefore, the isochorismate synthase activity is unperturbed by the generation of the D310E-variant, and the isochorismate-pyruvate lyase was very modestly increased. A variant salicylate synthase enzyme in which the wildtype glutamic acid is changed to the aspartic acid present in isochorismate synthase (E281D-Irp9) showed salicylate synthase catalytic efficiency that is 32-fold less than that of the wildtype enzyme (Table 3). The E281D-Irp9 variant showed an isochorismate-pyruvate lyase catalytic efficiency loss of 5-fold relative to the wildtype (Table 2). Taken together, these data would suggest that the isochorismate synthase activity is more perturbed than the isochorismate-pyruvate lyase activity in the E281D-Irp9 variant.
Double variant (D310E/A375T-PchA and E281D/T348A-Irp9) kinetic parameters
The double variants of PchA and Irp9 with changes at both non-conserved residues were also generated and their kinetic parameters determined. For PchA, the double variant had similar activity in the isochorismate synthase and isochorismate-pyruvate lyase assays as the A375T-PchA variant (Table 1, 2), suggesting that this variation is the more dominant and deleterious. The double variant of Irp9 appeared to have more than an additive effect of the two individual variants with isochorismate-pyruvate lyase catalytic efficiency three orders of magnitude less than WT-Irp9 (Table 2) and undetectable salicylate synthase activity.
Chorismate mutase activity
We did not detect chorismate mutase activity for PchA or Irp9 or any of the variants in the presence or absence of magnesium using the standard steady state chorismate mutase assay.
The rate of salicylate production by PchA and Irp9 and active site variants (progress curves)
Salicylate synthase activity had not been previously reported for PchA. Unexpectedly, over extended periods of time (up to 2000 s) we determined that the production of salicylate could be detected for all variants of PchA and Irp9 tested here. However, for only the WT- and E281D-Irp9 could we measure linear initial velocities for the production of salicylate for the determination of kinetic parameters (Table 3). Therefore, we investigated salicylate production using progress curves collected on a stopped-flow apparatus. When plotted on the logarithmic scale, WT-Irp9 showed a lag time of 2 seconds and produced 93% of the expected salicylate (Figure 4, closed circles) assuming complete conversion of chorismate to isochorismate to salicylate. E281D-Irp9 produced 81% of the expected salicylate after a lag of 10 s (closed squares). T348A-Irp9 (closed diamonds) produced 48% salicylate after a lag of 200 s, and E281D/T348A-Irp9 (closed triangles) only converted 15% of the chorismate to salicylate after a lag of 400 s. WT-PchA and the corresponding variants all showed 15–18% conversion to salicylate (open circles, squares and diamonds) with the exception of the double mutant (open triangles) which produced 34% salicylate, all producing the fluorescent species after 400 s. The question arose as to whether the fluorescent species was indeed salicylate, and if the chorismate remained unreacted or if a new product was being formed.
Figure 4. Progress curves.

Representative salicylate-versus-time progress curves for WT-Irp9 (●), E281D-Irp9 (■), T348A-Irp9 (◆), E281D/T348A-Irp9 (▲), WT-PchA (○), D310E-PchA (■), A375T-PchA (◇) and D310E/A375T-PchA (△) showing a lag phase before salicylate production from chorismate. Every 5th data point is displayed per trace for clarity.
Reaction Time Courses by NMR
The disappearance of substrate (chorismate) and the formation of intermediates and products were determined by 1H-NMR (Supplemental Figure S1) using a method akin to that used for the uncatalyzed reaction and for EntC(28–30) and analyzed to give relative concentrations which could be plotted as percentages over time (Figure 5). It should be noted that the starting chorismate substrate has a small amount of phenylpyruvate contamination (~6–7%) that can be seen in all of the time course experiments. The fluorescent species was confirmed as salicylate by NMR (Figure S1). Furthermore, three other compounds were observed to be formed: isochorismate and two new products, prephenate and phenylpyruvate. Prephenate is the product of the mutase reaction of chorismate, a reaction that may be performed nonenzymatically in aqueous solution. The prephenate is subsequently converted to phenylpyruvate over time, a reaction that may occur nonenzymatically (29). Since these two latter reactions occur nonenzymatically, the uncatalyzed reaction was recorded for these buffer and temperature conditions for direct comparison (Figure S1F and Figure 5F).
Figure 5. Kinetic analysis of the enzymatic conversion of chorismate.

by WT-PchA (A), D310E-PchA (B), D310E/A375T-PchA (C), WT-Irp9 (D), E281D/T348A-Irp9 (E), and the uncatalyzed reaction (F) in 50 mM Tris pD 7.9, 10 mM MgCl2 at 25 °C. The inset in part (F) shows a longer timeframe for comparison with PchA. The reactions were monitored by 1H-NMR spectroscopy (spectra are found in Supplemental Figure S1). Relative concentrations of chorismate (○), isochorismate (□), salicylate (◇), prephenate (■), and phenylpyruvate (◆) are plotted as a function of time. The lines connecting data points represent the curves determined by global fitting using KinTek Global Kinetic Explorer (32, 33).
The WT-PchA time course shows an equilibrium of chorismate (46% of reaction products at 960 min) and isochorismate (37%) that is reached within 15 min (Figure 5A) with a kobs for chorismate disappearance of 0.27 min−1 and a kobs for isochorismate formation of 0.29 min−1 (Table 4). The final ratio of chorismate to isochorismate is ~5:4, comparable to that reported for the isochorismate synthase EntC (30). Over the time course of the experiment, 9% of the chorismate is converted to prephenate with a rate of 0.00029 min−1 and 1% of the chorismate is converted to salicylate with a rate of 0.0013 min−1. Phenylpyruvate (7% final, up from 5% initial) was formed from prephenate at a rate of 0.00027 min−1. This value is 1,300-fold greater than the uncatalyzed reaction (0.00000021 min−1) and represents adventitious catalysis. Values for rate enhancement (kobs/kuncat) are found in Supplemental Table S1. These data agree with the previous conclusions that wildtype PchA is an isochorismate synthase that establishes an equilibrium between chorismate and isochorismate
D310E-PchA established an equilibrium between chorismate (14% of reaction products at 960 min) and isochorismate (10.5%) with a kobs for chorismate disappearance of 0.10 min−1 and a kobs for isochorismate formation of 0.12 min−1 (Figure 5B and Table 4). The final ratio of chorismate to isochorismate is ~4:3. Over the time course of the experiment, 14% of the chorismate is converted to prephenate with a rate of 0.010 min−1 and 4.5% of the chorismate is converted to salicylate with a rate of 0.0040 min−1. Phenylpyruvate (57% final, up from 6% initial) was formed from prephenate at a rate of 0.050 min−1. This value is 240,000-fold greater than the uncatalyzed reaction. Therefore, the variant does produce more salicylate than the native enzyme as proposed by the interchange hypothesis. However, the primary product being formed is phenylpyruvate from the prephenate intermediate, which is more suggestive of the permute hypothesis.
D310E/A375T-PchA established the equilibrium between chorismate (50% of reaction products at 960 min) and isochorismate (25%) much slower (Figure 5C) with a kobs for chorismate disappearance of 0.0018 min−1 and for isochorismate formation of 0.0017 min−1 (Table 4). The final ratio of chorismate to isochorismate is 2:1. Salicylate (4%) was produced very slowly at 0.00075 min−1. Prephenate (15%) was produced at 0.00027 min−1 and was attributed to the uncatalyzed reaction. Phenylpyruvate (6% final, up from 5% initial) was formed from prephenate at a rate of 0.0018 min−1. This value is 8,600-fold greater than the uncatalyzed reaction and represents adventitious catalysis. The isochorismate synthase reaction is sufficiently impaired by the generation of the PchA double variant that the uncatalyzed mutase reaction allows for an accumulation of prephenate relative to the wildtype enzyme.
WT-Irp9 produced primarily salicylate (82% of reaction products at 120 min) in agreement with the results seen in the progress curve. The kobs for salicylate formation was determined to be 0.23 min−1 (Table 4) with no isochorismate intermediate detected (Figure 5D). A prephenate intermediate is detected at early timepoints with phenylpyruvate forming at a rate of 0.063 min−1 to a final concentration of 14% at the end of the reaction time (up from an initial 5.5%). This value is 300,000-fold greater than the uncatalyzed reaction and represents adventitious catalysis. Only 5% of the chorismate in the absence of enzyme is converted to prephenate (uncatalyzed reaction) over the time course of the Irp9 reactions (120 min) with kobs of 0.0011 min−1 (Table 4) and minimal change in the phenylpyruvate concentration (Figure 5F). These results suggest that WT-Irp9 performs an adventitious reaction to convert chorismate to phenylpyruvate with a prephenate intermediate. Nevertheless, Irp9 is most efficient as a salicylate synthase, with no buildup of the isochorismate intermediate.
E281D/T348A-Irp9 produced 11% salicylate (of the reaction products at 120 min) (Figure 5E) in agreement with the progress curve. A kobs for salicylate formation of 0.056 min−1 was determined (Table 4). No isochorismate intermediate was detected in this reaction. Unexpectedly, the majority of the product was phenylpyruvate (85% final, up from 6% initial) with kobs for phenylpyruvate formation of 0.048 min−1. This value is 230,000-fold greater than the uncatalyzed reaction. The accumulation of the prephenate intermediate is evident in the early timepoints; however, a kobs for prephenate formation could not be reliably determined from these data. In contrast to the interchange hypothesis, the double variant does not become an isochorismate synthase (lose the isochorismate-pyruvate lyase activity selectively), but instead becomes a chorismate mutase-prephenate dehydratase as suggested by the permute hypothesis.
Global fit of NMR time course experiments
The NMR time course data in Figure 5 were simulated with KinTek Global Kinetic Explorer (32, 33) using the model shown in Figure 1. The exception is the uncatalyzed reaction, where only the k3 and k4 were modeled based on precedent(29). Indeed, the curves in the Figure 5 panels are the results of simulated time courses. The rate constants determined by global simulation (Table 5) are in good agreement with those determined experimentally
Table 5.
Rate constants from global fitting
| rate (min−1) | ||||||
|---|---|---|---|---|---|---|
| Irp9 | PchA | uncatalyzed | ||||
| WT | E281D/T348A | WT | D310E | D310E/A375T | ||
| k1 (chorismate → isochorismate) | 0.30 | 0.015 | 0.13 | 0.062 | 0.00098 | - |
| k−1 (isochorismate → chorismate) | 7.3 | 0.49 | 0.16 | 0.079 | 0.0020 | - |
| k2 (isochorismate → salicylate) | 16 | 1.2 | 0.000061 | 0.00028 | 0.00087 | - |
| k−2 (salicylate → isochorismate) | 0.032 | 0.0034 | 0.0021 | 0.0010 | 0.00022 | - |
| k3 (chorismate → prephenate) | 0.025 | 0.074 | 0.00051 | 0.0017 | 0.00023 | 0.00032 |
| k−3 (prephenate → chorismate) | 0.10 | 0.089 | 0.0024 | 0.0000000027 | 0.0011 | - |
| k4 (prephenate → phenylpyruvate) | 0.098 | 0.18 | 0.0027 | 0.036 | 0.00063 | 0.00011* |
| k−4 (phenylpyruvate → prephenate) | 0.0011 | 0.0040 | 0.0015 | 0.0098 | 0.00018 | - |
this value represents an upper limit. A value of 0.00000021 min−1 (as seen in the fitting of the kobs in Table 4) was fit equally as well.
Discussion
Isochorismate and Salicylate Synthases of the MST family
PchA and Irp9 are both involved in production of salicylate-capped siderophores, pyochelin (PchA) and yersiniabactin (Irp9) (Figure 2). PchA and Irp9 are putative structural homologues that both conduct the same first reaction: the conversion of chorismate to isochorismate (Figure 1A). Irp9 (15, 35, 36) subsequently converts the isochorismate to salicylate and pyruvate, whereas the production of salicylate in P. aeruginosa requires a second enzyme, an isochorismate-pyruvate lyase, to carry out the second reaction. This dichotomy is seen in other enzymes of this structural family (Figure 1B). EntC and MenF are isochorismate synthases: EntC (21, 37) is found in the pathway for the formation of the dihydroxybenzoate-capped siderophore enterobactin whereas MenF (13) is involved in the biosynthesis of the electron-carrier menaquinone (Vitamin K) (Figure 2). Both EntC and MenF are from Escherichia coli. MbtI is a salicylate synthase involved in salicylate-capped siderophore biosynthesis to form mycobactin (Mycobacterium tuberculosis) (14, 38). The inability to form salicylate is not surprising for EntC and MenF, because the isochorismate is subsequently converted to 2,3-dihydroxybenzoate (in two steps, enterobactin biosynthesis (39, 40)) or o-succinylbenzoate (in two steps, menaquinone biosynthesis (41–43)) for ultimate incorporation into metabolites that are not salicylate-containing. However, the inability to perform the lyase reaction is intriguing for PchA, since this means that P. aeruginosa requires a separate isochorismate-pyruvate lyase enzyme to form salicylate. Structures of MenF (13), EntC (21, 37), Irp9 (15) and MbtI (14, 38) have been determined. A structure of PchA has yet to be determined; however, the active site residues are highly conserved (Figure 3) (20).
The enzyme engineering hypotheses for lyase activity in MST enzymes
Inspection of the active sites of the structurally and functionally homologous members of the MST family that generate isochorismate, either as intermediate or product, showed two sites with trends consistent with the absence or presence of lyase activity (Figure 3). An alanine residue in the isochorismate synthases (lyase-deficient; A375 in PchA) is instead a threonine in the salicylate synthases (lyase-active; T348 in Irp9). The backbone carbonyl oxygen of this residue forms a hydrogen bond to the substrate hydroxyl in the salicylate and thought to be important in substrate orientation for lyase activity (13, 21). An aspartic acid or asparagine in the isochorismate synthases (lyase-deficient; D310 in PchA) is a glutamic acid in the salicylate synthases (lyase-active; E281 in Irp9). This amino acid chelates the catalytically-required magnesium ion through a water molecule (13, 15, 21, 37). The length of the sidechain has been hypothesized to be important in active site architecture, possibly allowing the formation of a reactive substrate conformation to drive the pericyclic lyase reaction (20). The simple, “interchange” hypothesis proposes that lyase activity could be instituted into the isochorismate synthase PchA and could be removed from the salicylate synthase Irp9 by interchanging these nonconserved active site residues. A second, more complex “permute” hypothesis proposes that the chorismate-utilizing enzymes are evolutionarily designed to convert chorismate into a variety of products, and active site mutagenesis may lead to the identification and/or augmentation of lesser adventitious activities.
Steady state kinetic parameters are inconclusive for testing these hypotheses
Steady state kinetic parameters for isochorismate synthase, isochorismate-pyruvate lyase and salicylate synthase activities were measured for the individual and double variants (Table 1, 2 and 3). The variation for binding the substrate hydroxyl (A375T in PchA or T348A in Irp9) showed a universal decrease for catalytic efficiency for the formation of isochorismate, as measured by isochorismate synthase or salicylate synthase activities. This was not the desired effect, as the interchange hypothesis suggested that sidechain swapping at these sites would alter lyase activity not isochorismate synthase activity. However, this result was not unexpected based on previous results which documented a similar change (loss of isochorismate synthase activity) in MenF and EntC (13, 21). In all cases, the mutational effect of the residue that chelates the magnesium (D310E in PchA or E281D in Irp9) had the less detrimental effects on catalytic efficiency. Indeed, the D310E-PchA showed isochorismate synthase catalytic efficiency that was within error of the wildtype value and a three-fold increase in the isochorismate-pyruvate lyase catalytic efficiency as proposed by the interchange hypothesis. The converse mutant in Irp9 (E281D) showed a diminished lyase catalytic efficiency, but showed a more significant decrease in salicylate synthase catalytic efficiency, suggesting that the isochorismate synthase activity is the more impaired of the two activities by this variation, in contrast to the interchange hypothesis.
The progress curves (Figure 4) made it evident that there were possibly more reaction products than merely chorismate, isochorismate and salicylate as proposed by the permute hypothesis. In the reaction carried out by the wildtype Irp9, not all of the substrate was converted to the salicylate product and in the reaction carried out by the wildtype PchA, a small amount of a product with fluorescent properties consistent for salicylate was being formed. Therefore, a more sensitive method for measuring the products than the simple steady state assays was required. Using NMR time courses similar to those used previously for the uncatalyzed reaction(28, 29) and for EntC (30), the relative concentrations of the products formed (expressed as percentages) and the rates at which the products were formed could be measured (Figures 5 and S1). Global analysis in KinTek Global Kinetic Explorer (32, 33) was used to determine rate constants using the model in Figure 1. In brief, k1 represents the isochorismate synthase reaction, k2 is the isochorismate-pyruvate lyase reaction, k3 is the chorismate mutase reaction and k4 is the prephenate dehydratase reaction. All reactions were allowed to be reversible in the model for the enzyme catalyzed reactions. Only k3 and k4 were modeled in the uncatalyzed reaction, as per precedent (28). The curves in Figure 5 were generated by the global analysis.
Adventitious chorismate mutase and prephenate dehydratase activity
Our results weigh in on the controversy as to whether enzymes of this class have chorismate mutase activity (14, 16).2 If the rate constants determined from the global analysis are used to define the rate enhancement compared to the uncatalyzed reaction, it is evident that both wildtype and the double variant of the salicylate synthase (Irp9) catalyze the chorismate mutase reaction (k3) 77 to 230-fold more efficiently than the uncatalyzed reaction (for example, wildtype 0.025 min−1/ uncatalyzed 0.00032 min−1 ≈ 77-fold rate enhancement; Supplemental Table S2). Moreover, the formation of phenylpyruvate from prephenate is catalyzed 890 (wildtype) to 1700-fold (double variant) faster than the enzyme-free reaction. In the wildtype enzyme, this means that the enzyme displays predominantly salicylate synthase activity, but does also have adventitious chorismate mutase-prephenate dehydratase activity. Conversely, the Irp9-double variant is predominantly a chorismate mutase-prephenate dehydratase with adventitious salicylate synthase activity.
A similar comparison of rate enhancement for the isochorismate synthase PchA indicates that this enzyme shows no chorismate mutase activity (~2-fold greater than the uncatalyzed reaction) but does have some adventitious prephenate dehydratase activity (24-fold enhancement). The D310E-PchA variant arguably has chorismate mutase activity (5-fold rate enhancement), and has significant prephenate dehydratase rate enhancement (330-fold). The double variant of PchA does not display activities other than those for the formation of isochorismate (no rate enhancement for isochorismate-pyruvate lyase or chorismate mutase) with a ~6-fold increase for prephenate dehydratase activity. Therefore, the wildtype PchA enzyme and the double variant are truly isochorismate synthase enzymes with the wildtype enzyme displaying nominal prephenate dehydratase activity. The D310E-PchA variant is an isochorismate synthase with considerable prephenate dehydratase activity.
Altered partitioning in the variant proteins
A critical look at partition ratios (Table 6) helps to explain the calculated rates and observed accumulation of products. There is one branch point and two commitment points in the model (Figure 1A). When chorismate binds, it may be converted to isochorismate (k1, isochorismate synthase activity) or it may be converted to prephenate (k3, chorismate mutase activity). This represents the branch point. All the enzymes and variants are strongly in favor of the generation of isochorismate (k1/k3 = 4 to 250) with the exception of the double variant of Irp9 (k1/k3 = 0.2). The first commitment point is the formation of either salicylate or the regeneration of chorismate from isochorismate (k2/k−1). The enzymes are divided at this junction as would be expected based on their lyase-active or lyase-deficient states: the PchA enzymes preferably regenerate chorismate (k2/k−1 = 0.00038 to 0.44), whereas the Irp9 variants preferentially form salicylate (k2/k−1 = 2.2 to 2.5). Finally, if prephenate is formed by a mutase reaction, there is a second commitment point for generation of phenylpyruvate or the regeneration of chorismate (k4/k−3). For the wildtype enzymes, these partition ratios are ~1. The most interesting value is that of the D310E-PchA variant that shows that the prephenate dehydratase activity of this enzyme is favored by 18,000,000 relative to the reformation of chorismate (while this value is error prone, it does show a strong commitment for dehydratase activity). If one considers the double variant of PchA only nominally catalytically active, this means that the variants of PchA (D310E) and Irp9 (E281D/T348A) tend toward chorismate mutase-prephenate dehydratase activity, but arrive there by different pathways. The D310E-PchA variant establishes an equilibrium between chorismate and isochorismate. If any chorismate is converted to prephenate then the prephenate is highly preferentially converted to phenylpyruvate. Conversely, for the E281D/T348A-Irp9, chorismate is more likely to be converted to prephenate (5-fold more likely) and then subsequently converted to phenylpyruvate (2-fold more likely).
Table 6.
Partition ratios calculated from global fitting
| k1/k3 | k2/k−1 | k4/k−3 | |
|---|---|---|---|
| PchA | |||
| WT | 250 | 0.00038 | 1.1 |
| D310E | 36 | 0.035 | 18,000,000 |
| D310E/A375T | 4.3 | 0.44 | 0.57 |
|
| |||
| Irp9 | |||
| WT | 12 | 2.2 | 0.98 |
| E281D/T348A | 0.2 | 2.5 | 2 |
|
| |||
| Key | |||
| >1, chorismate → isochorismate | >1, isochorismate → salicylate | >1, prephenate → phenylpyruvate | |
| <1, chorismate → prephenate | <1, isochorismate → chorismate | <1, prephenate → chorismate | |
Implications for acid-base chemistry in the MST enzymes
The permute hypothesis for these chorismate-utilizing enzymes suggests that these active sites are designed to permute chorismate and that variation in the active site may lead to development of adventitious activities. The mechanism of the isochorismate synthase activity has been previously defined as general acid-general base chemistry with a lysine general base and a glutamic acid general acid in reverse protonation states (Figure 6A) (18). Presumably, this is true for both the structurally conserved isochorismate and salicylate synthases. Prephenate dehydratase activity is a concerted decarboxylation reaction assisted by general acid catalysis for removal of the hydroxyl group (Figure 6B). Aromatization is hypothesized to be promoted by a forcing of planarity of the ring (44). One might imagine that a strategically placed general acid residue for the isochorismate synthase activity in the wildtype enzymes may serve a similar role in the variants for prephenate dehydratase activity (compare Figures 6A and 6B).
Figure 6. Catalytic mechanisms and transition states.

A) Isochorismate synthase mechanism (18). B) Prephenate dehydratase mechanism (44) . C) Isochorismate-pyruvate lyase transition state (19). D) Chorismate mutase transition state (45)
Implications for pericyclic reactions in MST enzymes
Similar arguments can be made for the lyase and mutase activities as were for the acid-base chemistry above. The isochorismate-pyruvate lyase activity is hypothesized to be a pericyclic reaction: quantitative hydrogen transfer from C2 to C9 has been demonstrated in the independent isochorismate-pyruvate lyase from Pseudomonas aeruginosa (PchB) (19) and experiments in the salicylate synthase MbtI (14) gave similar results. The chorismate mutase activity is likewise pericyclic, converting the linkage of the pyruvate tail to the ring from the C3 ether linkage to a C1-C9 linkage(45). Therefore, the transition states of the isochorismate-pyruvate lyase (Figure 6C) and chorismate mutase (Figure 6D) activities are achieved by folding of the pyruvate over the ring, aligning the C9 carbon either with the hydrogen attached to C2 (lyase) or the carbon of C1 (mutase). Since pericyclic reactions have a strong entropic/steric component to their energy barrier, it follows that enzymes that perform one reaction may have adventitious activity for the other. This has been documented previously for PchB (a lyase with adventitious mutase activity) (23, 25, 27, 46–48), and here for Irp9 (an enzyme with physiological lyase activity and adventitious mutase activity). The adventitious pericyclic reactions were more readily measured and sometimes augmented in the variants tested.
Conclusions
The simple interchange hypothesis that lyase activity could be instituted into the isochorismate synthase PchA and could be removed from the salicylate synthase Irp9 by interchanging the nonconserved active site residues was not borne out. Instead, the variants showed an augmentation of the adventitious chorismate mutase and prephenate dehydratase activities in agreement with the more complex, permute hypothesis. The sidechain of most importance was the residue that chelates the magnesium ion through a water molecule. This variation most likely moves the metal ion within the active site. Since the substrate carboxylate binds to the metal ion, subtle changes in the location of the ion may cause the formation of different transition states and the augmentation of adventitious activities.
Supplementary Material
Highlights.
Exchange of active site residues in related enzymes doesn’t interchange activities.
Exchange of active site residues augments adventitious or promiscuous activities.
Active site mutants in synthases augment mutase activity up to 12-fold.
Active site mutants in synthases augment dehydratase activity up to 240,000-fold.
Acknowledgments
This publication was made possible by funds from NIH grant number P20 RR016475 from the INBRE Program of the National Center for Research Resources, and by NIH grants numbered R01 AI77725 and K02 AI093675 from the National Institute for Allergy and Infectious Disease.
We are grateful to R. L. Schowen and K. A. Johnson for insightful discussions and suggestions and to A. S. Chilton and J. T. Douglas for technical assistance.
Abbreviations and Full Textual Notes
- EntC
isochorismate synthase from E. coli
- Irp9
salicylate synthase from Yersinia enterocolitica
- MbtI
salicylate synthase from Mycobacterium tuberculosis
- MenF
isochorismate synthase from E. coli
- MST
protein family containing proteins of menaquinone, siderophore or tryptophan biosynthesis
- PchA
isochorismate synthase from Pseudomonas aeruginosa
- PchB
isochorismate-pyruvate lyase from Pseudomonas aeruginosa
Footnotes
When measuring new activities in an active site evolutionarily designed for differing chemistry, one should consider the values of rate enhancement in terms of enzyme engineering projects such as directed evolution where an order of magnitude increase is significant (Aharoni et al., Nature Genetics 37, 73; Sarkar, Hauber, Hauber, Buchholz, Science 316, 1912), as opposed to standard enzyme catalytic efficiencies were a good enzyme is 105 M−1 s−1.
Chorismate mutase activity in MST enzymes has previously been attributed to a contaminant from the protein expression and purification process (Ziebart, Toney. Biochemistry 49, 2851). The products detected by NMR could be attributed to contamination of the variant enzymes by PheA, the chorismate mutase-prephenate dehydratase enzyme of the essential aromatic amino acid biosynthetic pathway, carried along during the purification process from the E. coli protein overexpression system. For PheA, the ratio of kcat for chorismate mutase to kcat of prephenate dehydratase is ~ 1 (Zhang, Wilson, Ganem. Biochemistry 39, 4722). In the data presented here, there are several variants for which the mutase activity is not greater than the uncatalyzed reaction, and for all of the variants the ratio for mutase (k3) to dehydratase (k4) from the global analysis ranges from 0.05 to 0.4. Because all of the enzymes are purified using the same protocol and because the ratio of the two activities should be constant regardless of concentration of the contaminating PheA, we propose that the mutase and dehydratase activities detected herein are not the result of contamination by proteins from the E. coli producer strains.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilks HM, Hart KW, Feeney R, Dunn CR, Muirhead H, Chia WN, Barstow DA, Atkinson T, Clarke AR, Holbrook JJ. A specific, highly active malate dehydrogenase by redesign of a lactate dehydrogenase framework. Science (New York, N Y. 1988;242:1541–1544. doi: 10.1126/science.3201242. [DOI] [PubMed] [Google Scholar]
- 2.Hedstrom L, Farr-Jones S, Kettner CA, Rutter WJ. Converting trypsin to chymotrypsin: ground-state binding does not determine substrate specificity. Biochemistry. 1994;33:8764–8769. doi: 10.1021/bi00195a018. [DOI] [PubMed] [Google Scholar]
- 3.Hedstrom L, Perona JJ, Rutter WJ. Converting trypsin to chymotrypsin: residue 172 is a substrate specificity determinant. Biochemistry. 1994;33:8757–8763. doi: 10.1021/bi00195a017. [DOI] [PubMed] [Google Scholar]
- 4.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science (New York, N Y. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 5.Jackson LK, Brooks HB, Osterman AL, Goldsmith EJ, Phillips MA. Altering the reaction specificity of eukaryotic ornithine decarboxylase. Biochemistry. 2000;39:11247–11257. doi: 10.1021/bi001209s. [DOI] [PubMed] [Google Scholar]
- 6.Bertoldi M, Castellani S, Voltattorni CB. Mutation of residues in the coenzyme binding pocket of Dopa decarboxylase. European journal of biochemistry / FEBS. 2001;268:2975–2981. doi: 10.1046/j.1432-1327.2001.02187.x. [DOI] [PubMed] [Google Scholar]
- 7.Bertoldi M, Gonsalvi M, Contestabile R, Voltattorni CB. Mutation of tyrosine 332 to phenylalanine converts dopa decarboxylase into a decarboxylation-dependent oxidative deaminase. The Journal of biological chemistry. 2002;277:36357–36362. doi: 10.1074/jbc.M204867200. [DOI] [PubMed] [Google Scholar]
- 8.Graber R, Kasper P, Malashkevich VN, Strop P, Gehring H, Jansonius JN, Christen P. Conversion of aspartate aminotransferase into an L-aspartate beta-decarboxylase by a triple active-site mutation. The Journal of biological chemistry. 1999;274:31203–31208. doi: 10.1074/jbc.274.44.31203. [DOI] [PubMed] [Google Scholar]
- 9.Joerger AC, Mayer S, Fersht AR. Mimicking natural evolution in vitro: an N-acetylneuraminate lyase mutant with an increased dihydrodipicolinate synthase activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5694–5699. doi: 10.1073/pnas.0531477100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. The ‘evolvability’ of promiscuous protein functions. Nature genetics. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar I, Hauber I, Hauber J, Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science (New York, N Y. 2007;316:1912–1915. doi: 10.1126/science.1141453. [DOI] [PubMed] [Google Scholar]
- 12.He Z, Stigers Lavoie KD, Bartlett PA, Toney MD. Conservation of mechanism in three chorismate-utilizing enzymes. J Am Chem Soc. 2004;126:2378–2385. doi: 10.1021/ja0389927. [DOI] [PubMed] [Google Scholar]
- 13.Kolappan S, Zwahlen J, Zhou R, Truglio JJ, Tonge PJ, Kisker C. Lysine 190 is the catalytic base in MenF, the menaquinone-specific isochorismate synthase from Escherichia coli: implications for an enzyme family. Biochemistry. 2007;46:946–953. doi: 10.1021/bi0608515. [DOI] [PubMed] [Google Scholar]
- 14.Zwahlen J, Kolappan S, Zhou R, Kisker C, Tonge PJ. Structure and mechanism of MbtI, the salicylate synthase from Mycobacterium tuberculosis. Biochemistry. 2007;46:954–964. doi: 10.1021/bi060852x. [DOI] [PubMed] [Google Scholar]
- 15.Kerbarh O, Chirgadze DY, Blundell TL, Abell C. Crystal structures of Yersinia enterocolitica salicylate synthase and its complex with the reaction products salicylate and pyruvate. J Mol Biol. 2006;357:524–534. doi: 10.1016/j.jmb.2005.12.078. [DOI] [PubMed] [Google Scholar]
- 16.Ziebart KT, Toney MD. Nucleophile specificity in anthranilate synthase, aminodeoxychorismate synthase, isochorismate synthase, and salicylate synthase. Biochemistry. 2010;49:2851–2859. doi: 10.1021/bi100021x. [DOI] [PubMed] [Google Scholar]
- 17.Ferrer S, Marti S, Moliner V, Tunon I, Bertran J. Understanding the different activities of highly promiscuous MbtI by computational methods. Phys Chem Chem Phys. 2012;14:3482–3489. doi: 10.1039/c2cp23149b. [DOI] [PubMed] [Google Scholar]
- 18.Meneely KM, Luo Q, Dhar P, Lamb AL. Lysine221 is the general base residue of the isochorismate synthase from Pseudomonas aeruginosa (PchA) in a reaction that is diffusion limited. Arch Biochem Biophys. doi: 10.1016/j.abb.2013.07.026. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeClue MS, Baldridge KK, Kunzler DE, Kast P, Hilvert D. Isochorismate Pyruvate Lyase: A Pericyclic Reaction Mechanism? J Am Chem Soc. 2005;127:15002–15003. doi: 10.1021/ja055871t. [DOI] [PubMed] [Google Scholar]
- 20.Lamb AL. Pericyclic reactions catalyzed by chorismate-utilizing enzymes. Biochemistry. 2011;50:7476–7483. doi: 10.1021/bi2009739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sridharan S, Howard N, Kerbarh O, Blaszczyk M, Abell C, Blundell TL. Crystal structure of Escherichia coli enterobactin-specific isochorismate synthase (EntC) bound to its reaction product isochorismate: implications for the enzyme mechanism and differential activity of chorismate-utilizing enzymes. J Mol Biol. 2010;397:290–300. doi: 10.1016/j.jmb.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Dosselaere F, Vanderleyden J. A metabolic node in action: chorismate-utilizing enzymes in microorganisms. Critical reviews in microbiology. 2001;27:75–131. doi: 10.1080/20014091096710. [DOI] [PubMed] [Google Scholar]
- 23.Zaitseva J, Lu J, Olechoski KL, Lamb AL. Two crystal structures of the isochorismate pyruvate lyase from Pseudomonas aeruginosa. J Biol Chem. 2006;281:33441–33449. doi: 10.1074/jbc.M605470200. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt K, Leistner E. Microbial Production of (+)-trans-Isochorismic Acid. Biotech & Bioeng. 1995;45:285–291. doi: 10.1002/bit.260450402. [DOI] [PubMed] [Google Scholar]
- 25.Luo Q, Olucha J, Lamb AL. Structure-function analyses of isochorismate-pyruvate lyase from Pseudomonas aeruginosa suggest differing catalytic mechanisms for the two pericyclic reactions of this bifunctional enzyme. Biochemistry. 2009;48:5239–5245. doi: 10.1021/bi900456e. [DOI] [PubMed] [Google Scholar]
- 26.Rieger CE, Turnbull JL. Small scale biosynthesis and purification of gram quantities of chorismic acid. Preparative biochemistry & biotechnology. 1996;26:67–76. doi: 10.1080/10826069608000051. [DOI] [PubMed] [Google Scholar]
- 27.Gaille C, Kast P, Haas D. Salicylate biosynthesis in Pseudomonas aeruginosa Purification and characterization of PchB, a novel bifunctional enzyme displaying isochorismate pyruvate-lyase and chorismate mutase activities. J Biol Chem. 2002;277:21768–21775. doi: 10.1074/jbc.M202410200. [DOI] [PubMed] [Google Scholar]
- 28.DeClue MS, Baldridge KK, Kast P, Hilvert D. Experimental and computational investigation of the uncatalyzed rearrangement and elimination reactions of isochorismate. J Am Chem Soc. 2006;128:2043–2051. doi: 10.1021/ja056714x. [DOI] [PubMed] [Google Scholar]
- 29.Wright SK, DeClue MS, Mandal A, Lee L, Wiest O, Cleland WW, Hilvert D. Isotope effects on the enzymatic and nonenzymatic reactions of chorismate. J Am Chem Soc. 2005;127:12957–12964. doi: 10.1021/ja052929v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Quinn N, Berchtold GA, Walsh CT. Overexpression, purification, and characterization of isochorismate synthase (EntC), the first enzyme involved in the biosynthesis of enterobactin from chorismate. Biochemistry. 1990;29:1417–1425. doi: 10.1021/bi00458a012. [DOI] [PubMed] [Google Scholar]
- 31.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibium and Steady State Enzyme Systems. John Wiley & Sons; New York: 1975. [Google Scholar]
- 32.Johnson KA, Simpson ZB, Blom T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Analytical biochemistry. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 33.Johnson KA, Simpson ZB, Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Analytical biochemistry. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Gaille C, Reimmann C, Haas D. Isochorismate synthase (PchA), the first and rate-limiting enzyme in salicylate biosynthesis of Pseudomonas aeruginosa. J Biol Chem. 2003;278:16893–16898. doi: 10.1074/jbc.M212324200. [DOI] [PubMed] [Google Scholar]
- 35.Kerbarh O, Ciulli A, Howard NI, Abell C. Salicylate biosynthesis: overexpression, purification, and characterization of Irp9, a bifunctional salicylate synthase from Yersinia enterocolitica. J Bacteriol. 2005;187:5061–5066. doi: 10.1128/JB.187.15.5061-5066.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelludat C, Brem D, Heesemann J. Irp9, encoded by the high-pathogenicity island of Yersinia enterocolitica, is able to convert chorismate into salicylate, the precursor of the siderophore yersiniabactin. J Bacteriol. 2003;185:5648–5653. doi: 10.1128/JB.185.18.5648-5653.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsons JF, Shi KM, Ladner JE. Structure of isochorismate synthase in complex with magnesium. Acta Crystallogr. 2008;D64:607–610. doi: 10.1107/S0907444908005477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrison AJ, Yu M, Gardenborg T, Middleditch M, Ramsay RJ, Baker EN, Lott JS. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J Bacteriol. 2006;188:6081–6091. doi: 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Duncan K, Walsh CT. Nucleotide sequence of a cluster of Escherichia coli enterobactin biosynthesis genes: identification of entA and purification of its product 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase. J Bacteriol. 1989;171:791–798. doi: 10.1128/jb.171.2.791-798.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rusnak F, Liu J, Quinn N, Berchtold GA, Walsh CT. Subcloning of the enterobactin biosynthetic gene entB: expression, purification, characterization, and substrate specificity of isochorismatase. Biochemistry. 1990;29:1425–1435. doi: 10.1021/bi00458a013. [DOI] [PubMed] [Google Scholar]
- 41.Palaniappan C, Sharma V, Hudspeth ME, Meganathan R. Menaquinone (vitamin K2) biosynthesis: evidence that the Escherichia coli menD gene encodes both 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylic acid synthase and alpha-ketoglutarate decarboxylase activities. J Bacteriol. 1992;174:8111–8118. doi: 10.1128/jb.174.24.8111-8118.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popp JL. Sequence and overexpression of the menD gene from Escherichia coli. J Bacteriol. 1989;171:4349–4354. doi: 10.1128/jb.171.8.4349-4354.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma V, Meganathan R, Hudspeth ME. Menaquinone (vitamin K2) biosynthesis: cloning, nucleotide sequence, and expression of the menC gene from Escherichia coli. J Bacteriol. 1993;175:4917–4921. doi: 10.1128/jb.175.15.4917-4921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Vleet J, Kleeb A, Kast P, Hilvert D, Cleland WW. 13C isotope effect on the reaction catalyzed by prephenate dehydratase. Biochim Biophys Acta. 2010;1804:752–754. doi: 10.1016/j.bbapap.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustin DJ, Mattei P, Kast P, Wiest O, Lee L, Cleland WW, Hilvert D. Heavy Atom Isotope Effects Reveal a Highly Polarized Transition State for Chorismate Mutase. J Am Chem Soc. 1999;121:1756–1757. [Google Scholar]
- 46.Luo Q, Olucha J, Lamb AL. Entropic and enthalpic components of catalysis in the mutase and lyase activities of Pseudomonas aeruginosa PchB. J Am Chem Soc. 2011;133:7229–7233. doi: 10.1021/ja202091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olucha J, Meneely KM, Lamb AL. Modification of Residue 42 of the Active Site Loop with a Lysine-Mimetic Side Chain Rescues Isochorismate-Pyruvate Lyase Activity in Pseudomonas aeruginosa PchB. Biochemistry. 2012 doi: 10.1021/bi300472n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olucha J, Ouellette AN, Luo Q, Lamb AL. pH dependence of catalysis by Pseudomonas aeruginosa isochorismate-pyruvate lyase: Implications for transition state stabilization and the role of lysine 42. Biochemistry. 2011;50:7198–7207. doi: 10.1021/bi200599j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeLano W. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.