

Hybridization to microarrays has been the standard for genome‐wide transcriptome analyses of prokaryotes in the past 10 years. Microarrays have several limitations, however, among which are a small dynamic range for detection of transcript levels due to problems with saturation, background noise, spot density and spot quality. Moreover, comparing different experiments requires complex normalization methods (Hinton et al., 2004) and comparing different strains requires designing pangenome arrays based on multiple sequenced genomes, leading to further problems in non‐specific or cross‐hybridization and complicated data analysis (Bayjanov et al., 2009). Most microarrays have a biased genome coverage, as they only contain a limited number of short probes for known or expected genes in sequenced genomes, and they rarely probe intergenic regions. Technological advances in array production and dropping costs have recently led to the design and use of high‐density tiling arrays based on overlapping short oligonucleotides covering both strands of entire genomes (Selinger et al., 2000; Mcgrath et al., 2007; Rasmussen et al., 2009; Toledo‐Arana et al., 2009). Tiling array and other studies have provided a first insight into far more complex transcriptomes than previously envisioned, including an ever‐expanding range of regulatory RNAs (Waters and Storz, 2009). To overcome the remaining limitations of microarrays, a totally new approach to whole‐transcriptome analysis was needed – and a much‐awaited breakthrough in DNA sequencing came to the rescue. Here, we describe the first whole‐transcriptome applications in prokaryotes and discover that a new treasure chest of regulation in prokaryotes is being opened.
Whole‐transcriptome sequencing
With the dawn of next generation (or deep) sequencing technologies in recent years (Ansorge, 2009; Metzker, 2010), their application to high‐depth sequencing of whole transcriptomes, a technique now referred to as RNA‐seq, has been explored (Morozova et al., 2009; Wang et al., 2009; Wilhelm and Landry, 2009). RNA‐seq requires a conversion of mRNA into cDNA by reverse transcription, followed by deep sequencing of this cDNA (Fig. 1A). RNA‐seq was initially only used for analysing eukaryotic mRNA, as prokaryote mRNA is less stable and lacks the poly(A) tail that is used for enrichment and reverse transcription priming in eukaryotes. But these technological difficulties are being overcome, as various methods for enrichment of prokaryote mRNA and appropriate cDNA library construction protocols have been developed, some generating strand‐specific libraries which provide valuable information about the orientation of transcripts.
Figure 1.
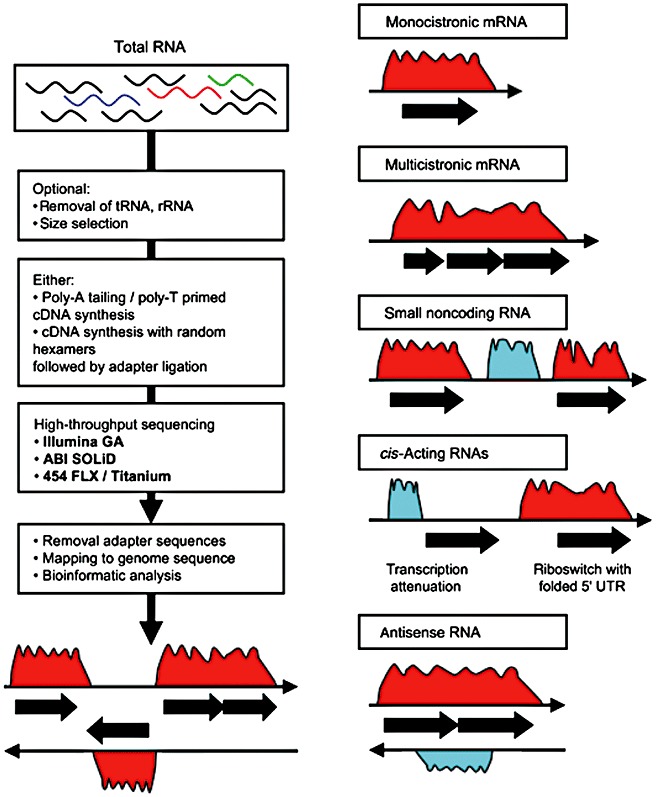
(Left panel) Flow diagram of the steps involved in microbial transcriptome sequencing. The starting material is a mix of RNA, followed by optional subtraction of tRNA and rRNA, generation of cDNA libraries, sequencing, bioinformatics and interpretation of cDNA sequencing read histograms. (Right panel) Schematic representation of transcriptome sequencing histograms. Examples are shown of monocistronic and polycistronic mRNAs, non‐coding RNA, cis‐acting RNAs, and antisense RNA. Black filled arrows represent annotated ORFs. Reprinted from van Vliet (2010). Copyright 2009, FEMS and Blackwell Publishing Ltd.
In June 2008, the first reports appeared of RNA sequencing of whole microbial transcriptomes, i.e. the yeasts Saccharomyces cerevisae (Nagalakshmi et al., 2008) and Schizosaccharomyces pombe (Wilhelm et al., 2008). Both studies demonstrated that most of the non‐repetitive sequence of the yeast genome is transcribed, and provided detailed information of novel genes, introns and their boundaries, 3′ and 5′ boundary mapping, 3′ end heterogeneity and overlapping genes, antisense RNA and more. Starting in 2009, several examples have been reported of prokaryote whole‐transcriptome analysis using tiling arrays and/or RNA‐seq, and these are summarized in Table 1. The first reviews of prokaryote transcriptome sequencing have just appeared (Croucher et al., 2009; van Vliet and Wren, 2009; Sorek and Cossart, 2010; van Vliet, 2010).
Table 1.
Whole‐transcriptome analysis of microbes.
| Technique | Corrected genes | New genes | ncRNA | Antisense RNA | Reference | |
|---|---|---|---|---|---|---|
| Bacteria | ||||||
| Mycoplasma pneumoniae | TA, RNAseq, spotted arrays | 5 | 4 | 108 | 89 | Guell et al. (2009) |
| Salmonella enterica sv Typhi | ssRNA‐seq | 40 | Perkins et al. (2009) | |||
| Chlamydia trachomatis L2b | RNA‐seq | 5 | 41 | 25 | Albrecht et al. (2009) | |
| Listeria monocytogenes EGD‐e | TA | 5 | 45 | 7 | Toledo‐Arana et al. (2009) | |
| Listeria monocytogenes 10403S | RNA‐seq | 67 | Oliver et al. (2009) | |||
| Burkholderia cenocepacia | RNA‐seq | 13 | Yoder‐Himes et al. (2009) | |||
| Bacillus anthracis Sterne 34eF2 | RNA‐seq | 11 | 57 | Passalacqua et al. (2009) | ||
| Bacillus subtilis 168 | TA | 119 | 84 | 127 | Rasmussen et al. (2009) | |
| Vibrio cholerae | RNA‐seqa | 520 | 127 | Liu et al. (2009) | ||
| Archaea | ||||||
| Sulfolobus solfataricus P2 | RNA‐seq | 162 | 80 | 310 | 185 | Wurtzel et al. (2010) |
| Halobacterium salinarum | TA | 61 | 10 | 61 | Koide et al. (2009) | |
| Eukaryotes | ||||||
| Schizosaccharomyces pombe | TA, RNA‐seq | 75 | 26 | 427 | 37 | Wilhelm et al. (2008) |
| Saccharomyces cerevisiae | RNA‐seq | 64 | 487 | Nagalakshmi et al. (2008) |
Enriched for only sRNAs of 14–200 nt.
TA, tiling array; RNAseq, cDNA sequencing; ss, strand‐specific; ncRNA, non‐coding RNA.
Novel general features discovered
Numerous new insights into genomic elements, gene expression and complexity of regulation are emerging from these new high‐throughput and high‐resolution studies of microbial transcriptomes (Fig. 1B).
Gene structure/length, novel genes
Gene annotation has always been fraught with difficulties and is not a trivial exercise. Most gene‐finding algorithms miss or miss‐annotate small protein‐encoding genes and non‐coding RNAs (together called sRNAs), but tiling arrays and RNA‐seq can readily identify these genes (Figs 2 and 3). The high resolution of these techniques allows transcription start sites (TSS) to be mapped with single‐base pair resolution. Moreover, gene structure can be corrected (Table 1), as many gene starts are found to be downstream of the automatically predicted start of largest possible ORFs, e.g. in Sulfolobus solfataricus (Wurtzel et al., 2010).
Figure 2.
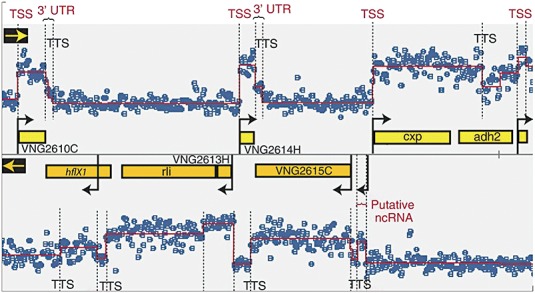
Transcriptome structure in H. salinarum determined with high‐density tiling arrays (60‐mer overlapping probes). Segment of genome map with signal intensity of total RNA is shown. Each blue dot represents probe intensity (in log2 scale) in the forward (upper panel) or reverse strand (lower panel). The overlaid red line is the result of a segmentation algorithm that was applied to determine transcription start sites (TSS and black arrows), transcription termination sites (TTS), untranslated regions in mRNAs (3′ UTR) and putative non‐coding RNAs. Reprinted and adapted from Koide et al. (2009). Copyright 2009, EMBO and Macmillan Publishers Limited.
Figure 3.
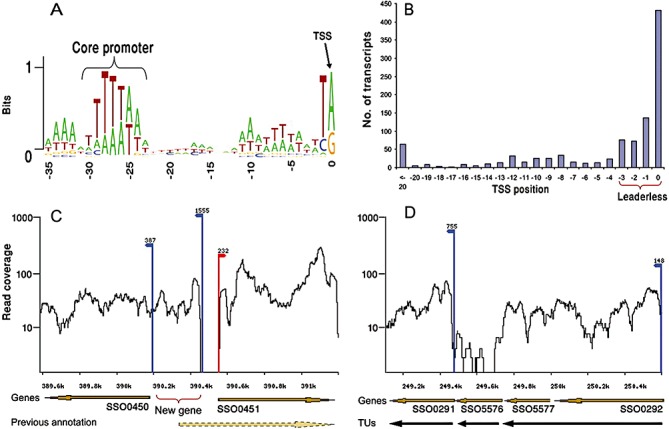
The structure of the S. solfataricus transcriptome determined by RNA‐seq. A. Core promoter. B. Distribution of mapped TSS (transcription start site) positions relative to the ORF ATG codon. C. Example of correction of gene annotations. Transcriptome data indicate that gene SSO0451 actually is 228 bp shorter, and that a new small gene is encoded on the reverse strand. D. Refinement of operon definition. Transcriptome data show either 2 or 3 separate transcriptional units (TU), instead of the predicted 1 TU. Red arrow indicates TSS on forward strand, and blue arrows indicate TSS on reverse strand. Reprinted from Wurtzel et al. (2010). Copyright 2009, Cold Spring Harbor Laboratory Press.
Untranslated regions
Whole‐transcriptome mapping can identify contiguous expression extending into flanking regions of a protein‐encoding gene, indicative of 5′ or 3′ untranslated regions (UTRs). Long 5′ UTRs are often indicative of upstream regulatory elements, such as riboswitches (Toledo‐Arana et al., 2009). Archaea have much shorter or no 5′ UTRs compared with bacteria (Koide et al., 2009; Wurtzel et al., 2010), suggesting alternative modes of regulation. Long 3′ UTRs could affect expression of downstream genes or genes on the opposite strand, as found in archaea (Brenneis and Soppa, 2009).
Operon structures
Whole‐transcriptome data allow operons to be better defined, and the first experimentally determined operon maps show that 60–70% of bacterial genes are transcribed as operons, but only 30–40% in archaea. Staircase‐like expression within operons appears to be common (Guell et al., 2009).
Whole‐transcriptome analysis of Mycoplasma pneumoniae, using a mixture of tiling arrays, deep sequencing and 137 different growth conditions, showed that there is context‐dependent modulation of operon structure (Guell et al., 2009). This involves repression or activation of operon internal genes as well as genes located at the operon ends. This adds a whole new level of complexity to gene regulation. Similar ‘conditional operons’ were found in Halobacterium salinarum (Koide et al., 2009).
Non‐coding RNAs
Non‐coding RNAs (ncRNA), typically 50–500 nt long, can play important regulatory roles in prokaryotic physiology, such as virulence, stress response and quorum sensing. These ncRNAs have been largely overlooked in prokaryote genome annotation, since they are very difficult to detect with existing gene‐prediction software (Meyer, 2008; Livny and Waldor, 2009). Many act by binding to target 5′ UTR by base pairing, resulting in inhibition of translation or mRNA degradation. Whole‐transcriptome analysis of several prokaryotes has now identified large numbers of ncRNAs (Table 1), some of which are induced during niche switching, such as in Burkholderia cenocepacia (Yoder‐Himes et al., 2009).
Antisense RNA
Cis‐antisense RNA was previously thought to be extremely rare in prokaryotes, but whole‐transcriptome analysis has recently detected hundreds of antisense transcripts in bacteria and archaea (Table 1). Some of these have been experimentally shown to downregulate their sense counterparts (Toledo‐Arana et al., 2009). This is an area in which much is still to be discovered, as cis‐antisense may be a common form of regulation in prokaryotes.
Validation and comparing techniques
The ultimate goal is to obtain a complete and bias‐free view on microbial transcriptomes. The question remains in how far RNA‐seq has the potential to provide such a view. Clearly, RNA‐seq has a number of advantages above microarray technology, since RNA‐seq offers both a single‐base resolution and a high‐mapping resolution. RNA‐seq is especially suited to identify novel transcripts, alternative splice variants and non‐coding RNA (Marioni et al., 2008; Mortazavi et al., 2008; Nagalakshmi et al., 2008; Wilhelm et al., 2008).
However, some studies indicate that RNA‐seq is also not bias‐free (Marioni et al., 2008; Mortazavi et al., 2008). In recent studies that compared expression levels measured using both (tiling) microarrays and RNA‐seq, expression levels between the two technologies show reasonably good correlation (ranging from 0.62 to 0.75) (Marioni et al., 2008; Mortazavi et al., 2008; Fu et al., 2009), especially when comparison is restricted to protein‐coding gene loci (Sasidharan et al., 2009a,b). It should be noted that in order to compare expression levels from tiling microarray and RNA‐seq, one has to consider the different data types of the two technologies. Comparison of results may depend on the procedure applied to convert continuous expression levels from tiling microarray into a ‘digital’ signal (Sasidharan et al., 2009a,b). Correlating expression levels from both technologies to proteomics data shows that RNA‐seq provides a better estimate not only of absolute transcript levels but also of protein levels (Fu et al., 2009).
As demonstrated in a recent study on M. pneumoniae, combining various experimental data types can provide a more complete view on a transcriptome than using tiling arrays or RNA‐seq alone (Guell et al., 2009). They report that in some cases (in particular for lowly expressed genes), RNA‐seq data alone were not sufficient to unambiguously define operon boundaries. However, the single‐base resolution of RNA‐seq allows more precise prediction of promoter locations (Guell et al., 2009).
Future
Deep RNA sequencing provides clear advantages over the conventional (tiling) micro array technology. It allows transcriptome analysis of the entire nucleotide sequence of the genome, it is very sensitive, it offers a large dynamic range, and it allows accurate determination of boundaries (e.g. TSS, 3′/5′ ends, exons). However, RNA‐seq is not completely bias‐free. Nearly all studies to date have used some sort of enrichment procedure for mRNA, inherently leading to some bias. In many recent studies this enrichment step is being skipped, as the enormous volume of cDNA sequence data holds enough information, even if mRNA comprises only a few % of the total RNA. Just throw away 95–98% of your sequence data!
The conversion of RNA into complementary DNA (cDNA) may also lead to bias. Recently, a new method was developed that measures RNA levels directly without this conversion step (Ozsolak et al., 2009). The method is based on direct sequencing of RNA and is an extension of single‐molecule DNA sequencing technology (Braslavsky et al., 2003; Harris et al., 2008). The direct method uses RNA directly as a template for nucleotide incorporation by a modified DNA polymerase with reverse transcriptase activity. Under optimal conditions the method yields sequences in the range of 20–40 nucleotides in length, with a total raw base error rate of approximately 4%. These read lengths and error rates are sufficient to align sequences to reference genomes (Ozsolak et al., 2009).
What does the future hold for sequencing and RNA‐seq? There is no doubt that the revolution that has occurred in our ability to sequence and profile RNA from the days of a single ‘Southern blot’ to microarray RNA dot‐blot hybridization and Q‐PCR to RNA‐seq has been exciting, informative and rapid. In the future we will need to miniaturize as we move to single‐cell sequencing and transcriptomics. How will this be achieved? IBM is working on nanotechnology (‘The DNA transistor’; for a video see http://www.youtube.com/watch?v=wvclP3GySUY) to enable even more rapid, accurate and cheap genome sequencing (patent US200828191A1). DNA, or in fact any charged polymer, can be made to move through nanopores, and detection of the bases moving through the pore is possible. In fact the DNA moves through the pore too quickly and needs to be slowed down to be readable. So in the not too distant future, we may see that the genome sequence, transcriptome and regulome of a single cell will all be determined before the first coffee break of the day.
Acknowledgments
This project was carried out within the research programmes of the Kluyver Centre for Genomics of Industrial Fermentation and the Netherlands Bioinformatics Centre, which are part of the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research. TT is funded by the HAN University of Applied Sciences.
References
- Albrecht M., Sharma C.M., Reinhardt R., Vogel J., Rudel T. Deep sequencing‐based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge W.J. Next‐generation DNA sequencing techniques. Nature Biotechnology. 2009;25:195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Bayjanov J.R., Wels M., Starrenburg M., Van Hylckama Vlieg J.E., Siezen R.J., Molenaar D. PanCGH: a genotype‐calling algorithm for pangenome CGH data. Bioinformatics. 2009;25:309–314. doi: 10.1093/bioinformatics/btn632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braslavsky I., Hebert B., Kartalov E., Quake S.R. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci USA. 2003;100:3960–3964. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis M., Soppa J. Regulation of translation in haloarchaea: 5′‐ and 3′‐UTRs are essential and have to functionally interact in vivo. Plos One. 2009;4:e4484. doi: 10.1371/journal.pone.0004484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher N.J., Fookes M.C., Perkins T.T., Turner D.J., Marguerat S.B., Keane T. A simple method for directional transcriptome sequencing using Illumina technology. Nucleic Acids Res. 2009;37:e148. doi: 10.1093/nar/gkp811. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X., Fu N., Guo S., Yan Z., Xu Y., Hu H. Estimating accuracy of RNA‐Seq and microarrays with proteomics. BMC Genomics. 2009;10:161. doi: 10.1186/1471-2164-10-161. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guell M., Van Noort V., Yus E., Chen W.H., Leigh‐Bell J., Michalodimitrakis K. Transcriptome complexity in a genome‐reduced bacterium. Science. 2009;326:1268–1271. doi: 10.1126/science.1176951. et al. [DOI] [PubMed] [Google Scholar]
- Harris T.D., Buzby P.R., Babcock H., Beer E., Bowers J., Braslavsky I. Single‐molecule DNA sequencing of a viral genome. Science. 2008;320:106–109. doi: 10.1126/science.1150427. et al. [DOI] [PubMed] [Google Scholar]
- Hinton J.C., Hautefort I., Eriksson S., Thompson A., Rhen M. Benefits and pitfalls of using microarrays to monitor bacterial gene expression during infection. Curr Opin Microbiol. 2004;7:277–282. doi: 10.1016/j.mib.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Koide T., Reiss D.J., Bare J.C., Pang W.L., Facciotti M.T., Schmid A.K. Prevalence of transcription promoters within archaeal operons and coding sequences. Mol Syst Biol. 2009;5:285. doi: 10.1038/msb.2009.42. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.M., Livny J., Lawrence M.S., Kimball M.D., Waldor M.K., Camilli A. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res. 2009;37:e46. doi: 10.1093/nar/gkp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livny J., Waldor M.K. Mining regulatory 5′UTRs from cDNA deep sequencing datasets. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y. RNA‐seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath P.T., Lee H., Zhang L., Iniesta A.A., Hottes A.K., Tan M.H. High‐throughput identification of transcription start sites, conserved promoter motifs and predicted regulons. Nat Biotechnol. 2007;25:584–592. doi: 10.1038/nbt1294. et al. [DOI] [PubMed] [Google Scholar]
- Metzker M.L. Sequencing technologies – the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Meyer I.M. Predicting novel RNA‐RNA interactions. Curr Opin Struct Biol. 2008;18:387–393. doi: 10.1016/j.sbi.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Morozova O., Hirst M., Marra M.A. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet. 2009;10:135–151. doi: 10.1146/annurev-genom-082908-145957. [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver H.F., Orsi R.H., Ponnala L., Keich U., Wang W., Sun Q. Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B‐dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics. 2009;10:641. doi: 10.1186/1471-2164-10-641. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F., Platt A.R., Jones D.R., Reifenberger J.G., Sass L.E., McInerney P. Direct RNA sequencing. Nature. 2009;461:814–818. doi: 10.1038/nature08390. et al. [DOI] [PubMed] [Google Scholar]
- Passalacqua K.D., Varadarajan A., Ondov B.D., Okou D.T., Zwick M.E., Bergman N.H. Structure and complexity of a bacterial transcriptome. J Bacteriol. 2009;191:3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins T.T., Kingsley R.A., Fookes M.C., Gardner P.P., James K.D., Yu L. A strand‐specific RNA‐Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. Plos Genet. 2009;5:e1000569. doi: 10.1371/journal.pgen.1000569. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S., Nielsen H.B., Jarmer H. The transcriptionally active regions in the genome of Bacillus subtilis. Mol Microbiol. 2009;73:1043–1057. doi: 10.1111/j.1365-2958.2009.06830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan R., Agarwal A., Rozowsky J., Gerstein M. An approach to compare genome tiling microarray and MPSS sequencing data for transcript mapping. BMC Res Notes. 2009a;2:211. doi: 10.1186/1756-0500-2-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan R., Agarwal A., Rozowsky J., Gerstein M. An approach to comparing tiling array and high throughput sequencing technologies for genomic transcript mapping. BMC Res Notes. 2009b;2:150. doi: 10.1186/1756-0500-2-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinger D.W., Cheung K.J., Mei R., Johansson E.M., Richmond C.S., Blattner F.R. RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nat Biotechnol. 2000;18:1262–1268. doi: 10.1038/82367. et al. [DOI] [PubMed] [Google Scholar]
- Sorek R., Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet. 2010;11:9–16. doi: 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- Toledo‐Arana A., Dussurget O., Nikitas G., Sesto N., Guet‐Revillet H., Balestrino D. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. et al. [DOI] [PubMed] [Google Scholar]
- Van Vliet A.H. Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol Lett. 2010;302:1–7. doi: 10.1111/j.1574-6968.2009.01767.x. [DOI] [PubMed] [Google Scholar]
- Van Vliet A.H., Wren B.W. New levels of sophistication in the transcriptional landscape of bacteria. Genome Biol. 2009;10:233. doi: 10.1186/gb-2009-10-8-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters L.S., Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm B.T., Landry J.R. RNA‐Seq‐quantitative measurement of expression through massively parallel RNA‐sequencing. Methods. 2009;48:249–257. doi: 10.1016/j.ymeth.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Wilhelm B.T., Marguerat S., Watt S., Schubert F., Wood V., Goodhead I. Dynamic repertoire of a eukaryotic transcriptome surveyed at single‐nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. et al. [DOI] [PubMed] [Google Scholar]
- Wurtzel O., Sapra R., Chen F., Zhu Y., Simmons B.A., Sorek R. A single‐base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder‐Himes D.R., Chain P.S., Zhu Y., Wurtzel O., Rubin E.M., Tiedje J.M., Sorek R. Mapping the Burkholderia cenocepacia niche response via high‐throughput sequencing. Proc Natl Acad Sci USA. 2009;106:3976–3981. doi: 10.1073/pnas.0813403106. [DOI] [PMC free article] [PubMed] [Google Scholar]