

Abstract
Acute pulmonary infection by Streptococcus pneumoniae is characterized by high bacterial numbers in the lung, a robust alveolar influx of polymorphonuclear cells (PMNs) and a risk of systemic spread of the bacterium. We investigated host-mediators of S. pneumoniae-induced PMN migration and the role of inflammation in septicemia following pneumococcal lung infection. Hepoxilin A3 (HXA3) is a PMN chemoattractant and a metabolite of the 12-lipoxygenase (12-LOX) pathway. We observed that S. pneumoniae infection induced the production of 12-lipoxygenase in cultured pulmonary epithelium and in the lungs of infected mice. Inhibition of the 12- LOX pathway prevented pathogen-induced PMN transepithelial migration in vitro and dramatically reduced lung inflammation upon high-dose pulmonary challenge with S. pneumoniae in vivo, thus implicating HXA3 in pneumococcus-induced pulmonary inflammation. PMN basolateral-to-apical transmigration in vitro significantly increased apical-to-basolateral transepithelial migration of bacteria. Mice suppressed in the expression of 12-lipoxygenase exhibited little or no bacteremia and survived an otherwise lethal pulmonary challenge. Our data suggest that pneumococcal pulmonary inflammation is required for high level bacteremia and systemic infection, partly by disrupting lung epithelium through 12-LOX-dependent HXA3 production and subsequent PMN transepithelial migration.
INTRODUCTION
Streptococcus pneumoniae (pneumococcus), the most common cause of community-acquired bacterial pneumonia, is a Gram-positive pathogen that asymptomatically colonizes the nasopharynx of up to 50% of adults but is also capable of causing lethal disease. Local infection can result in sinusitis and otitis media (1, 2), while migration of pneumococci to lower respiratory tract may result in robust bacterial growth in the lung. Indeed, pneumonia is one of the most important manifestations of invasive pneumococcal disease. Entry of S. pneumoniae from alveolar spaces into bloodstream can result in life-threatening septicemia, as a pneumococcal antiphagocytic polysaccharide capsule diminishes bloodstream clearance and fosters high-level bacterial growth (3). Invasive pneumococcal infections result in approximately one million deaths annually worldwide (4).
The massive influx of polymorphonuclear leukocytes (PMNs, aka neutrophils) into pulmonary alveolar spaces is a hallmark of pneumococcal pneumonia (5, 6). PMNs are integral to innate immunity, but poorly controlled inflammation can result in tissue destruction, obstruction of gaseous exchange and ultimately lung failure (7, 8). Mobilization of PMNs from the circulation to the mucosal surface during pathogenic insult is a complex multistep process, involving PMN interactions with multiple tissue components such as endothelium, interstitium, and epithelium (8, 9, 10). This process requires a fine-tuned coordination of cytokine networks, multiple receptor-ligand interactions, as well as the temporal and localized secretion of various PMN chemokines and chemoattractants, including IL-8 (CXCL8), CXCL5, and PGP, the CXCR ligand derived from extracellular matrix degradation (8, 11-14, 15, 16). Despite significant progress in this area of study, our understanding of the molecular mechanisms governing each step of this highly complex process remains incomplete. For example, although IL-8 is a well-documented neutrophil chemoattractant known to be induced in cultured respiratory epithelial cells and to play an important role in pulmonary inflammation during lung infection (17), it is unclear if IL-8 production by lung epithelial cells is sufficient for mediating transepithelial PMN migration (18, 19). In fact, several model systems indicate that IL-8 is basolaterally secreted at mucosal surfaces, and thus may play an important role in recruiting PMNs from the vasculature to interstitial spaces where a second distinct signal would be required to guide PMNs across the epithelium to the airspace (18). Furthermore, IL-8, as well as a number of other potentially important molecules, such as selectins, VLA-4, PECAM-1, CD11/CD18, and ICAM-1, are dispensable for PMN migration in the lung, implying that PMN recruitment as a consequence of S. pneumoniae infections is subject to novel regulatory processes (20-23).
Eicosanoids, including cyclooxygenase and lipoxygenase-catalyzed products of arachidonic acid, are lipids affecting inflammation of the lung and other tissues (24). One such compound is hepoxilin A3 (HXA3), a hydroxy epoxide derived from arachidonic acid via the 12-lipoxygenase (12-LOX) pathway (10, 25, 26). HXA3 has been shown to play a role in a number of biological processes, ranging from glucose-dependent insulin secretion to vascular permeability (25-27). Recent studies have also shown that this molecule is capable of activating PMNs and promoting PMN migration across intestinal epithelia in response to Salmonella enterica serovar Typhimurium or Shigella flexneri (25, 28), as well as across pulmonary epithelium in response to Pseudomonas aeruginosa (18, 29). The polarized secretion of HXA3 from the apical side of mucosal epithelia creates a chemoattractant gradient that ultimately directs the PMNs to cross the epithelial barrier (25). In this role, it is inferred that HXA3 serves a critical function as the “gate keeper” of the mucosal epithelium, since it emanates from the site of infection to establish a chemotactic gradient that guides PMNs across mucosal surfaces, the final step in PMN recruitment to the mucosal lumen.
In progressing from pneumonia to life-threatening septicemia, pneumococci must translocate from alveolar space into bloodstream and in doing so, must breach the epithelial barrier. The purpose of the current study was to identify the critical chemoattractant(s) promoting PMN migration across the epithelium during acute pulmonary infection and to test whether this inflammatory process contributes to the retrograde migration of bacteria and systemic disease.
MATERIALS AND METHODS
Bacteria
S. pneumoniae strains TIGR4 (serotype 4), D39 (serotype 2), G54 (serotype 19F) and R6 (the capsular mutant of D39) were grown in Todd-Hewitt broth (BD Biosciences) supplemented with 0.5% yeast extract and used at late log phase. The capsular mutant (ΔcpsA-L::spec) of TIGR4 was a gift from Andrew Camilli, Tufts University, Boston, MA, and was grown and used in a manner similar to the wild type TIGR4 strain. For mice experiments, bacteria were stored at −80°C in media with 25% (v/v) glycerol, and thawed, and diluted in PBS to appropriate concentrations, as required. Bacterial number in stocks was confirmed by plating serial dilutions on blood agar. B. subtilis strain 168 was grown overnight at 37°C in Luria broth. When required, S. pneumoniae strains were heat killed at 65 °C for 1h (30).
Growth and maintenance of epithelial cells
Human pulmonary mucoepidermoid carcinoma-derived NCI-H292 (H292) cells were grown on the underside of collagen-coated Transwell filters (0.33-cm2, Corning Life Sciences) in RPMI 1640 medium (ATCC) with 2 mM L-glutamine, 10% FBS, and 100 U penicillin/streptomycin. As the transepithelial resistance of lung epithelial monolayers are typically very low (31), integrity of monolayers was assessed by horseradish peroxidase assay, as described (18).
Animals
C57BL/6J mice and Alox12/15 knockout (Alox15−/−) mice (B6.129S2-ALOX15tm1Fun/J) were obtained from Jackson Laboratories. All animal experiments were performed in accordance with Tufts University Animal Care and Use Committee approved protocols.
PMN transmigration assay
PMNs were isolated from whole blood obtained from healthy human volunteers using previously described protocols involving HBSS without calcium and magnesium (18, 32). PMN transmigration assay was performed following a previously described protocol, using HBSS (containing calcium and magnesium, termed “HBSS+Ca/Mg”) (32), but with bacterial infection and PMN migration times of 2½h each. For PMN migration in response to lipid extracts (generated as stated below), extracts were re-constituted in 600μl of HBSS+Ca/Mg and added to the apical side of uninfected monolayers. For all assays, formylated Met-Leu-Phe (“fMLP”) and HBSS+Ca/Mg were used as positive and negative controls, respectively.
Pneumococcal translocation across epithelial monolayer
Apical sides of H292 monolayers were infected with S. pneumoniae (1×107 cfu in 1ml HBSS+Ca/Mg), while 200 μl HBSS+Ca/Mg was added to the basolateral side. After 2½h at 37°C, 1×106 PMNs (in 100μl HBSS+Ca/Mg) were added to the basolateral side. In separate control experiments, PMNs were added to the apical rather than the basolateral side of the monolayers. Aliquots (50μl) were removed from the basolateral chambers of wells after PMNs were added and serial dilutions were plated on blood agar. Previous experiments have shown that S. pneumoniae cannot grow significantly in HBSS+Ca/Mg over the time periods tested here.
RT-PCR and Western blotting
Total RNA was isolated from infected or uninfected H292 cells, and semi-quantitative RT-PCR for ALOX12 and ALOX15 was performed using specific primers (29). GAPDH was used as internal control. Whole cell lysates were prepared from infected monolayers using the protocol of Hurley et al., 2004 (18), and probed with either anti-12-LOX or anti-15-LOX Ab (Santa Cruz). Band intensities were quantified after normalization to GAPDH.
Lipid extraction
Monolayers were infected with TIGR4 for 2 and 3 hours, washed, and incubated in HBSS+Ca/Mg for 1-3 hrs at 37°C, 5% CO2. The conditioned buffer was collected and stored at −80°C. Whenever required, monolayers were pretreated with 50 μM CDC or 1μM baicalein for 3h and 24h, respectively. Samples were size-fractionated by hydrophobic surface chromatography and eluted with methanol. Eluted fractions were vacuum dried and their activity was analyzed by PMN migration assay through uninfected H292 monolayers, as stated above.
Treatment of monolayers with inhibitors
Cinnamyl-3,4-dihydroxy- α-cyanocinnamate (CDC), baicalein (bcn), and caffeic acid (CA) were obtained from Enzo Life Sciences, Plymouth Meeting, PA. Cells were pretreated with caffeic acid for 1h; with CDC for 3h; with cycloheximide (Sigma) for 18h; and with baicalein for 24h. Drug concentrations were maintained during infection. To minimize potential direct effects of the inhibitors, drugs were removed by washing before adding PMNs.
Binding assay
Bacteria were added to the apical side of H292 monolayers in 1ml HBSS+Ca/Mg, while the basolateral chamber received 100μl HBSS+Ca/Mg. After incubation at 37°C, 5% CO2 for 40 min, monolayers were washed and incubated in trypsin-EDTA for 1h. Serial dilutions were plated on blood agar to quantitate bound bacteria. Alternatively, after washing, the monolayers were fixed in paraformaldehyde (4% w/v in PBS) for 1h, and stained with 0.007% crystal violet overnight. Filters were cut out and number of bound bacteria was counted in five random fields of vision.
Determination of barrier integrity
Compromise of barrier integrity due to bacteria-elicited PMN migration was measured as described (44). The apical side of H292 monolayers was infected with S. pneumoniae and 1×106 PMNs were added to the basolateral side. Twenty μg/ml FITC-dextran (MW 40 kDa) was added to the apical side and FITC-dextran migration to the basolateral side was measured by quantitating fluorescence of basolateral media at the indicated times.
Murine infection studies
Mice were intratracheally challenged with TIGR4 (2× 105 cfu in 50 μl PBS). Control mice received PBS. Effect of 12/15-lipoxygenase on S. pneumoniae-induced inflammation was studied in C57BL/6 (wild type) mice treated with CDC, or Alox15−/− mice; and compared to inflammation in infected wild type, untreated mice. CDC (8 mg/kg, in DMSO) was injected intraperitoneally at 24h, 18h, 3h, and 2h preinfection and then 2h, 3h, 18h, and 24h postinfection.
For histological studies, mice were euthanized at 48h postinfection, then whole lungs were isolated, fixed in 10% formalin (Sigma-Aldrich), and paraffin embedded. Lung blocks were sectioned at 5μm and adhered to silanized slides. Three mice per group were analyzed. Three sections from each mouse were stained with hematoxylin/eosin and analyzed using a Nikon Eclipse E800 microscope, fitted with a Canon EOS 30D camera. For analysis of bronchoalveolar lavage fluid (BALF), mice were euthanized at 48h postinfection; BALF was collected by washing the lungs twice with 1ml PBS via a cannula. BALF was centrifuged in a cytospin (150×g, 10 min, 4°C), cells were stained with Hemacolor staining kit (EMD Biochemicals Ltd, Gibbstown, NJ) and counted for differential analysis. For analysis of 12-LOX and 15-LOX levels in the lungs, equal amounts of lung homogenates were run on SDS-PAGE and immunoblotted with either anti-12-LOX or anti-15-LOX Ab (Santa Cruz). Immunoblotting with anti-tubulin provided a loading control. KC and MIP-2 levels in blood were determined using commercial ELISA kits (Sigma) following the manufacturer’s instructions. To enumerate the bacterial load in the lung at 48h, whole lungs were isolated, homogenized in PBS and serial dilutions were plated on blood agar. Serial dilutions of BALF were plated on blood agar to enumerate bacteria in the airways.
Mice were also monitored for sickness and survival over 7 days. For survival assays, six animals per group were analyzed. Following infection, mice were monitored for sickness each day and moribund animals were euthanized as per a protocol approved by the Tufts University Animal Care and Use Committee. Progression of bacteremia was monitored by plating dilutions of tail blood on blood agar plates every 24h post-infection.
Flow Cytometry
For flow cytometry studies, anti-Ly-6G-PE (clone 1A8), anti-CD11c-FITC (clone N418), anti-F4-80-PE-Cy7 (clone BM8) and anti-TCRβ-APC (clone H57-597) were obtained from BD Biosciences. Mice were euthanized at 48h post-infection and BALF was collected as stated above. Lung tissues were digested with 1 mg/ml Type II collagenase (Worthington) and 50 U/ml DNase (Worthington) to obtain a single-cell suspension as previously described (52). Cells present in the digested lungs and BALF were stained on ice for 30 min with relevant MAbs and then washed. Following this, the cells were run through a FACSCalibur flow cytometer (BD Biosciences) and the fluorescence intensities of the stained cells were determined. Collected data were analyzed using FlowJo software (Tree Star, Inc.) to determine the numbers of PMNs (Ly6G+), T cells (TCRβ+), macrophages (F4/80+) and DC (F4/80−, CD11c+) in both the lung digests and BALF.
Statistical analyses
Statistical analysis was carried out using GraphPad Prism program (GraphPad Software, San Diego, CA). Log-rank (Mantel-Cox) test was performed for analysis of survival curves. For comparison between two groups, Mann-Whitney test was performed. P values <0.05 were considered significant in all cases.
RESULTS
Diverse serotypes of S. pneumoniae elicit PMN migration in vitro in a manner promoted by the polysaccharide capsule
To investigate how S. pneumoniae induces an acute inflammatory response in the lung, we established an in vitro model of PMN migration using polarized monolayers of human mucoepidermoid pulmonary carcinoma cell line NCI-H292 (“H292”). This cell line forms polarized barriers when grown on permeable filters and has been used extensively for PMN transepithelial migration studies (33, 34). Using this model, we examined three pneumococcal isolates, capsular serotype 4 strain TIGR4, serotype 2 strain D39 and serotype 19F strain G54 (35, 36, 37). Whereas infection with the soil bacterium B. subtilis, which bound efficiently to the monolayers (data not shown), resulted in barely detectable PMN migration, each pneumococcal strain elicited strong PMN transmigration response (Fig. 1A; p< 0.0001 compared to B. subtilis). Although D39 bound to epithelial cells somewhat more efficiently than either of the other pneumococcal strains (data not shown), TIGR4 generated the strongest PMN migration response, with a dose of 1×107 cfu/monolayer (i.e., a multiplicity of infection of approximately 10) resulting in a maximal response. The PMN migration response to TIGR4 infection occurred in a time-dependent manner, reaching a maximum at 2.5h (Fig. 1B).
Fig. 1.
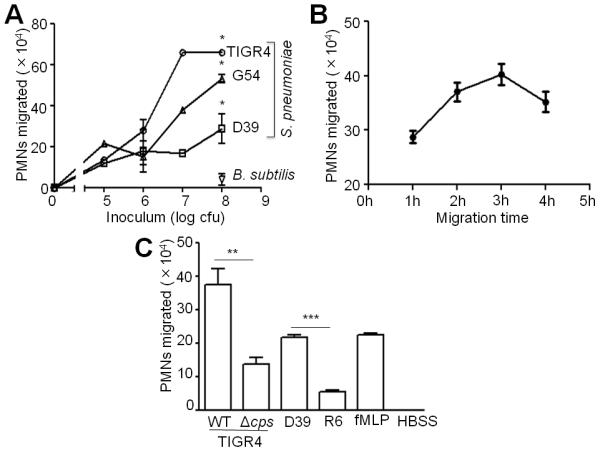
Diverse serotypes of S. pneumoniae elicit robust PMN migration in vitro in a dose, time and capsule-dependent manner. A, H292 monolayers were infected with the indicated doses of S. pneumoniae or B. subtilis.1×106 PMNs were added to the basolateral side and PMN migration to the apical side was quantified by MPO assay. Shown is a representative of two experiments. B, Apical sides of H292 monolayers were infected with 1×107 S. pneumoniae TIGR4. 1×106 PMNs were added to the basolateral side, and allowed to migrate for the indicated time periods. PMN migration to the apical side was quantified by MPO assay. Shown is a representative of two experiments. C, H292 monolayers were infected with 1×107 S. pneumoniae TIGR4, its capsular mutant (Δcps), S. pneumoniae D39, or its capsular mutant R6. Basolateral-to-apical PMN migration was quantified by MPO assay. Positive and negative control wells received formylated-Met-Leu-Phe (“fMLP”) and HBSS+Ca/Mg, respectively. Shown is a representative of two experiments.
Given that one difference in the S. pneumoniae strains tested here is their capsular polysaccharide serotype, we examined whether the capsule promotes PMN migration. The capsule-deficient TIGR4 and D39 strains bound to the polarized monolayers indistinguishably when compared to their respective wild type strains (data not shown), but induced levels of PMN migration two- (p<0.01) and 3.5-fold (p<0.0001) lower than their wild type parental strains, respectively (Fig. 1C), suggesting that the pneumococcal capsule directly or indirectly promotes PMN transmigration in vitro. The observation that the capsular mutants did not completely abrogate PMN transepithelial migration suggests that multiple bacterial factors contribute to the PMN transmigration response.
Bacterial viability appeared to be essential for PMN migration because heat-killed bacteria induced 280-fold less PMN migration (p<0.0001) (Figure 2A), in spite of binding to H292 cells as efficiently as the live bacteria (data not shown). Pretreatment of H292 monolayers with cycloheximide resulted in a 26-fold decrease (p<0.0001) in PMN migration, suggesting that de novo host protein synthesis was required (Fig. 2B). The dual requirements of bacterial viability and host cell protein synthesis suggest that this inflammatory response involves an active dialogue between microbe and host.
Fig. 2.
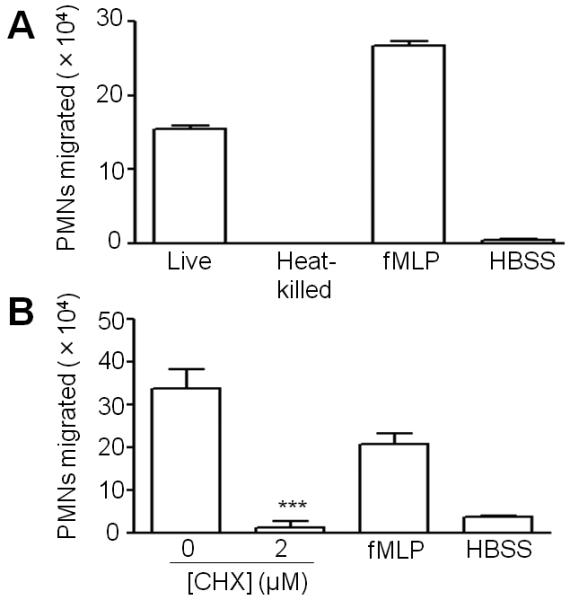
Live bacteria and de novo protein synthesis by the host cells are required for S. pneumoniae-induced PMN migration. A, Apical sides of H292 monolayers were infected with either live or heat-killed S. pneumoniae TIGR4. PMNs were added basolaterally and PMN migration to the apical side was quantified by MPO assay. fMLP and HBSS+Ca/Mg were used as positive and negative controls, respectively. Binding of live and heat-killed bacteria to H292 monolayers, when assessed by viable counts or microscopically, were statistically indistinguishable. Shown is a representative of two experiments. B, Protein synthesis in H292 monolayers was blocked by treatment with cycloheximide before infection with S. pneumoniae TIGR4 (1×107/monolayer), and PMN migration was assessed as described in Materials and Methods. Shown is a representative of two experiments.
12-lipoxygenases are induced upon pneumococcal infection of pulmonary epithelial monolayers and required for PMN transmigration
Since the eicosanoid HXA3 promotes PMN migration across polarized monolayers of human alveolar adenocarcinoma cells in response to Pseudomonas aeruginosa (18, 29), we sought to determine the significance of lipid-mediated signaling in the PMN transepithelial migration in response to S. pneumoniae infection. Purified HXA3 promoted PMN migration across H292 monolayers (data not shown). In addition, lipids from apical supernatants of S. pneumoniae-infected H292 cells exhibited significantly (p<0.0001) more chemotactic activity than lipids from uninfected cells (Fig. 3A).
Fig. 3.
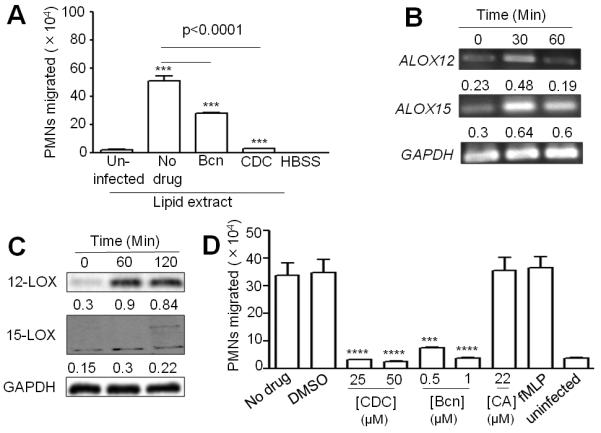
12-lipoxygenase is induced upon pneumococcal infection of pulmonary epithelial monolayers and is required for PMN transmigration. A, Mock-treated H292 monolayers, or monolayers pretreated with 1μM baicalein or 50 μM CDC, were infected with TIGR4. After infection, lipid extracts were prepared from the conditioned buffer, as stated in Materials and Methods. PMN migration in response to extracted lipids was measured as stated before. Shown is a representative of two experiments; B, Semi-quantitative RT-PCR analyses of ALOX12 and ALOX15 levels were performed using total RNA from infected or uninfected H292 cells, using - specific primers. GAPDH was used as the internal control. Shown is a representative gel picture from three independent experiments. Band intensities normalized to the band intensities of GAPDH, which was taken as 1, are presented below each band. Data represent means from three independent experiments. C, Lysates of H292 monolayers infected with TIGR4 were analyzed by immunoblotting using anti- ALOX12, ALOX15 or GAPDH antibodies. Band intensities were normalized to the band intensities of GAPDH and their values are given below each blot. Shown are the means from two independent experiments. D, H292 monolayers were pretreated with the indicated doses (μM) of CDC, baicalein (“Bcn”), or caffeic acid (“CA”) prior to infection with TIGR4. PMNs were added to the basolateral side and migration was quantified by MPO assay. fMLP, HBSS+Ca/Mg or DMSO were used as positive, negative and solvent controls, respectively. Shown is a representative of two experiments.
12-lipoxygenase is central to HXA3 synthesis (38), and of the three functional human isoforms of 12-lipoxygenase (i.e. 15-LOX-1, 12R-LOX and the platelet-type p12-LOX), 15-LOX-1 has been shown to contribute to HXA3-mediated PMN migration across the intestinal epithelium (28). 15-LOX-1, which also displays 15-lipoxygenase activity, is highly expressed in airway epithelium and may contribute significantly to HXA3 generation during pulmonary inflammation (39, 40). Transcriptional analysis showed that ALOX15, which encodes 15-LOX-1, as well as ALOX12, which encodes p12 LOX, were elevated within 30 minutes of infection of H292 monolayers with S. pneumoniae (Fig. 3B). Western blot analysis of lysates from infected cells at 1 and 2h post-infection showed increased levels of the corresponding proteins (Fig. 3C).
Consistent with a role for HXA3 as an essential chemoattractant in this process, pretreatment of H292 monolayers with either of the 12-lipoxygenase inhibitors CDC or baicalein (18, 25), which did not alter bacterial growth or bacterial binding to the monolayers (data not shown), significantly reduced S. pneumoniae-induced PMN migration (Fig. 3D). In contrast, caffeic acid, a 5-lipoxygenase inhibitor that prevents the synthesis of another bioactive lipid, LTB4 (18, 25), or the solvent control (DMSO), had no inhibitory effect on PMN transepithelial migration (Fig. 3D). Neither CDC nor baicalein adversely influenced PMN migration in response to formylated Met-Leu-Phe (fMLP; data not shown). As predicted, both CDC and baicalein diminished the chemoattractant activity of lipid extracted from supernatants of infected H292 cells (Fig. 3A). These data suggest that infection of H292 cells by S. pneumoniae leads to induction of 12-lipoxygenases, possibly leading to the production of HXA3, and blocking 12-lipoxygenase activity by using pharmacological inhibitors abrogates PMN migration across H292 monolayers.
12-lipoxygenase activity promotes migration of PMNs into the airways in the absence of infection
To test whether 12-lipoxygenase plays a role in the recruitment of PMNs in the airways under basal, i.e. uninfected, conditions, we measured airway PMNs in C57BL/6 mice that were pharmacologically inhibited for 12-lipoxygenase activity by CDC treatment, or in Alox15−/− mice, which are deficient in leukocyte-type 12/15-LOX. Whereas human 15-LOX-1 displays greater 15-lipoxygenase than 12-lipoxygenase activity, its murine homolog, leukocyte-type 12/15-LOX, primarily acts as a 12-lipoxygenase (39, 40). Notably, the numbers of PMNs in bronchoalveolar lavage fluid (BALF) of CDC-treated C57BL/6 mice or Alox15−/− mice were six- or three-fold lower than the PMNs in the BALF of untreated C57BL/6 mice, respectively (Fig 4). These differences were not due to differences in total pulmonary PMNs, which were indistinguishable among the three experimental groups, indicating that in the absence of exogenous insult, 12-lipoxygenase plays a specific role in recruitment of PMNs into the alveoli.
Fig. 4.
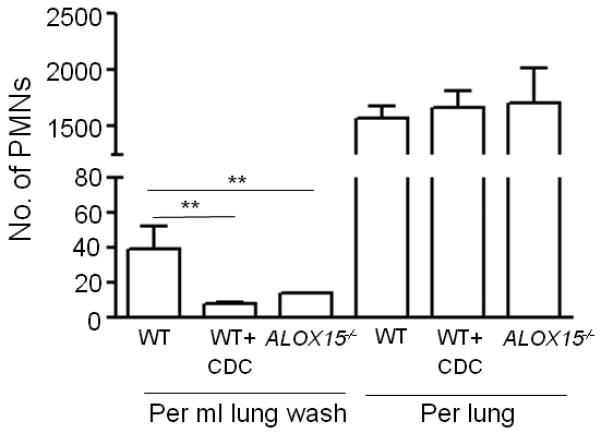
12-lipoxygenase activity is required for alveolar neutrophil recruitment even during absence of S. pneumoniae infection. Wild type C57BL/6 mice, with or without CDC treatment, or Alox15−/− mice were sacrificed and PMNs present in BALF or lungs were analyzed by flow cytometry as described in Materials and Methods. Shown is a representative of two independent experiments.
12-lipoxygenase activity is required for pulmonary inflammation during acute S. pneumoniae infection
Infection of cultured H292 pulmonary epithelial cells with S. pneumoniae resulted in an induction of 12- and 15-LOX proteins (Fig. 3C), so we tested whether the levels of these proteins increase in a well-established murine acute infection model (22). In fact, western blot analysis of lung homogenates revealed that both 12- and 15-LOX were induced upon infection of C57BL/6 mice with S. pneumoniae TIGR4 (Fig. 5). 12- and 15-LOX immunoblotting revealed multiple bands, which may represent the products of splice variants.
Fig. 5.
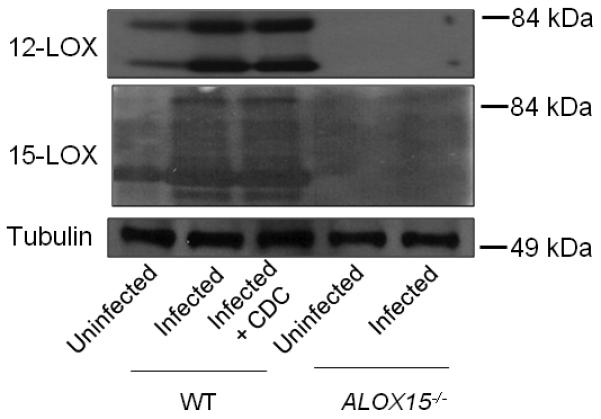
12-lipoxygenase is induced in vivo upon pneumococcal infection. Wild type C57BL/6 mice (with or without CDC pretreatment) or Alox15−/− mice were intratracheally challenged with 2×105 S. pneumoniae TIGR4. Control mice received only PBS. Mice were euthanized, lung homogenates were immunoblotted and probed using anti-ALOX12, ALOX15 or tubulin antibodies. Shown is a representative figure from two independent experiments. Corresponding molecular weight markers are denoted.
Upon finding that 12-LOX is induced during pneumococcal pneumonia in mice, we tested whether 12-lipoxygenase activity is essential for eliciting PMN transepithelial migration during acute infection. We infected untreated wild type (WT) C57BL/6 mice, C57BL/6 mice treated with the 12-lipoxygenase inhibitor CDC, or Alox15−/− mice, with S. pneumoniae TIGR4. Hematoxylin and eosin (H&E) staining of the lung 48h post-infection showed that S. pneumoniae infection of untreated WT mice elicited a robust acute inflammatory response, with congested vasculature and alveolar infiltration by PMNs and erythrocytes (compare Fig. 6A(i) and (ii)). Consistent with this histological assessment, flow cytometry of pulmonary single cell suspensions revealed six-fold greater numbers of PMNs in lung tissue of infected WT mice compared to the uninfected mice (Fig. 6B, top). Infection was also associated with a two-fold greater number of pulmonary macrophages, but no increase in dendritic or T cells (Fig. 6B, top). The broncheoalveolar lavage fluid contained dramatically higher numbers of inflammatory cells, including dendritic and T cells as well as PMNs and macrophages, indicating that a broad population of inflammatory cells migrate into airway spaces during acute pneumococcal infection (Fig. 6B, bottom).
Fig. 6.
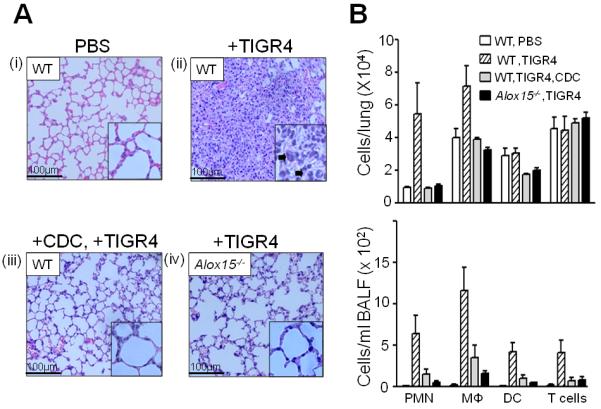
12-lipoxygenase activity is required for pulmonary inflammation during S. pneumoniae infection of mice. Untreated [“+TIGR4”; panel A(ii)] or CDC-treated [“+CDC, +TIGR4”; panel A(iii)] wild type (“WT”) C57BL/6 mice, or Alox15−/− [panel A(iv)] mice were inoculated intratracheally with TIGR4, or with PBS alone [panel A(i)], as described in Materials and Methods. Mice were sacrificed, and hematoxylin and eosin stained lung sections (at 20× magnification) were examined by light microscopy. Inset: lung sections at 40× magnification; arrowheads denote PMNs. Figures are representative of two independent experiments. Bronchoalveolar lavage fluid (BALF) was isolated and cytospin preparations were prepared. PMNs were counted from Wright-Giemsa stained preparations by observation at 100×. Average number of PMNs per 5 fields of observation is noted in the corresponding histology figures. For flow cytometry, BALF and lungs from infected WT (with or without CDC treatment) or Alox15−/− mice were collected. Cells present in the digested lungs and BALF were stained with relevant MAbs and the fluorescence intensities of the stained cells were determined by flow cytometry. Collected data were analyzed to determine the numbers of PMNs, macrophages (Mɸ), dendritic cells or T cells in the lungs (B, top) and in the BALF (B, bottom).
Infection-associated inflammation was dependent on 12-lipoxygenase activity: pharmacologic inhibition of 12-lipoxygenase activity by treatment with CDC or genetic ablation of leukocyte-type 12/15-LOX dramatically reduced pulmonary inflammation, as assessed by histology (compare Fig. 6A(i) vs. (iii) or (iv)). Similarly, analysis of pulmonary single cell suspensions by flow cytometry showed that the PMN and macrophage responses in lung tissue of CDC-treated WT mice or Alox15−/− mice were entirely blunted (Fig. 6B, top). Similarly, analysis of inflammatory cells in BALF demonstrated that airway infiltration by inflammatory cells was almost entirely dependent on 12-lipoxygenase activity (Fig. 6B, bottom). Consistent with these results, although serum levels of the two inflammatory cytokines, KC and MIP-2, increased five-to six-fold during pneumococcal infection of each of the three groups of mice tested, their levels were not affected by 12-lipoxygenase activity (data not shown). Interestingly, 12-lipoxygenase-mediated inflammation did not promote host defense because the numbers of bacteria in either lung homogenate or BALF at 48h post-infection were indistinguishable in wild type, CDC-treated and Alox15−/− mice (data not shown). Thus, during acute S. pneumoniae lung infection, 12-lipoxygenase (and by implication HXA3) is required for a robust innate inflammatory response that does not appear to promote microbial clearance, perhaps in part due to the pneumococcal production of an antiphagocytic capsule (41).
S. pneumoniae-elicited PMN migration compromises epithelial barrier integrity and facilitates pneumococcal transepithelial migration
Pneumococcal entry into the circulation is a critical event that may foreshadow the development of infection to a life-threatening phase. Inflammatory signaling may promote pneumococcal penetration of epithelial monolayers (42, 43, 45). Our finding that 12-lipoxygenase is essential for PMN transepithelial migration and pulmonary inflammation during infection provided a means to investigate the role of PMN migration in disruption of epithelial barrier integrity and transmigration of S. pneumoniae. To assess the barrier function of H292 monolayers, we measured the apical-to-basolateral flux of the paracellular marker FITC-labeled dextran (44) following treatment with S. pneumoniae and/or PMNs. While PMNs alone did not compromise the epithelial barrier, apical infection with S. pneumoniae resulted in a significant increase in FITC-dextran flux (Fig. 7A), consistent with previous studies (45). Notably however, addition of PMNs to the basolateral surface of infected H292 monolayers resulted in an approximately two-fold increase in FITC-dextran movement across monolayers (Fig. 7A), suggesting that S. pneumoniae-induced PMN migration significantly alters the barrier function of H292 monolayers.
Fig. 7.
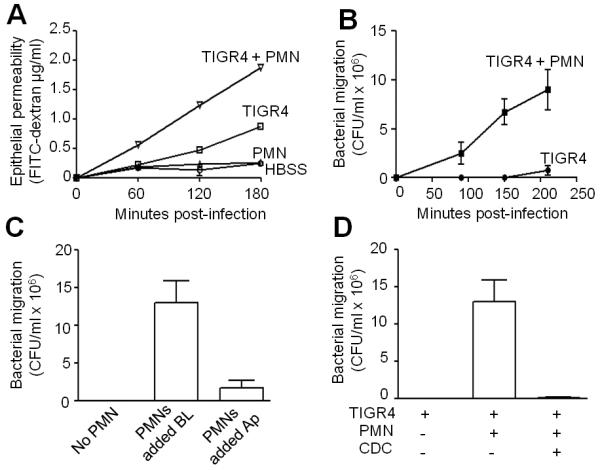
S. pneumoniae-elicited PMN migration compromises epithelial barrier integrity and facilitates pneumococcal transepithelial migration. A, Compromise in barrier integrity due to S. pneumoniae -elicited PMN migration was measured using the migration of fluorescein isothiocyanate (FITC) labelled-dextran (MW 40kDa, Sigma). Infection and PMN migration times were 2½h each. Control monolayers received either bacteria or PMNs. Shown is a representative of two experiments. B, Apical sides of H292 monolayers were infected with TIGR4, and PMNs, or HBSS+Ca/Mg alone, were added the basolateral side. Transepithelial migration of bacteria was quantitated as stated in Materials and Methods. Shown is a representative of two experiments. C, Apical sides of H292 monolayers were infected with TIGR4, and PMNs were added to either basolateral or apical side of the monolayers. Control monolayers received HBSS+Ca/Mg instead of PMNs. Transepithelial migration of bacteria was quantitated as stated in Materials and Methods. Shown is a representative of two experiments. D, H292 monolayers were pretreated with CDC, and the apical side was infected with TIGR4. PMNs, or HBSS+Ca/Mg alone, were added the basolateral side. Bacterial migration was determined as stated in Material and Methods. Shown is a representative of two experiments.
To assess the extent to which this PMN-associated compromise in barrier function facilitates apical-to-basolateral bacterial translocation across the airway epithelium, we quantified viable bacteria in the basolateral chamber after the basolateral addition of PMNs. In the absence of PMNs, S. pneumoniae exhibited barely detectable movement across the monolayer (Fig. 7B). In contrast, upon the basolateral addition of PMNs, the rate of pneumococcal transepithelial migration increased by one to two orders of magnitude (Fig. 7B). The addition of PMNs to the apical side of H292 monolayers had only a minor effect on the apparent apical-to-basolateral bacterial migration, suggesting that transepithelial PMN migration was critical for bacterial penetration of the epithelial monolayer (Fig. 7C). Indeed, pretreatment of monolayers with CDC abrogated PMN-associated bacterial penetration of the H292 monolayers (Fig. 7D). Thus, the simplest model to explain these findings is that the basolateral-to-apical movement of PMNs across the monolayer results in a transient disruption of cellular junctions, permitting the retrograde movement of bacteria across the monolayer.
12-LOX-mediated inflammation promotes bacteremia and lethality during experimental infection
To determine if the 12-lipoxygenase pathway promotes invasive disease during infection, we monitored S. pneumoniae bacteremia by measuring CFU in tail vein blood after intratracheal inoculation of untreated mice, mice treated with the 12-lipoxygenase inhibitor CDC, or Alox15−/− mice, inoculated with a high dose of infection. At 24h post-infection, mock-treated WT mice suffered moderate (>104/ml) bacteremia, and by 48h exhibited high level (> 106/ml) bacteremia (Fig. 8A). By day three post-infection, a time point associated with > 106 TIGR4/ml of blood, approximately half of untreated mice succumbed to infection or were moribund and thus euthanized (Fig. 8B). One day later, no untreated mice were alive. In contrast, infection of CDC-treated WT mice resulted in no detectable bacteremia at any time point (Fig. 8A). Alox15−/− mice suffered only low-level (< 5 × 103/ml) bacteremia at 24 and 48h post-infection, and were devoid of bloodstream bacteria by 72h. The failure of S. pneumoniae to achieve high-level bacteremia in the CDC-treated or Alox15−/− mice was associated with uniform survival through the termination of the experiment at 7 days post-infection (Fig. 8B).
Fig. 8.
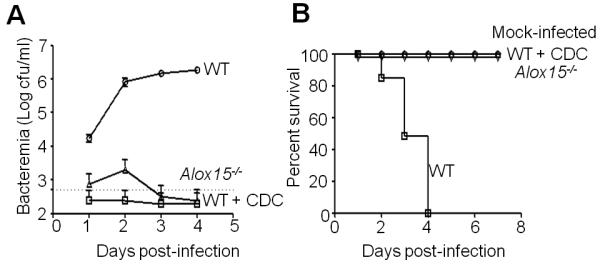
12-LOX mediated inflammation promotes bacteremia and lethality during S. pneumoniae infection of mice. Mock-treated wild type C57BL/6 mice (“WT”), CDC-treated wild type mice, or Alox15−/− mice were intratracheally inoculated with TIGR4 (see Materials and Methods). A control group of wild type mice received PBS. Six mice per group were used for the experiment. A, The cfu in the murine blood was determined at the specified time points. Shown is a representative of two experiments. B, The survival of mice was monitored over a 7 day post-infection period. Shown is a representative of two experiments.
DISCUSSION
The potent lipid chemoattractant HXA3 (10, 25) has been demonstrated to mediate intestinal inflammation in response to several bacterial pathogens (25, 28), and to promote pulmonary inflammation in response to Pseudomonas infection (18, 29). We found that the addition of purified HXA3 to the apical side of polarized H292 pulmonary epithelial cell monolayers elicited basolateral-to-apical migration of PMNs (data not shown), and that S. pneumoniae infection of H292 cells increased the expression of 12-lipoxygenase, the enzyme required for HXA3 synthesis. In an in vitro inflammation model, inhibition of 12-lipoxygenase diminished PMN transmigration across H292 monolayers. Importantly, pharmacological inhibition of 12-lipoxygenase or genetic ablation of Alox15 diminished the levels of airway inflammatory cells in uninfected mice, and dramatically diminished pulmonary inflammation after intratracheal challenge with S. pneumoniae. Thus, although our results do not rule out a role for other bioactive products of the 12-lipoxygenase pathway or the additional regulation of HXA3 production or secretion by pneumococcal infection (28, 38), they indicate that 12-lipoxygenase, and likely HXA3, play an important role in the migration of PMNs into the airway under both basal conditions and during acute S. pneumoniae infection.
We observed that infected mice deficient in 12-lipoxygenase activity by chemical inhibition or genetic ablation demonstrated not just lower levels of PMNs in the alveolar space, but also significantly lower numbers of total pulmonary PMNs. In addition, upon high dose S. pneumoniae pulmonary challenge, 12-lipoxygenase activity was required to achieve maximal numbers of macrophages in the lung, and maximal numbers of macrophages, dendritic cells and T cells in the airways. Thus, although our in vitro studies have exclusively assayed transmigration of PMNs across epithelium, 12-lipoxygenase may play a broad role in pulmonary inflammation. It is possible that unique features of the pulmonary architecture, e.g., a minimal subepithelial space separating the epithelium and endothelium, may foster a particularly prominent role for HXA3 in egress of PMNs from the pulmonary vasculature. Additionally, 12-lipoxygenase-mediated PMN migration across airway epithelium may enhance other events, e.g., a reduction in barrier integrity leading to the invasion of bacteria or extravasation of inflammatory cells, resulting in more generalized inflammation.
PMNs are an integral component of innate immunity, yet inhibition of PMN migration had no effect on the bacterial load in the lung or the airway fluid, at least in the time frame observed in our murine model. These results are consistent with the previous finding that PMN-depletion resulted in diminished pulmonary histopathology and bacteremia, as well as enhanced survival, during murine infection (46). Virulent S. pneumoniae, including the TIGR4 strain utilized in our animal infections, encodes a thick polysaccharide capsule that inhibits phagocytosis that may limit the degree of PMN-mediated clearance in this infection model (41).
The evolution of lung infection to life-threatening septicemia involves pneumococcal entry into the circulation upon breaching of the mucosal barrier, a step that has been modeled in vitro using polarized epithelial monolayers (43, 45). Pneumococci have been reported to bind E-cadherin, which participates in cell-cell adherence, and this binding activity might localize the bacterium to cell junctions as a step in bacterial transmigration (53). In addition, several studies suggest that bacterial transmigration involves a host-bacterium crosstalk that results in the paracellular movement of pneumococci across the monolayer. For example, pneumococci can cross cultured epithelial and endothelial monolayers by binding and activating host plasminogen, which subsequently cleaves critical components of the cellular junctions (43). A common finding is that inflammatory signaling contributes to barrier compromise, e.g., pneumococcal activation of the pattern recognition receptors leads to inflammatory signaling and diminished production of critical tight junction components (42, 45).
We found that induction of 12-lipoxygenase/HXA3, when coupled to the addition of PMNs to the basolateral surface of H292 cells, greatly increased bacterial transepithelial migration. HXA3 is capable of activating PMNs (10), but the dramatic increase in apical-to-basolateral bacterial transmigration depended on the concomitant basolateral-to-apical PMN transmigration: abrogation of PMN transmigration, either by the addition of PMNs to the apical rather than basolateral epithelial surface or by chemical inhibition of 12-lipoxygenase, diminished bacterial movement across monolayers. These results reinforce previous studies indicating that the transient disruption of epithelial barrier function can facilitate the translocation of toxins or pathogens across polarized epithelial monolayers (45, 47).
Importantly, just as disruption of HXA3 synthesis blocked bacterial transmigration in vitro, we found that inhibition of 12-lipoxygenase activity or genetic ablation of Alox15 in mice virtually eliminated bacteremia following intratracheal challenge. This finding suggests that PMN transmigration across respiratory epithelium may promote retrograde movement of pneumococci into the circulation, although it is also possible that acute pulmonary inflammation promotes systemic disease in additional ways, such as inflammatory damage to mucosal surface or seeding of the bloodstream by bacteria in (the enhanced) lymphatic drainage (7, 48, 49). Remarkably, the dramatic decrease in bacteremia corresponded to an equally dramatic effect on survival, and the virtual lack of demonstrable disease in mice unable to generate a 12-lipoxygenase-mediated acute inflammatory response provides stark evidence that a robust innate immune response contributes to, and indeed is essential for, lethality during experimental pneumococcal infection.
That the nonpathogenic bacterium B. subtilis did not elicit PMN transmigration in our in vitro model suggests that the induction of HXA3 production and/or secretion by respiratory epithelial cells is pathogen-specific. S. pneumoniae strains deficient in the synthesis of the polysaccharide capsule, a major virulence factor, induced reduced levels of PMN migration in vitro, suggesting that the pneumococcal capsule may be one of the bacterial factors that contribute to inflammation. It is possible that the polysaccharide capsule directly signals epithelial cells to produce and/or secrete HXA3. Alternatively, capsule production may indirectly promote 12-lipoxygenase induction by increasing the production or surface expression of the putative critical bacterial factor(s), or by enhancing the longevity of bacteria in the presence of PMNs added to the assay (54). Identification of S. pneumoniae factors that contribute to this host cell response would provide both a potential avenue to distinguish strains with low or high invasive potential as well as bacterial products that, when targeted, might diminish systemic disease. Finally, this study supports the possibility that anti-inflammatory agents, in particular those that block 12-lipoxygenase-mediated PMN migration, may provide a strategy to combat invasive pneumococcal infection. Given that 12-lipoxygenase-mediated inflammation appears central to other pulmonary disorders, such as asthma and acute lung injury (50, 51), inhibitors of these inflammatory pathways may have broad therapeutic value.
ACKNOWLEDGEMENTS
The authors thank Andrew Camilli, Chelsea Witte, Neil Greene and Erik Boll for discussion and Yushuan Lai, Blanca Corral Castroviejo and Stacie Clark for technical assistance. We also thank Lauren Richey for her assistance with mouse pathology.
This work was supported in part by American Lung Association Senior Research Training Fellowship RT 194942 N (to R. B.) and by the NIH (EY022208 to K.G.). B.A.M. is supported by the National Institutes of Health Grants DK56754 and DK33506, as well as the Crohn’s and Colitis Association of America. B.P.H is supported by the Cystic Fibrosis Foundation and NIH/NIAID.
REFERENCES
- 1.Hava DL, Hemsley CJ, Camilli A. Transcriptional regulation in the Streptococcus pneumoniae rlrA pathogenicity islet by RlrA. J. Bacteriol. 2003;185:413–421. doi: 10.1128/JB.185.2.413-421.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peltola VT, Boyd KL, McAuley JL, Rehg JE, McCullers JA. Bacterial sinusitis and otitis media following influenza virus infection in ferrets. Infect. Immun. 2006;74:2562–2567. doi: 10.1128/IAI.74.5.2562-2567.2006. JA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch JP, 3rd, Zhanel GG. Streptococcus pneumoniae: epidemiology and risk factors, evolution of antimicrobial resistance, and impact of vaccines. Curr. Opin. Pulm. Med. 2010;16:217–225. doi: 10.1097/MCP.0b013e3283385653. [DOI] [PubMed] [Google Scholar]
- 4.Mulholland K. Strategies for the control of pneumococcal diseases. Vaccine. 1999;17(Suppl 1):S79–84. doi: 10.1016/s0264-410x(99)00112-7. [DOI] [PubMed] [Google Scholar]
- 5.Doerschuk CM, Markos J, Coxson HO, English D, Hogg JC. Quantitation of neutrophil migration in acute bacterial pneumonia in rabbits. J. Appl. Physiol. 1994;77:2593–2599. doi: 10.1152/jappl.1994.77.6.2593. [DOI] [PubMed] [Google Scholar]
- 6.Moreland JG, Bailey G, Nauseef WM, Weiss JP. Organism-specific neutrophil-endothelial cell interactions in response to Escherichia coli, Streptococcus pneumoniae, and Staphylococcus aureus. J. Immunol. 2004;172:426–432. doi: 10.4049/jimmunol.172.1.426. JP. [DOI] [PubMed] [Google Scholar]
- 7.Weiss SJ. Tissue destruction by neutrophils. N. Engl. J. Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 8.Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 2009;77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol. Rev. 2003a;83:309–336. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 10.McCormick BA. Bacterial-induced hepoxilin A3 secretion as a pro-inflammatory mediator. FEBS J. 2007;274:3513–3518. doi: 10.1111/j.1742-4658.2007.05911.x. [DOI] [PubMed] [Google Scholar]
- 11.Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am. J. Respir. Cell. Mol. Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect. Immun. 2009;77:5300–5310. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prince AS, Mizgerd JP, Wiener-Kronish J, Bhattacharya J. Cell signaling underlying the pathophysiology of pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;291:L297–L300. doi: 10.1152/ajplung.00138.2006. [DOI] [PubMed] [Google Scholar]
- 14.Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L285–290. doi: 10.1152/ajplung.00461.2000. [DOI] [PubMed] [Google Scholar]
- 15.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 16.Smart SJ, Casale TB. Pulmonary epithelial cells facilitate TNF-alpha-induced neutrophil chemotaxis. A role for cytokine networking. J. Immunol. 1994;152:4087–4094. [PubMed] [Google Scholar]
- 17.Kunkel SL, Standiford T, Kasahara K, Strieter RM. Interleukin-8 (IL-8): the major neutrophil chemotactic factor in the lung. Exp Lung Res. 1991;17:17–23. doi: 10.3109/01902149109063278. [DOI] [PubMed] [Google Scholar]
- 18.Hurley BP, Siccardi D, Mrsny RJ, McCormick BA. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J. Immunol. 2004;173:5712–5720. doi: 10.4049/jimmunol.173.9.5712. [DOI] [PubMed] [Google Scholar]
- 19.Jahn HU, Krüll M, Wuppermann F, Klucken AC, Rosseau S, Seybold J, Hegemann JH, Jantos CA, Suttorp N. Infection and activation of airway epithelial cells by Chlamydia pneumoniae. J. Infect. Dis. 2000;182:1678–1687. doi: 10.1086/317608. [DOI] [PubMed] [Google Scholar]
- 20.Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J. Immunol. 2005;175:7530–7535. doi: 10.4049/jimmunol.175.11.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doyle NA, Bhagwan SD, Meek BB, Kutkoski GJ, Steeber DA, Tedder TF, Doerschuk CM. Neutrophil margination, sequestration, and emigration in the lungs of L-selectin-deficient mice. J. Clin. Invest. 1997;99:526–533. doi: 10.1172/JCI119189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizgerd JP, Meek BB, Kutkoski GJ, Bullard DC, Beaudet AL, Doerschuk CM. Selectins and neutrophil traffic: margination and Streptococcus pneumoniae-induced emigration in murine lungs. J. Exp. Med. 1996;184:639–645. doi: 10.1084/jem.184.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tasaka S, Qin L, Saijo A, Albelda SM, DeLisser HM, Doerschuk CM. Platelet endothelial cell adhesion molecule-1 in neutrophil emigration during acute bacterial pneumonia in mice and rats. Am. J. Respir. Crit. Care Med. 2003;167:164–170. doi: 10.1164/rccm.2202011. [DOI] [PubMed] [Google Scholar]
- 24.Frank JA, Matthay MA. Leukotrienes in acute lung injury: a potential therapeutic target? Am. J. Respir. Crit. Care Med. 2005;172:261–262. doi: 10.1164/rccm.2505008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, McCormick BA. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc. Natl. Acad. Sci. USA. 2004;101:7421–7426. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace-Asciak CR, Martin JM. Hepoxilin, a new family of insulin secretagogues formed by intact rat pancreatic islets. 1984 doi: 10.1016/0262-1746(84)90069-6. [DOI] [PubMed] [Google Scholar]
- 27.Laneuville O, Corey EJ, Couture R, Pace-Asciak CR. Hepoxilin A3 increases vascular permeability in the rat skin. Eicosanoids. 1991;4:95–97. [PubMed] [Google Scholar]
- 28.Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infect. Immun. 2008;76:3614–3627. doi: 10.1128/IAI.00407-08. BP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamang DL, Pirzai W, Priebe GP, Traficante DC, Pier GB, Falck JR, Morisseau C, Hammock BD, McCormick BA, Gronert K, Hurley BP. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J. Immunol. 2012;189:4960–4969. doi: 10.4049/jimmunol.1201922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns T, Zhong Z, Steinitz M, Pirofski LA. Modulation of polymorphonuclear cell interleukin-8 secretion by human monoclonal antibodies to type 8 pneumococcal capsular polysaccharide. Infect. Immun. 2003b;71:6775–6783. doi: 10.1128/IAI.71.12.6775-6783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mason RJ, Williams MC. Phospholipid composition and ultrastructure of A549 cells and other cultured pulmonary epithelial cells of presumed type II cell origin. Biochim. Biophys. Acta. 1980;617:36–50. doi: 10.1016/0005-2760(80)90222-2. [DOI] [PubMed] [Google Scholar]
- 32.McCormick BA, Hofman PM, Kim J, Carnes DK, Miller SI, Madara JL. Surface attachment of Salmonella typhimurium to intestinal epithelia imprints the subepithelial matrix with gradients chemotactic for neutrophils. J. Cell. Biol. 1995;131:1599–1608. doi: 10.1083/jcb.131.6.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Schilfgaarde M, van Alphen L, Eijk P, Everts V, Dankert J. Paracytosis of Haemophilus influenzae through cell layers of NCI-H292 lung epithelial cells. Infect. Immun. 1995;63:4729–4737. doi: 10.1128/iai.63.12.4729-4737.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galka F, Wai SN, Kusch H, Engelmann S, Hecker M, Schmeck B, Hippenstiel S, Uhlin BE, Steinert M. Proteomic characterization of the whole secretome of Legionella pneumophila and functional analysis of outer membrane vesicles. Infect. Immun. 2008;76:1825–1836. doi: 10.1128/IAI.01396-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;293:498–506. doi: 10.1126/science.1061217. RJ. [DOI] [PubMed] [Google Scholar]
- 36.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 2007;189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dopazo J, Mendoza A, Herrero J, Caldara F, Humbert Y, Friedli L, Guerrier M, Grand-Schenk E, Gandin C, de Francesco M, et al. Annotated draft genomic sequence from a Streptococcus pneumoniae type 19F clinical isolate. Microb. Drug Resist. 2001;7:99–125. doi: 10.1089/10766290152044995. M. [DOI] [PubMed] [Google Scholar]
- 38.Hurley BP, Williams NL, McCormick BA. Involvement of phospholipase A2 in Pseudomonas aeruginosa-mediated PMN transepithelial migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L703–L709. doi: 10.1152/ajplung.00390.2005. [DOI] [PubMed] [Google Scholar]
- 39.Kuhn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002;68-69:263–290. doi: 10.1016/s0090-6980(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 40.Kuhn H, O’Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006;45:334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 2010;78:704–715. doi: 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beisswenger C, Coyne CB, Shchepetov M, Weiser JN. Role of p38 MAP kinase and transforming growth factor-beta signaling in transepithelial migration of invasive bacterial pathogens. J. Biol. Chem. 2007;282:28700–28708. doi: 10.1074/jbc.M703576200. [DOI] [PubMed] [Google Scholar]
- 43.Attali C, Durmort C, Vernet T, Di Guilmi AM. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect. Immun. 2008;76:5350–5356. doi: 10.1128/IAI.00184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tafazoli F, Zeng CQ, Estes MK, Magnusson KE, Svensson L. NSP4 enterotoxin of rotavirus induces paracellular leakage in polarized epithelial cells. J. Virol. 2001;75:1540–1546. doi: 10.1128/JVI.75.3.1540-1546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarke TB, Francella N, Huegel A, Weiser JN. Invasive bacterial pathogens exploit TLR-mediated downregulation of tight junction components to facilitate translocation across the epithelium. Cell Host Microbe. 2011;9:404–414. doi: 10.1016/j.chom.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marks M, Burns T, Abadi M, Seyoum B, Thornton J, Tuomanen E, Pirofski LA. Influence of neutropenia on the course of serotype 8 pneumococcal pneumonia in mice. Infect. Immun. 2007;75:1586–1597. doi: 10.1128/IAI.01579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rankin S, Isberg RR, Leong JM. The integrin-binding domain of invasin is sufficient to allow bacterial entry into mammalian cells. Infect. Immun. 1992;60:3909–3912. doi: 10.1128/iai.60.9.3909-3912.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pang LM, Mellins RB, Rodriguez-Martinez F. Effect of acute lymphatic obstruction on fluid accumulation in the chest in dogs. J. Appl. Physiol. 1975;39:985–989. doi: 10.1152/jappl.1975.39.6.985. [DOI] [PubMed] [Google Scholar]
- 49.Marco AJ, Domingo M, Ruberte J, Carretero A, Briones V, Dominguez L. Lymphatic drainage of Listeria monocytogenes and Indian ink inoculated in the peritoneal cavity of the mouse. Lab. Anim. 1992;26:200–205. doi: 10.1258/002367792780740549. [DOI] [PubMed] [Google Scholar]
- 50.Andersson CK, Claesson HE, Rydell-Tormanen K, Swedmark S, Hallgren A, Erjefalt JS. Mice lacking 12/15-lipoxygenase have attenuated airway allergic inflammation and remodeling. Am. J. Respir. Cell. Mol. Biol. 2008;39:648–656. doi: 10.1165/rcmb.2007-0443OC. [DOI] [PubMed] [Google Scholar]
- 51.Rossaint J, Nadler JL, Ley K, Zarbock A. Eliminating or blocking 12/15-lipoxygenase reduces neutrophil recruitment in mouse models of acute lung injury. Crit. Care. 2012;16:R166. doi: 10.1186/cc11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hollifield M, Ghanem EB, de Villiers WJS, Garvy BA. Scavenger receptor A dampens induction of inflammation in response to the fungal pathogen Pneumocystis carinii. Infect. Immun. 2007;75:3999–4005. doi: 10.1128/IAI.00393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderton JM, Rajam G, Romero-Steiner S, Summer S, Kowalczyk AP, Carlone GM, Sampson JS, Ades EW. E-cadherin is a receptor for the common protein pneumococcal surface adhesin A (PsaA) of Streptococcus pneumoniae. Microb Pathog. 2007;42:225–236a. doi: 10.1016/j.micpath.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 54.Weinberger DM, Trzciński K, Lu YJ, Bogaert D, Brandes A, Galagan J, Anderson PW, Malley R, Lipsitch M. Pneumococcal capsular polysaccharide structure predicts serotype prevalence. PLoS Pathog. 2009;5:e1000476. doi: 10.1371/journal.ppat.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]