

Abstract
Levels of the neurotrophic cytokine S100β and the proinflammatory cytokine interleukin-6 (IL-6) are both elevated in Alzheimer’s brain, and both have been implicated in β-amyloid plaque formation and progression. We used RT-PCR and electrophoretic mobility shift assay to assess S100β induction of IL-6 expression and the role of κB-dependent transcription in this induction in neuron-enriched cultures and in neuron–glia mixed cultures from fetal rat cortex. S100β (10 or 100 ng/ml × 24 h) increased IL-6 mRNA levels two- and fivefold, respectively (p < 0.05 in each case), and S100β (100–1,000 ng/ml) induced increases in medium levels of biologically active IL-6 (30–80%). Combined in situ hybridization and immunohistochemistry preparations localized IL-6 mRNA to neurons in these cultures. S100β induction of IL-6 expression correlated with an increase in DNA binding activity specific for a κB element and was inhibited (75%) by suppression of κB binding with double-stranded “decoy” oligonucleotides. The low levels of S100β required to induce IL-6 overexpression in neurons, shown here, suggest that overexpression of S100β induces neuronal expression of IL-6 and of IL-6-induced neurodegenerative cascades in Alzheimer’s disease.
Keywords: S100β, Interleukin-6, Nuclear factor κB, Neuronal culture, Alzheimer’s disease
Interleukin-6 (IL-6) is a proinflammatory cytokine that is synthesized by astrocytes (Frei et al., 1989; Benveniste et al, 1990; Lee et al., 1993) and microglia (Frei et al., 1989; Lee et al., 1993). Neurons are also capable of synthesizing IL-6 (Schobitz et al., 1992; Gadient and Otten, 1994; Ringheim et al., 1995) in response to injury-related stimuli (Murphy et al., 1995), suggesting a role for neurons in intercellular cytokine signaling and coordination of injury responses. In various nonneuronal cell types IL-6 expression is regulated through the transcription factor nuclear factor κB (NFκB).
SI00β, an astrocyte-derived neuronotrophic cytokine that is up-regulated in response to injury, has been shown to stimulate neurite outgrowth (Kligman and Marshak, 1985), increase neuronal cytoplasmic free calcium levels (Barger and Van Eldik, 1992), and increase neuronal synthesis of the β-amyloid precursor protein (Li et al., 1998; for reviews, see Van Eldik and Zimmer, 1988; Barger et al., 1992). S100β can also induce expression of inducible nitric oxide synthase in astrocytes through NFκB (Hu et al., 1997).
The common role of S100β and IL-6 in injury responses suggests linkage or coordination of their respective expressions. In the present study we show for the first time direct regulation of neuronal IL-6 expression by S100β. These findings may have implications for pathogenic mechanisms of neurodegeneration in Alzheimer’s disease. IL-6 levels are elevated in CSF (Blum-Degen et al., 1995) and in brain tissue (Bauer et al., 1991; Huell et al., 1995) in Alzheimer’s disease, and IL-6 has been implicated in β-amyloid plaque formation (Huell et al., 1995). Tissue levels of biologically active S100β and of S100β mRNA are elevated in Alzheimer’s disease (Marshak et al., 1991), and S100β overexpression correlates both with the density of neuritic plaques (Sheng et al., 1994) and with the quantity of dystrophic neurites overexpressing βAPP in individual neuritic plaques (Mrak et al., 1996). Overexpression of S100β, with consequent trophic and toxic effects on neurons and their processes, has been proposed as an important pathogenic mechanism in the development of neuritic and neurofibrillary pathological changes in Alzheimer’s disease (Mrak et al., 1995; Griffin et al., 1998).
MATERIALS AND METHODS
Cortical cell culture
Cortical neuron-enriched cultures were derived from cerebral cortex of fetal (embryonic day 18) Sprague–Dawley rats, as previously described (Li et al., 1998). In brief, neocortical tissue was stripped of meninges and digested with 0.125% trypsin (GIBCO, Grand Island, NY, U.S.A.) and 0.01% DNase (Sigma, St. Louis, MO, U.S.A.) in calcium- and magnesium-free Hanks’ buffer solution (GIBCO) for 15–20 min at 37°C. Tissue was minced in 10% fetal bovine serum (GIBCO) in Dulbecco’s minimal essential medium (GIBCO), and cells were dissociated by trituration.
Cells were plated (a) in 35-mm-diameter dishes at 5 × 105 per plate (for immunocytochemistry or in situ hybridization), (b) at 1 × 106 per plate (for RNA isolation) in 24-well plates at 1 × 105 cells per well (secreted IL-6 bioactivity assays), or (c) in 60-mm-diameter dishes at 2 × 106 per plate [for electrophoretic mobility shift assay (EMSA)]. All culture plates were precoated with 50 μg/ml poly-D-lysine hydrobromide (70,000–150,000 mol wt; Sigma). Cultures were kept at 37°C in a humidified atmosphere, containing 5% CO2, for 24 h, after which the culture medium was replaced with serum-free medium containing Neurobasal medium (GIBCO) supplemented with B27 (GIBCO). Half of the medium volume was exchanged with fresh serum-free medium every 3 days. Cultures were exposed to 10−5 M cytosine arabinoside at day 5 for 24 h to inhibit nonneuronal growth. Experiments were performed on cultures after 10–14 days in vitro, at which time synthetic S100β [a generous gift from Linda Van Eldik, Northwestern University, Chicago, IL, U.S.A. (Winningham-Major et al., 1989)] was added to experimental cultures; parallel control cultures were left untreated.
For immunocytochemistry combined with in situ hybridization, glia–neuron cultures (“mixed cultures”) were used. These were derived from cerebral cortex of fetal (embryonic day 18) Sprague–Dawley rats, as described above, except the cultures were seeded at low density (5 × l05 per 35-mm-diameter dish) and were not exposed to cytosine arabinoside.
RNA isolation
Total cellular RNA was extracted from 10 cultures of each treatment group (for each experimental point), using TriRe-agent RNA isolation reagent (Molecular Research Center, Cincinnati, OH, U.S.A.) according to the manufacturer’s instructions, with the addition of phenol–chloroform–isoamyl alcohol extraction to increase RNA purity. RNA integrity was verified on agarose gels with ethidium bromide staining and quantified by spectrophotometric absorbance at 260 nm. RNA samples used in this study had 260/280 nm absorbance ratios of 1.7–2.0.
RT reaction and PCR amplification
RT of 1 μg samples of total RNA was performed using the Advantage RT-for-PCR Kit (Clontech, Palo Alto, CA, U.S.A.) according to the manufacturer’s instructions. In brief, the RNA and 1 μl of oligo(dT) primer (20 μM) were incubated at 70°C for 5 min before addition of 5 × reaction buffer (4 μl) with 0.5 mM deoxynucleotide triphosphates, 20 units of RNasin, and Moloney murine leukemia virus reverse transcriptase (200 units). The resulting 20 μl of reaction mixture was incubated at 42°C for 60 min and then at 95°C for 5 min. For comparisons of mRNA levels among different RNA samples, RT was performed simultaneously using reagents from a single master mix.
PCR amplification used reagents from Clontech. For IL-6 PCR, the forward primer was located at nucleotides 81–102 (5′CAA GAG ACT TCC AGC CAG TTG C 3′), whereas the reverse primer was located at nucleotides 694–670 (5′ TTG CCG AGT AGA CCT CAT AGT GAC C 3′), yielding an amplified product of 614 bp. For glyceraldehyde-3-phosphate dehydrogenase (G3PDH) amplification, the forward primer was located at nucleotides 550–569 (5′ ACC ACA GTC CAT GCC ATC AC 3′), whereas the reverse primer was located at nucleotide 982–1,001 (5′ TCC ACC ACC CTG TTG CTG TA 3′), yielding an amplified product of 452 bp. For normal (noncompetitive) PCR, the 1 μl of RT product of each sample was placed in a reaction with a master mix containing both IL-6 and G3PDH primers: The forward and reverse primers (20 μ M; 1 μl each) were combined with 5 μl of 10 × PCR buffer, 1 μl of 10 mM deoxynucleotide triphosphates, and 1 μl of Taq polymerase and diluted to a final volume of 50 μl Amplification was performed through 28 cycles of 94°C for 1 min, 62°C for 45 s, and 72°C for 45 s. The PCR procedure was stopped by final extension for 10 min at 72°C. Equal volumes of the reaction mixture from each sample were loaded on a 1.5% agarose gel with ethidium bromide staining. Fluorescent images were digitized, and IL-6 levels were normalized with the signals from corresponding G3PDH bands using NIH Image version 1.60 software.
Quantification of IL-6 mRNA was refined using a competitive PCR approach with known concentrations of a competitor MIMIC DNA (533 bp) generated using two hybrid (mimic) primers (forward, 5′ CAA GAG ACT TCC AGC CAG TTG CAA GTG AAA TCT CCT CCG 3′; reverse, 5′ TTG CCG AGT AGA CCT CAT AGT GAC CAT GGC AGT AGA TGG 3′; Ransom Hill Bioscience, Ramona, CA, U.S.A.; the under-bars denotes specific primer sequences for rat IL-6) from unrelated DNA fragment supplied in the MIMIC kit (Clontech). Then the competitive PCR was performed by adding 1 μl of RT product of each sample, respectively, to the PCR assay with serially diluted MIMIC DNA (10−7–10−22 M per reaction) and 1 μl each of the specific forward and reverse sequences of rat IL-6 primers (10 μM). The relative concentration of IL-6 mRNA RT product was estimated from the competitor concentrations that gave equal intensities of IL-6 (614-bp) and MIMIC (533-bp) products.
Secreted IL-6 bioactivity assay
Secreted IL-6 bioactivity in serum-free medium was measured using an IL-6-dependent 7TD1 cell line (American Type Culture Collection, Rockville, MD, U.S.A.) and a [3H]thymidine microproliferation assay (Nordan et al., 1987). Neuron-conditioned medium was cleared by centrifugation and stored at − 80°C until assayed. 7TD1 cells were seeded at a concentration of 5 × 104 cells per well in 100 μ1 of culture medium (RPMI 1640, consisting of 2% heat-inactivated fetal bovine serum, 50 mM β-mercaptoethanol, 50 μg/ml streptomycin, and 50 U/ml penicillin) in 96-well plates. Cultures were incubated in triplicate with 100 μl of a 1:6 dilution of each neuron-conditioned medium sample. Parallel 7TD1 cultures were treated with serial dilutions of purified IL-6 (R&D Systems, Minneapolis, MN, U.S.A.) to standardize the response. After 36 h, cultures were exposed to [3H]thymidine (0.5 μCi per well; specific activity, 6.7 Ci/mmol; ICN) for an additional 9 h. Cells were harvested onto glass fiber filters with an automated cell harvester and counted for radioactivity in a liquid scintillation counter. Purified S100β alone did not increase [3H]thy-midine incorporation in 7TD1 cells, excluding possible mito-genic effects arising from residual S100β remaining from treatment of the neuronal cultures.
EMSA
Nuclear extracts of treated cells were prepared and assayed as previously described (Barger and Harman, 1997). In brief, cultures were lysed in a buffer containing Nonidet P-40 (Fluka, Ronkonkoma, NY, U.S.A.), and nuclei were collected by centrifugation, washed to remove detergent, and extracted in a high-salt buffer. Salt concentrations were reduced by dilution before DNA-binding reactions. In these reactions, 2 μg of nuclear protein was incubated with 32P-labeled double-stranded oligonucleotide probe containing the immunoglobulin/HIV-κB enhancer sequence (Promega, Madison, WI, U.S.A.). For competition assays to determine binding specificity, a 30–300-fold molar excess of unlabeled, double-stranded oligonucleotide was added during parallel binding reactions. The unlabeled competitor was the immunoglobulin/HIV-κB probe itself, an oligonucleotide containing the putative κB site from the rat IL-6 promoter (5′ A AT GTG GGA TTT TCC CAT GA 3′), or a mutant κB sequence deficient in NFκB binding (5′ ACT TGA GCA GAC TTT CCC AGG C 3′) (each duplexed with the complementary strand). Complexes were resolved by nondena-turing gel electrophoresis (6%) and visualized by autoradiography.
“Decoy” oligonucleotide treatments
Double-stranded oligonucleotides containing a κB enhancer sequence (5′ CCA AGG GGA CTT TCC ATG G 3′) or a control oligonucleotide of scrambled sequence (5′ GAC CAT GTC GTG CAG TAG C 3′) were obtained by combining single-stranded, phosphothioated oliogonucleotide (Oligos Etc., Wilsonville, OR, U.S.A.) with equimolar amounts of their complementary strands, heating to 85°C, and cooling slowly in a heating block. The annealed oligonucleotides were quantified by absorbance at 260 nm.
Decoy or control oligonucleotides (1 μM) were combined with 47 μg/ml Lipofectamine (GIBCO) to allow formation of liposome complexes. This mixture then was diluted 10-fold into the existing culture medium. After 3 h, the culture medium was completely replaced with fresh medium containing various concentrations of S100β. After 24 h of incubation with S100β, RNA was extracted for RT-PCR, nuclear proteins were extracted for EMSA, or in situ hybridization was performed.
Immunocytochemistry
For morphological characterization, cells grown in 35-mm-diameter dishes were rinsed once in phosphate-buffered saline (PBS) and fixed for 30 min with 4% paraformaldehyde in PBS (pH 7.4). Cells then were rinsed once in PBS, permeabilized for 15 min with 0.2% Triton X-100 in PBS, blocked in 1% normal sheep serum in PBS for 30 min, and incubated with appropriate primary antibodies overnight at 4°C in PBS containing 1% normal sheep serum as previously described (Sheng et al., 1997). Mouse monoclonal anti-microtubule-associated protein (MAP) antibodies (MAP2; 1:500; Sigma) were used to identify neurons, rabbit polyclonal anti-glial fibrillary acidic protein (GFAP) antibodies (1:2,000; Dako, Carpinteria, CA, U.S.A.) were used to identify astrocytes, and monoclonal anti-Mac 1 antibody (1:100; Scios, Sunnyvale, CA, U.S.A.) was used to identify microglia. The primary antibodies were detected with the appropriate anti-rabbit or anti-mouse IgG, using a Vector (Burlingame, CA, U.S.A.) ABC kit with diaminobenzidine or Vector Red kit for color development. Dual immunohistochemistry was performed as previously described (Sheng et al., 1997).
In situ hybridization
To generate sense and antisense riboprobes, the 1.16-kb fragment excised from the human IL-6 cDNA clone was sub-cloned into pBluescript-SK+ at the EcoRl site. Sense RNA probe was generated from HinndIII-linearized plasmid and T3 polymerase, and the antisense probe was synthesized from BamHI-linearized plasmid and T7 polymerase, using an RNA Digoxigenin Label Kit (Boehringer-Mannheim) according to the kit manufacturer’s instructions. The size of the labeled riboprobes was checked by comparison with RNA standards.
Hybridization was performed as previously described (Li et al., 1998). Fixed cells were washed once with PBS and incubated with 0.2% Triton X-100 in PBS for 15 min. Cultures were prehybridized at 37°C for 1 h with a solution containing 50% formamide, 5% dextran sulfate, 0.02% Ficoll, 0.02% bovine serum albumin, 0.02% polyvinylpyrrolidone, 500 μg/ml tRNA, and 500 μg/ml sheared salmon sperm DNA. Hybridization with RNA probes was carried out by adding either digoxigenin-labeled sense or antisense riboprobe to the above prehybridization solution (to a final concentration of 0.5 μg of RNA probe/ml). Dishes were sealed with tape and allowed to incubate overnight at 42°C. Following hybridization, cells were washed with 2× saline-sodium citrate (SSC) for 30 min, incubated in RNase A solution for 30 min at 37°C, washed twice in 1× SSC, and washed once in 0.5× SSC. Colorimetric detection was performed according to the manufacturer’s instructions and was terminated after 2 h by washing for 5 min in 10 mM Tris-HCl and 1 mM EDTA (pH 8.0) at room temperature.
In mixed cultures, combined in situ hybridization and fluorescence immunocytochemistry were used to differentiate neurons expressing IL-6 from other cell types that also express IL-6. In situ hybridization of IL-6 mRNA was followed by immunohistochemical localization of neurons with MAP2 antibody (1:500 in PBS at 4°C overnight) through visualization using fluorescein-labeled rabbit anti-mouse IgG antibody (Vector).
Statistics
Statistical significance of differences between experimental treatments and controls was assessed using Student’s t test for unpaired data.
RESULTS
Composition of cortical neuron-enriched cultures
Our fetal rat cortical neuron-enriched cultures consisted primarily of neurons (illustrated in Fig. 1A) with small numbers of astrocytes (illustrated in Fig. 1B). This predominant neuronal component was immunoreactive for MAP2, whereas GFAP-immunoreactive astrocytes constituted <5% of cells, and Mac-1-immunoreactive microglia constituted <1% of cells (data not shown).
FIG. 1.

Immunocytochemical identification of cell types and in situ hybridization results show S100β induction of IL-6 mRNA. A: MAP2-immunoreactive neurons in primary rat fetal cortical culture. B: MAP2/GFAP dual-label immunocytochemical preparation shows astrocytes (red, arrow) and neurons (brown). C: IL-6 mRNA expression by S100β-treated (100 ng/ml × 24 h) cultured cortical neurons. D: IL-6 mRNA is not expressed in untreated (0 ng of S100β) “control” sister cultures. E and F: Colocalization of IL-6 mRNA expression (E) and MAP2 immunoreactivity (F) within individual neurons in cortical neuron–glia (mixed cultures) treated with S100β (100 ng/ml × 24 h). A–D, bar = 100 μm; E and F, bar = 50 μm.
S100β induction of expression of biologically active IL-6 in neuron-enriched cultures
The levels of biologically active IL-6 in culture medium of cortical neuron-enriched cultures increased in a dose-dependent manner following incubation of the cultures with S100β for 24 h (Fig. 2). These increases were significant at doses of ≥100 ng of S100β/ml (p < 0.02), with an 80% increase resulting from treatment with 1 μg of S 100β/ml (p < 0.001). IL-6 levels in culture medium were determined using a bioassay in which the end point is [3H]thymidine uptake by 7TD1 IL-6-dependent cells (Fig. 2). These results were confirmed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (data not shown). Treatment of 7TD1 IL-6-dependent cells with S100β had no effect on [3H]thymidine incorporation (data not shown).
FIG. 2.
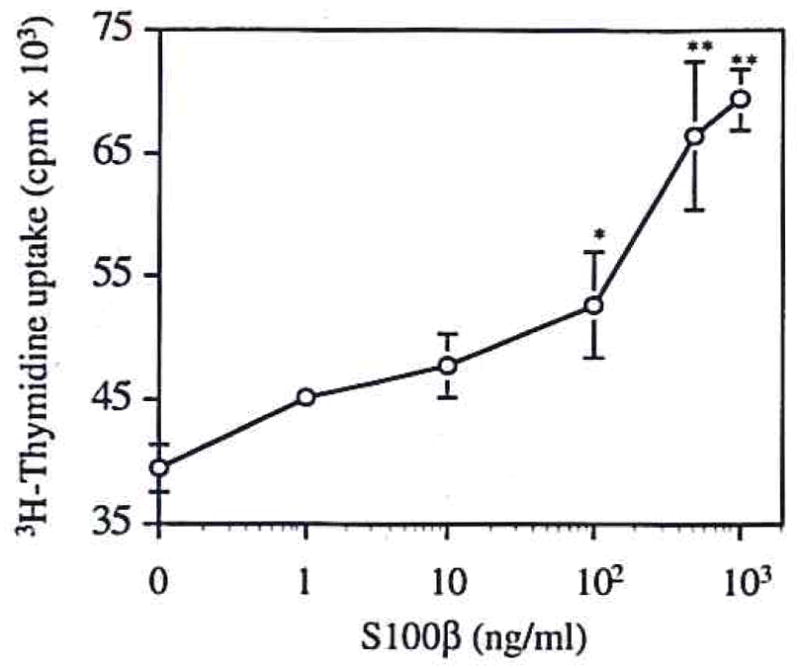
S100β induction of IL-6 secretion in neuronal cultures. S100β (10–1,000 ng/ml × 24 h) induces dose-dependent increases of IL-6 production. Results are given as [3H]thymidine uptake by an IL-6-dependent 7TD1 cell line incubated with conditioned medium from neuronal cultures treated with the given concentrations of S100β. Data are mean ± SEM (bars) values for four replicates. *p < 0.05, **p < 0.001 versus control.
S100β induction of IL-6 mRNA expression in neuron-enriched cultures
S100β treatment increased IL-6 mRNA expression in cortical neuron-enriched cultures (Fig. 3). Significant increases in IL-6 mRNA were seen after 12 h of treatment with 100 ng of S100β/ml and after 24 h of treatment with 10 ng of S100β/ml (Fig. 3B). The potential contribution of endotoxin or other contaminants was excluded through the use of controls in which S100β was applied after preab-sorption with anti-S100β antibody (Fig. 3A) or in the presence of polymyxin B (data not shown).
FIG. 3.

RT-PCR shows S100β induction of IL-6 mRNA expression in neuronal cultures. A: Cortical neurons were incubated for 24 h either with S100β (100 ng/ml) that had been preabsorbed with anti-S100β antibody (lanes 2–4) or with S100β alone at the indicated doses. G3PDH mRNA was used to verify equal quantities of starting cDNA for each sample. B: Densitometric analysis of ethidium bromide-stained gels. IL-6 mRNA levels relative to G3PDH mRNA levels are expressed relative to values in control (Con) cultures. Data are mean ± SEM (bars) values for six to eight individual samples, **p < 0.01, significantly different from corresponding Con value. C and D: Competitive RT-PCR, performed on aliquots of samples used for analysis shown in lanes 2 and 8 of A. There is an ~10-fold difference in IL-6 mRNA expression between S100β-treated (D) and control (C) cultures.
For quantitative analysis of IL-6 mRNA levels, we used a competitive PCR approach that allows estimation of the absolute levels of IL-6 cDNA obtained from culture mRNA samples (Fig. 3C and D). The principal advantage of this procedure is obviation of the potential for nonlinear relationships between samples containing different amounts of input cDNA. These measurements confirmed the increases, shown by quantitative RT-PCR, in IL-6 mRNA expression relative to G3PDH mRNA expression in S100β-treated cultures.
Localization of IL-6 mRNA expression to neurons in neuron-enriched and in mixed cultures
In situ hybridization with antisense IL-6 cRNA probe showed progressive increases in the relative numbers of culture cells expressing IL-6 mRNA following treatment with S100β (100 ng/ml) for ≥12 h (data not shown). At 24 h (Fig. 1C) there were significantly greater numbers of IL-6-expressing cells in S100β-treated cultures (66 ± 5% of total cells, mean ± SD) compared with control cultures (9 ± 2%). Control (0 ng of S100β/ml) cultures showed only scattered, weakly IL-6-positive cells (Fig. 1D) at all time points, and both control and treated cultures hybridized with sense IL-6 RNA showed only weak background labeling (data not shown).
Sparsely seeded neuron-glia cultures (mixed cultures) were used to establish the identity of IL-6-expressing cells. In situ hybridization (for IL-6 mRNA) combined with MAP2 immunocytochemistry (to identify specifically neuronal processes) showed colocalization of IL-6 mRNA (in the perikarya) and MAP2 immunoreactivity (in the processes) of individual neurons (Fig. 1E and F) following treatment with 100 ng of S100β/ml. IL-6 mRNA expression was not restricted to neurons, as it was also evident in MAP2-negative (nonneuronal) cells (Fig. 1E). However, pretreatment of these mixed cultures with the interleukin-1 receptor antagonist had no effect on the expression of biologically active IL-6, suggesting that S100β-mediated actions were not dependent on indirect actions of S100β on the expression of interleukin-1, another potent regulator of IL-6 (p = 0.8).
Role of κB-binding activity in regulation of IL-6 by S100β
The IL-6 gene promoter in rats has a κB enhancer (−63 to −54) that binds NFκB and thus regulates rat IL-6 expression in several nonneuronal cell types (Franchimont et al., 1997). Furthermore, S100β has been shown to elevate the expression of another NFκB-regulated gene, inducible nitric oxide synthase (Hu et al., 1997; Akama et al., 1998), in astrocytes. To determine whether a similar mechanism was involved in the regulation of IL-6 by S100β, we treated neuronal cultures with S100β in the presence of κB-containing oligonucleotides, which can interfere with NFκB activity by acting as a “decoy” for the transcription factor (Morishita et al, 1997). Addition of the κB decoy significantly reduced the effect of S100β on IL-6 mRNA expression (75% decrease; Fig. 4A, B, and D), whereas scrambled (control) NFκB DNA had no effect (Fig. 4A–C). Moreover, decoy NFκB DNA inhibited S100β-mediated IL-6 mRNA expression in neurons in situ (21 ± 7% of neurons expressed IL-6 mRNA vs. 66 ± 5% without decoy DNA; p < 0.001; data not shown), whereas scrambled NFκB DNA had little effect on S100β induction of IL-6 mRNA expression in neurons in situ (66 ± 4% of the cells expressed IL-6 mRNA; data not shown), suggesting a role for NFκB-mediated transcription in stimulation of neuronal IL-6 synthesis by S100β.
FIG. 4.

Suppression of κB DNA binding blocks S100β induction of IL-6 mRNA expression in cortical neuron cultures. A: An example of RT-PCR results. Lanes 1 and 2 represent IL-6 mRNA levels following incubation with control medium. Lanes 3 and 4 represent IL-6 mRNA levels following treatment with S100β (100 ng/ml). Lanes 5 and 6 represent IL-6 mRNA levels following simultaneous treatment with S100β (100 ng/ml) and control (Con) DNA (100 nM). Lanes 7 and 8 represent IL-6 mRNA levels following simultaneous treatment with S100β (100 ng/ml) and “decoy” (Dec) DNA (100 nM). B: Densitometric analysis of ethidium bromide-stained gels. IL-6 mRNA levels relative to G3PDH mRNA levels are expressed relative to values in control cultures. Data are mean ± SEM (bars) values for four individual samples. Indicated values are significantly different: *p < 0.01. C and D: Competitive RT-PCR, performed on aliquots of samples used for analysis shown in lanes 5 and 7 of A. There is an ~10-fold difference in IL-6 mRNA expression between S100β+ control DNA-treated (C) and S100β+ Dec DNA-treated (D) cultures.
S100 β did induce a κB-dependent DNA-binding factor in primary cultures of cortical neurons, as detected by EMSA analysis (Fig. 5A), but the activity detected corresponded to a non–Rel κB-binding factor that is distinct from classically defined NFκB (RelA/p50 heterodimer). We have referred elsewhere to this DNA-binding factor as neuronal κB-binding factor (NKBF), based on its prominence in neurons. This neuronal DNA binding factor differs from NFκB in antigenicity, native and denatured electrophoretic mobility, and DNA sequence preference (Moerman et al, 1999). Treatment with 10 or 30 ng/ml S100β enhanced NKBF activity in a dose-dependent manner (Fig. 5A). This enhancement could be competed out by increasing concentrations of κB (Fig. 5B, lanes κB). To confirm that κB binding is relevant to IL-6 regulation, we tested its ability to bind the κB element in the rat IL-6 promoter (Fig. 5B, lanes rIL-6p). Specificity of the competition was demonstrated through the lack of competition observed with a mutated κB sequence (Fig. 5B, lanes mut).
FIG. 5.

EMSA demonstration of S100 β induction of κB binding factor. Nuclear extracts from primary cortical cultures were assayed by EMSA for DNA-binding activity specific for the κB enhancer element. A: Cultures were treated for 90 min with the indicated concentrations of S100β (in ng/ml). In the lane marked “C,” the 30 ng/ml sample was assayed in the presence of a 200-fold excess unlabeled probe. B: A nuclear extract from S100β-treated cultures was assayed for binding to the κB sequence in the absence of competitor (−) or in the presence of a 30-, 100-, or 300-fold molar excess of the following competitors: double-stranded oligonucleotides that were the same sequence as the probe (κB); a mutant κB (mut); or a consensus κB site from the rat IL-6 promoter (rlL-6p).
DISCUSSION
S100β, a neurite growth-promoting cytokine, and IL-6, a proinflammatory cytokine, are both overex-pressed in Alzheimer’s disease, and, based on their established actions, both have been implicated in Alzheimer’s pathogenesis (Griffin et al., 1989, 1998; Vandenabeele and Fiers, 1991; Bauer et al., 1992). Immuno-reactivity for IL-6 is found in β-amyloid plaques in Alzheimer’s disease (Bauer et al., 1992), but the cells of origin of this IL-6 are yet to be identified. Here, we show that one of these cytokines, S100β, regulates neuronal synthesis and release of the other, IL-6, through activation of a specific neuronal nuclear κB-binding factor.
The κB-binding factor NFκB plays an important role in the activation of the IL-6 gene in various cells of the immune system (Libermann and Baltimore, 1990; Matsusaka et al., 1993), in fibroblasts (Zhang et al., 1990), and in astrocytes (Sparacio et al., 1992). The decoy DNA approach (Tanaka et al., 1994) used here has been shown to suppress effectively κB-mediated transcription and has been used to provide evidence for the involvement of NFκB in various in vitro systems (Sharma and Narayanan, 1996) and also in vivo (Morishita et al., 1997). We have previously demonstrated that this approach can be used to inhibit the elevation of a novel κB-binding activity induced in cortical neurons by either tumor necrosis factor (Barger et al., 1995) or dehydroepiandosterone (Mao and Barger, 1998). The activity of this novel factor is now attributed to NKBF (Mao et al., 1999; Moerman et al., 1999), which appears to be responsible for the effects of S100β on neuronal IL-6 expression. In nonneuronal cell types, IL-6 synthesis is regulated by NFκB (Sparacio et al., 1992), and our observation of S100β-induced IL-6 mRNA expression in nonneuronal cells (presumably glia, as illustrated in Fig. 1E) may be mediated by this mechanism. In contrast, our results suggest that S100β-induced expression of IL-6 in neurons is mediated through NKBF, a similar but not identical transcription factor. The specific binding of NKBF to the IL-6 promoter κB element, together with the effects of the κB decoy treatment, provides evidence that this κB-binding factor acts as a physiologically relevant transcription factor for neuronal IL-6 expression induced by S100β.
In Alzheimer’s disease, the levels of expression of S100β correlate directly with the density of neuritic β-amyloid plaques (Sheng et al., 1994). Overexpression of both S100β (Mrak et al., 1996) and IL-6 (Bauer et al, 1991) is present in early stages of neuritic β-amyloid plaque development. These findings, together with the neurite growth-promoting properties of S100β (Marshak et al., 1991), and S100β induction of β-amyloid precursor protein expression (Li et al., 1998), have been cited as implicating S100β in the conversion of diffuse amyloid deposits into neuritic plaques through promotion of proliferation of dystrophic neurites, overexpressing β-amyloid precursor protein, in these plaques (Mrak et al., 1996; Griffin et al., 1998). We have previously shown that S100β induces neurite outgrowth from neurons in enriched cultures similar to those used for this study (Li et al., 1998). The S100β-driven up-regulation of IL-6 shown here suggests that S100β-induced expression of biologically active IL-6 may be an important pathogenic link in the glial-neuronal interactions that promote progression of neuropathological changes in Alzheimer’s disease.
Acknowledgments
This work was supported in part by grants AG12411 and NS35872 from the National Institutes of Health, the Alzheimer’s Association, and the Donald W. Reynolds Foundation. The authors thank Mr. Richard A. Jones for technical support, Dr. Jin G. Sheng for expert advice, and Ms. Pam Free for secretarial support.
Abbreviations used
- EMSA
electrophoretic mobility shift assay
- GFAP
glial fibrillary acidic protein
- G3PDH
glyceraldehyde-3-phos-phate dehydrogenase
- IL-6
interleukin 6
- MAP
microtubule-associated protein
- NFκB
nuclear factor κB
- NKBF
neuronal κB-binding factor
- PBS
phosphate-buffered saline
- SSC
saline–sodium citrate
References
- Abraham CR, Shirahama T, Potter H. α1Antichymo-trypsin is associated solely with amyloid deposits containing the β-protein. Amyloid and cell localization of α1 arantichymotrypsin. Neurobiol Aging. 1990;11:123–129. doi: 10.1016/0197-4580(90)90045-2. [DOI] [PubMed] [Google Scholar]
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc Natl Acad Sci USA. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Harman AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apoli-poprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- Barger SW, Van Eldik LJ. S100β stimulates calcium fluxes in glial and neuronal cells. J Biol Chem. 1992;267:9689–9694. [PubMed] [Google Scholar]
- Barger SW, Wolchok SR, Van Eldik LJ. Disulfide-linked S100β dimers and signal transduction. Biochim Biophys Acta. 1992;1160:105–112. doi: 10.1016/0167-4838(92)90043-d. [DOI] [PubMed] [Google Scholar]
- Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors α and β protect neurons against amyloid β-peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Volk B, Berger M. Interleukin-6 and alpha2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 1991;285:111–114. doi: 10.1016/0014-5793(91)80737-n. [DOI] [PubMed] [Google Scholar]
- Bauer J, Strauss S, Volk B, Berger M. IL-6-mediated events in Alzheimer’s disease pathology. Immunol Today. 1992;12:422. doi: 10.1016/0167-5699(91)90148-M. [DOI] [PubMed] [Google Scholar]
- Benveniste EN, Sparacio SM, Norris JG, Grenett HE„, Fuller GM. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol. 1990;30:201–212. doi: 10.1016/0165-5728(90)90104-u. [DOI] [PubMed] [Google Scholar]
- Blum-Degen D, Muller T, Kuhn W„ Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neuroxci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- Franchimont N, Rydziel S, Canalis E. Interleukin 6 is autoregulated by transcriptional mechanisms in cultures of rat osteoblastic cells. J Clin Invest. 1997;100:1797–1803. doi: 10.1172/JCI119707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei K, Malipiero UV, Leist TP, Zinkernagel RM, Schwab ME, Fontana A. On the cellular source and function of interleukin-6 produced in the central nervous system in viral diseases. Eur J Immunol. 1989;19:689–694. doi: 10.1002/eji.1830190418. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten U. Identification of interleukin-6-expressing neurons in the cerebellum and hippocampus of normal adult rats. Neurosci Lett. 1994;182:243–246. doi: 10.1016/0304-3940(94)90807-9. [DOI] [PubMed] [Google Scholar]
- Ganter U, Strauss S, Jonas U, Weidemann A, Beyreuther K, Volk B, Berger M, Bauer J. α2-Macroglobulin synthesis in interleukin-6-stimulated human neuronal (SH-SY5Y neuroblastoma) cells. Potential significance for the processing of Alzheimer β-amyloid precursor protein. FEBS Lett. 1991;282:127–131. doi: 10.1016/0014-5793(91)80460-k. [DOI] [PubMed] [Google Scholar]
- Griffin WST, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C. Brain interleukin-1 and S100 immunoreactivity are elevated in Down syndrome and Alzheimer’s disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WST, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Ferreira A, Van Eldik LJ. S100β induces neuronal cell death through nitric oxide release from astrocytes. J Neurochem. 1997;69:2294–2301. doi: 10.1046/j.1471-4159.1997.69062294.x. [DOI] [PubMed] [Google Scholar]
- Huell M„ Strauss S, Volk B, Berger M, Bauer J. Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol (Berl) 1995;89:544–551. doi: 10.1007/BF00571510. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-κB is activated in primary neurons by amyloid β peptides and in neurons surrounding early plaques from patients with Alzheimer’s disease. Proc Natl Acad Sci USA. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Ota T, Matsuoka Y, Nomura Y, Taniguchi T. Alteration of transcription factors NF-κB and STAT1 in Alzheimer’s disease brains. Newrosci Lett. 1997;237:17–20. doi: 10.1016/s0304-3940(97)00797-0. [DOI] [PubMed] [Google Scholar]
- Kligman D, Marshak DR. Purification and characterization of a neurite extension factor from bovine brain. Proc Natl Acad Sci USA. 1985;82:7136–7139. doi: 10.1073/pnas.82.20.7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Liu W, Dickson DW, Brosnan CE, Berman JW. Cytokine production by human fetal microglia and astrocytes. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- Li Y, Wang J, Sheng JG, Liu L, Barger SW, Jones RA, Van Eldik LJ, Mrak RE, Griffin WST. S100β increases levels of β-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–1428. doi: 10.1046/j.1471-4159.1998.71041421.x. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Barger SW. Neuroprotection by dehydroepiandro-sterone-sulfate: role of an NFκB-like factor. Neuroreport. 1998;9:759–763. doi: 10.1097/00001756-199803090-00036. [DOI] [PubMed] [Google Scholar]
- Mao X, Moerman AM, Lucas MM, Barger SW. Inhibition of the activity of a neuronal κB-binding factor by glutamate. J Neurochem. 1999;73:1851–1858. [PubMed] [Google Scholar]
- Marshak DR, Pesce SA, Stanley LC, Griffin WST. Increased S100β neurotrophic activity in Alzheimer disease temporal lobe. Neurobiol Aging. 1991;13:1–7. doi: 10.1016/0197-4580(92)90002-f. [DOI] [PubMed] [Google Scholar]
- Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, Akira S. Transcription factors NF-IL6 and NF-κB synergistically activate transcription of the inflammatory cytokines interleukin 6 and interleukin 8. Proc Natl Acad Sci USA. 1993;90:10193–10197. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman AM, Mao X, Lucas MM, Barger SW. Characterization of a neuronal κB-binding factor distinct from NF-κB. Mol Brain Res. 1999;67:303–315. doi: 10.1016/s0169-328x(99)00091-1. [DOI] [PubMed] [Google Scholar]
- Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Sheng JG, Griffin WST. Glial cytokines in Alzheimer’s disease: review and pathogenic implications. Hum Pathol. 1995;26:816–823. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Sheng JG, Griffin WST. Correlation of astrocytic S100β expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:273–279. doi: 10.1097/00005072-199603000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P, Grondin J, Altares M. Induction of interleukin-6 in axotomized sensory neurons. J Neurosci. 1995;15:5130–5136. doi: 10.1523/JNEUROSCI.15-07-05130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordan RP, Pumphrey JG, Rudikoff S. Purification and NH2-terminal sequence of a plasmacytoma growth factor derived from the murine macrophage cell line P388D1. J Immunol. 1987;139:813–817. [PubMed] [Google Scholar]
- Ringheim GE, Burgher KD, Heroux JA. Interleukin-6 mRNA expression by cortical neurons in culture: evidence for neuronal sources of interleukin-6 production in the brain. J Neu-roimmunol. 1995;63:113–123. doi: 10.1016/0165-5728(95)00134-4. [DOI] [PubMed] [Google Scholar]
- Schobitz BD, Voorhuis AM, De Kloet ER. Localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Neurosci Lett. 1992;136:189–192. doi: 10.1016/0304-3940(92)90046-a. [DOI] [PubMed] [Google Scholar]
- Sharma HW, Narayanan R. The NF-κB transcription in oncogenesis. Anticancer Res. 1996;16:589–596. [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WST. S100β protein expression in Alzheimer’s disease: potential role in the pathogenesis of neuritic plaques. J Neurosci Res. 1994;39:398–404. doi: 10.1002/jnr.490390406. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WST. Glial-neuronal interactions in Alzheimer’s disease: progressive association of IL-lα+ microglia and S100β+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997;56:285–290. [PubMed] [Google Scholar]
- Sparacio SM, Zhang Y, Vilcek J, Benveniste EN. Cytokine regulation of interleukin-6 gene expression in astrocytes involves activation of an NFκB-like nuclear protein. J Neuroim-munol. 1992;39:231–242. doi: 10.1016/0165-5728(92)90257-l. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Vickart P, Bertrand JR, Rayner B, Morvan F, Imback JL, Paulin D, Malvy C. Sequence-specific interaction of α-β-anomeric double-stranded DNA with the p50 subunit of NF-κB: application to the decoy approach. Nucleic Acids Res. 1994;22:3069–3074. doi: 10.1093/nar/22.15.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Fiers W. Is amyloidogenesis during Alzheimer’s disease due to an IL-l/IL-6-mediated acute phase response in the brain? Immunol Today. 1991;12:217–219. doi: 10.1016/0167-5699(91)90032-O. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Zimmer DB. Mechanisms of action of the S100 family of calcium modulated proteins. In: Gerday Ch, Gilles R, Bolis L., editors. Calcium and Calcium Binding Proteins. Springer-Verlag; Berlin: 1988. pp. 114–127. [Google Scholar]
- Winningham-Major F, Staecker JL, Barger SW, Coats S, Van Eldik LJ. Neurite extension and neuronal survival activities of recombinant S100β proteins that differ in the content and position of cysteine residues. J Biol Chem. 1989;109:3063–3071. doi: 10.1083/jcb.109.6.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lin JX, Vilcek J. Interleukin-6 induction by tumor necrosis factor and interleukin-1 in human fibroblasts involves activation of a nuclear factor binding to a κB-like sequence. Mol Cell Biol. 1990;101:3818–3823. doi: 10.1128/mcb.10.7.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]