

Abstract
Diverse cellular processes relevant to cancer progression are regulated by the acetylation status of proteins. Among such processes is chromatin remodeling via histone proteins, controlled by opposing histone deacetylase (HDAC) and histone acetyltransferase (HAT) enzymes. Histone deacetylase inhibitors (HDACi) show great promise in preclinical cancer models, but clinical trials treating solid tumors have failed to improve patient survival. This is due in part to an inability of HDACi to effectively accumulate in cancerous cells. To address this problem we designed HDACi with secondary pharmacophores to facilitate selective accumulation in malignant cells. We present the first example of HDACi compounds targeted to prostate tumors by equipping them with the additional ability to bind the androgen receptor (AR) with non-steroidal antiandrogen moieties. Leads among these new dual-acting molecules bind to the AR and halt AR transcriptional activity at lower concentrations than clinical antiandrogens. They inhibit key isoforms of HDAC with low nanomolar potency. Fluorescent microscopy reveals varying degrees of AR nuclear localization in response to these compounds that correlates with their HDAC activity. These biological properties translate into potent anticancer activity against hormone dependent (AR+) LNCaP and to a lesser extent against hormone independent (AR−) DU145 prostate cancer, while having greatly reduced toxicity in non-cancerous cells. This illustrates that engaging multiple biological targets with a single chemical probe can achieve both potent and cell-type selective responses.
Keywords: Histone Deacetylase inhibitors, Synthesis, Prostate Cancer, Androgen Receptor, Antiandrogens, Selective Cancer Therapy, Dual Targeting Compounds
Introduction
Among the next generation of potential therapies for cancer are histone deacetylase inhibitors (HDACi), a chemical class of small molecules targeting the epigenetically regulated histone code.1–3 Carcinogenesis is frequently associated with aberrant acetylation status of proteins, and inhibiting HDAC enzymes can induce cancer cell death by increasing acetylation of nuclear (e.g. histone)4–5 and/or many non-histone proteins including α-tubulin,6 p53,7–8 and E2F.9 HDACi have stimulated much enthusiasm in oncology recently with over 500 cancer clinical trials initiated to date, resulting in two clinically approved drugs, SAHA (vorinostat, Figure 1b) and FK228 (romidepsin).10 Despite their success in blood malignancies, current HDACi have serious limitations in solid tumors. For instance, National Cancer Institute Trial 6862 of SAHA in men with advanced prostate cancer resulted in toxicities that required early termination of therapy for all patients.11 All new HDACi agents introduced into the clinic follow the same single-target paradigm. Their inability to significantly accumulate in solid tumors, combined with their rapid excretion and off-target toxicity, are significant contributors to their failure to translate into efficacy against solid tumors. Therefore, preclinical evaluation of new molecules in this class will need to focus on improving cell type selectivity and enhancing tumor tissue distribution.
Figure 1.

Design of dual-targeting AR-HDACi compounds. (a) The androgen receptor (AR) resides in the cytoplasm chaperoned by HSP90 in complex with dynein on microtubules, ready to transport to the nucleus upon ligand binding. Structures are shown of AR antagonists cyanonilutamide 1, nilutamide 2 and bicalutamide 3. (b) Histone acetyltransferase (HAT) and histone deacetylase (HDAC) alternatively attach or remove acetyl on lysine residues of histone side chains, remodeling of chromatin architecture and resulting in epigenetic gene regulation. HDAC activity can be disrupted by chemical probes such as SAHA (a prototypical HDACi). (c) Antiandrogen bicalutamide (green) and representative AR-HDACi compound (blue) docked into the ligand binding domain of an apo AR homology model. (d) AR-HDACi docked into the active site of HDAC2 with the targeting cap-group (blue) recognizing the surface at the entrance of the pocket, the linker (gray) traversing the tunnel to the catalytic zinc chelated by the hydroxamic acid zinc binding group (red). (e) A representative dual-targeting compound showing three pharmacophoric sections (targeting cap group, linker, and zinc binding group).
In order to address these needs in the context of prostate cancer (PCa), we have created HDACi that are designed to selectively accumulate into malignant prostate cells. To achieve this, we chose to incorporate into a prototypical HDACi pharmacophore a non-steroidal antiandrogen scaffold based on cyanonilutamide 1 (Figure 1a) which targets PCa via the androgen receptor (AR). The AR binds its natural agonist ligand dihydrotestosterone (DHT), causing conformational changes that initiate translocation to the nucleus. Once in the nucleus, transcription of genes is promoted via a coactivator protein assembly.8 Clinical success in treating PCa has been achieved by inhibiting this important growth axis via antiandrogens, especially the non-steroidal ligands nilutamide 2 and bicalutamide 3 (Figure 1a) because they do not have the broad off-target effects of steroidal analogues.12 PCa frequently advances to the much more lethal castration-resistant prostate cancer (CRPC), becoming resistant to these therapies by overexpressing ARs.13,14 Therapies designed to use AR for cancer cell delivery stand to benefit from the fact that expression levels of AR is about six-fold higher in castration resistant as compared to hormone-sensitive prostate cancer.15 Appending antiandrogen moieties are expected to endow HDACi with favorable tumor accumulation in vivo because antiandrogens exhibit PCa tumor to blood plasma ratios as high as 259 to 1.16 Many antiandrogens cause AR to localize to the nucleus (Figure 1a),17 and could therefore promote nuclear transport of AR-HDACi (Figure 1e).18
We have synthesized and screened a series of these dual-targeting compounds and showed that they 1) engage the AR and 2) inhibit histone deacetylase (HDAC) enzymes resulting in therapeutic impact. It is instructive to emphasize that these are not designed to hit both targets simultaneously, but rather are designed to engage the first target (AR), accumulate selectively, and then be released to engage the second target (HDAC). Indeed, our design hypothesis is supported by the data from these molecules which show binding to AR, potent inhibition of HDAC, and selective antiproliferative activity in AR dependent PCa cells.
RESULTS AND DISCUSSION
AR-HDACi Design and Synthesis
We used 1,2,3-triazole as a connection moiety between the targeting cap group and the linker group containing the zinc chelating hydroxamate (Figure 1e) as our previous studies have revealed that the triazole moiety enhanced HDACi activity.19 This design approach enabled the joining of the linker and the targeting cap group using the Cu(I)-catalyzed Huisgen cycloaddition between appropriate azides and terminal alkynes in the penultimate step of our synthesis (Scheme 1). We investigated the suitability of two different N3-modified hydantoins – aryl alkyne 7 and alkyl alkyne 8 – as head groups for the proposed bifunctional agents. Alkyl and aryl extensions from the N3 of the hydantoin ring have resulted in potent antiandrogens.20,21 Predicted binding modes suggested that modification through this position would not interfere with key interactions in the AR ligand binding domain (Figure 1c), allowing for the SAHA-like moiety to extend towards the exterior of the receptor. Indeed this has been the modification of choice for other dual-targeting approaches utilizing the anti-androgen nilutamide scaffolds with tubulin inhibitors (colchicine),22 DNA-intercalators (doxorubicin), 23 and nanoparticle delivery.24 These binding moieties are distinct from steroidal scaffolds (e.g. testosterone and DHT) which were the first to be utilized in bifunctional AR targeting approaches.25
Scheme 1.

Synthesis of Antiandrogen Equipped HDACi Compounds a
aReagents and conditions: (a) MsCl, TEA, THF, yield = 87%. (b) TsCl. yield = 95%. (c) NaH, THF, yield = 85% for 7, 89% for 8. (d) NaN3, 70°C, yield = 90–96%. (e) KOH, yield = quantitative. (f) EDC or TBTU coupling reagent, yield = 56–91%. (g) CuI, DIPEA, DMSO, argon, yield = 82–98%. (h) TFA:DCM (0.2:10), TIPS, yield = 60–90%.
The synthesis was carried out successfully to give the first two compounds (14c and 15c, linker length n = 5, Scheme 1), both of which exhibited nanomolar inhibition of HDAC (discussed below). Chain length is also a critical variable in optimizing the positioning of the zinc binding group (ZBG) for HDAC inhibition profiles. Therefore, the success of the n = 5 conjugates prompted us to explore the biological effects of chain-length variation, totaling six from each cap group, n = 3, 4, 5, 6, 7, and 8 (compounds 14a–b, 14d–e, 15a–b and 15d–e).
The synthesis of the alkyne cap groups (7 and 8, Scheme 1) was achieved with a straightforward alkylation of the cyanonilutamide 1 with mesylated (5) and tosylated (6) alkynes respectively. The series of azide-alkyl-O-trityl hydroximates 11a–f were obtained by coupling the azido acids with O-trityl hydroxylamine. The two pieces were then brought together using the regioselective Cu(I) catalyzed cycloaddition of an azide to a terminal alkyne, forming the 1,2,3 triazole ring.26, 27 The deprotection of the trityl group in trifluoroacetic acid (TFA)/triisopropyl silane (TIPS) mixture afforded the compounds.
HDAC Inhibition Study
The compounds’ enzyme inhibition activity was evaluated against HDAC isoforms 1, 6, and 8 and compared to SAHA (Figure 2). All compounds possess potent anti-HDAC activity with 14d showing superior activity relative to SAHA against all HDAC isoforms tested. Additionally, these AR-HDACi conjugates proved to be more selective for HDAC6. The aryl cap conjugates (14a–f) showed greater HDAC inhibition activity than the alkyl cap conjugates (15a–f). We observed that both conjugate sets showed greatest inhibition activity with a linker length of 5 – 6. This observation is in agreement with the previous structure activity relationship (SAR) studies which revealed that a hydrophobic spacer of 5–6 carbons is essential for optimal inhibitory activity of hydroxamate HDACi.28 Both phenyl and alkyl derivative with short linker lengths (less than n = 4) have decreased HDAC1 and HDAC6 inhibition, likely due to their inability to traverse the hydrophobic tunnel, leading to the enzyme active sites, for proper zinc ion chelation. To validate intracellular HDAC inhibition, we probed the acetylation status of α-tubulin (a cytoplasmic HDAC6 substrate) with Western blot analysis.29 Indeed, we found a dose dependent increase in acetylated tubulin (Figure 2b). Acetylation was more pronounced than SAHA for the most potent HDAC6 inhibitor 14d, agreeing with the cell-free HDACi assay.
Figure 2.

(a) Inhibition profile of AR-HDACi against HDAC isoforms 1, 6 and 8. (b) Intracellular HDAC inhibition of representative compounds probed in DU145 via acetylation of α-tubulin, a cytoplasmic HDAC6 substrate (note that SAHA and 15b were dosed at 10 μM).
AR Binding Affinity and Antagonist Activity
Androgen receptor binding affinity measured by competition with [3H] mibolerone (Table 1) revealed the benzyl head group (aryl nilutamide HDACi, 14a–f) to be 4-fold stronger (on average) than the alkyl head group (alkyl nilutamide HDACi, 15a–f).30 The extra entropic penalty associated with the increased number of rotatable bonds in the alkyl linker (as compared to the aryl) may be the cause of their decrease binding affinity. We were encouraged to find that every conjugate in the aryl series (14a–f) showed greater affinity than cyanonilutamide (1), bicalutamide (3) and enzalutamide. Although the alkyl series (15a–f) were generally less active, we saw a strong correlation between the linker lengths and potency, as increase in linker lengths caused increase AR binding affinity (Table 1). The increase in potency as the chain length increases from 15a to 15f trends closely (R2 = 0.864) with the increasing hydrophobicity (logP) of the alkyl series (Supplemental Table 1). Conversely, the aryl nilutamide derivatives showed no such trend (R2 = 0.0295, Supplemental Figure 1). The linker length-dependence of AR-binding could result from the alkyl linked 15a–f gaining affinity from increased hydrophobic contacts, whereas the peak of AR-binding affinity in the aryl nilutamide series when n = 6, 7 (compounds 14d and 14e) could be the consequence of optimal positioning of the distal hydroxamate to a set of hydrogen bond partners (tryptophan-741 and histidine-874, Figure 4d–e)
Table 1.
AR and sex hormone-binding globulin (SHBG) binding affinitya
| analogue | chain length | AR
|
SHBG | |
|---|---|---|---|---|
| IC50 (μM) | Ki (μM) | RBA (%) | ||
| 14a | n = 3 | 1.08 | 0.72 | 105 ± 16.3 |
| 14b | n = 4 | 1.00 | 0.67 | 104 ± 12.3 |
| 14c | n = 5 | 1.20 | 0.80 | 102 ± 12.9 |
| 14d | n = 6 | 0.69 | 0.46 | 107 ± 10.3 |
| 14e | n = 7 | 0.65 | 0.44 | 106 ± 9.1 |
| 14f | n = 8 | 1.59 | 1.06 | 102 ± 2.0 |
| 15a | n = 3 | 5.93 | 3.96 | 109 ± 4.7 |
| 15b | n = 4 | 6.73 | 4.48 | 92.0 ± 5.1 |
| 15c | n = 5 | 4.02 | 2.68 | 105 ± 3.8 |
| 15d | n = 6 | 3.91 | 2.60 | 104 ± 1.1 |
| 15e | n = 7 | 3.55 | 2.37 | 107 ± 3.4 |
| 15f | n = 8 | 1.43 | 0.96 | 106 ± 9.8 |
| cyanonilutamide (1) | 1.91 | 1.28 | 98.0 ± 4.6 | |
| bicalutamide (3) | 3.69 | 2.46 | NT | |
| enzalutamide | 2.03 | 1.35 | NT | |
| testosterone | 0.0047 | 0.0032 | 3.0 ± 0.9 | |
IC50 values from 4–5 concentrations tested in duplicate. SHBG assays were performed at 33 μM, and are an average of three independent experiments. RBA, relative binding affinity (% [3H]DHT that remains bound, relative to control); NT, not tested.
Figure 4.

(a) Crystal structure (PDB:2AM9) of testosterone (green) inducing an agonist conformation of helix-12 (H12) that enables coactivator binding, which when blocked results in antagonist conformations (b) with H12 replacing the coactivator binding surface. Small molecules able to recruit corepressor proteins (c) can actively silence AR genes, resulting in strong antagonist or inverse agonist activity. (d) Molecular modeling of antagonist AR-HDACi 14d (purple) shown as sticks with hydrogen bonds to tryptophan-741, histidine-874 and the main chain of kinked helix-4 (H4). Overlapped on 14d is the structure of testosterone (green). (e) 14d shown as spheres filling the cleft between H3, H4, H11 and H12 modeled in the antagonist position. (f) Inverse agonist 14b shown as spheres stabilizing corepressor peptide.
These results revealed that attachment of HDAC inhibition moieties to nilutamide antiandrogens did not abolish their interaction with AR, and even enhanced it.
Binding of small molecules to the AR may result in either agonist or antagonist activity. This is the result of differences in the protein surface topology induced by the small molecules. We therefore evaluated the effect of these AR-HDACi conjugates on AR transcriptional activity to decipher the consequence of their AR interaction on AR activity (Figure 3). For agonist activity, the ligand-induced conformation changes enable the recruitment of co-activator complexes required for DNA binding and transcriptional activity (Figure 4a) while the binding of these co-activator complexes is disrupted by small molecules with AR antagonist activity (Figure 4b).31
Figure 3.
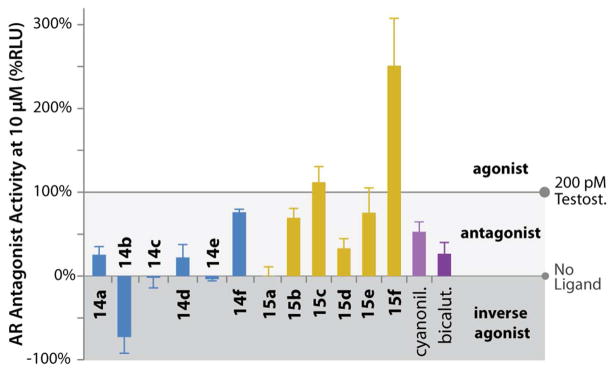
Antagonist activity (%RLU for 10 μM). All compounds competed against 200 pM testosterone.
To assess the androgenic or anti androgenic activity of the AR-HDACi conjugates, we evaluated transactivation of an AR response element upstream of a minimal promoter controlling a luciferase reporter gene in HEK 293T cells. When dosed at 10 μM, along with 200 pM testosterone, many of the AR-HDACi conjugates showed excellent AR antagonist activity. Compounds 14a, 14c–e and 15a were as much or more potent than bicalutamide, while 14b showed a surprising ability to reduce activity lower than basal levels, characteristic of an inverse agonist (Figure 3). Overall, the aryl-nilutamide derivatives (14a–f) were more potent antagonists than the alkyl-nilutamide conjugates (15a–f), correlating with the general trend seen with RBA (Table 1). Within each series, there is no linear correlation between linker lengths, RBA and antagonist activity; a result that is consistent with prior observations on non-steroidal antiandrogens.32 The increased potency of these compounds against AR is by itself a significant finding, regardless of their ability to function as HDACi.
Sex Hormone Binding Globulin Binding
Endogenous steroids that bind the androgen receptor (e.g. testosterone and DHT) are transported through the blood via sex hormone-binding globulin (SHBG) in humans.33 Because SHBG is not present in mice or rats, a lack of strong interaction with SHBG is important to ensure future use of those species for predictive absorption, distribution, metabolism, and excretion (ADME) analysis and preclinical in vivo cancer models. Therefore we screened the ability of all compounds to compete with the high affinity ligand [3H]DHT for SHBG (Table 1). Indeed, as expected from SHBG substrate requirements, none of the AR-HDACi conjugates showed appreciable binding to SHBG at concentrations as high as 33 μM. Thus, SHBG binding is expected not to play any role in the bioactivity of this class of compounds, and will not confound pharmacokinetics in either mice or humans.34
Molecular Modeling of Androgen Receptor Binding
Critical to biological activity of the steroid super family of nuclear receptors is the ligand-induced conformational changes of an otherwise floppy helix-12 (H12, at the C-terminus of the LBD). Crystal structures of androgens inducing an agonist conformation of the AR (Figure 4a) reveal H12 closing over the steroid binding pocket, creating a stable surface onto which coactivators can bind to initiate AR target genes. Although no crystal structures exist for the AR in antagonist forms, protein coordinates from homologous receptors (such as the estrogen receptor) bound to antagonists show H12 displaced (Supplemental Figure 2).35 Therefore, in order to understand the structural basis of AR antagonist activity, we performed molecular docking analyses of the AR-HDACi conjugates on an apo AR homology model that has H12 displaced from the normal agonist position (Figure 4b).36 Docked structures (such as representative 14d, Figure 4) show the cyano-nilutamide portion of the conjugates fitting into the binding pocket in a similar fashion as bicalutamide.
Almost all low energy conformations of the aryl-nilutamide series 14a–f had the benzyl-triazole portion fitting into the groove between H3 and tryptophan-741 (Figure 4d) that is otherwise occupied by H12 in agonist conformations. The alkyl linker of 14d filled the hydrophobic cavity shaped by tryptophan-741, leucine-712 and isoleucine-737 (Figure 4d), with the hydroxamic acid forming up to five hydrogen bonds with tryptophan-741, histidine-874 and the exposed amide backbone of H4 (glutamine-738). The HDACi portion of the molecule could therefore prevent agonist conformations of H12.
The unique ability of 14b to reduce the AR transcriptional activity lower than the basal level indicated an inverse agonist binding mode that could occur from the stable association of AR with corepressor proteins (Figure 4c, f),37 in a fashion similar to inverse agonist crystal structure of estrogen-related receptor-γ (ERRγ, PDB:2GPV)38 or retinoic acid receptor-α (RARα, PDB: 3KMZ). We therefore built a corepressor-bound model of the apo-AR complexed with corepressor peptide (CoRNR) extracted from ERα crystal structure (PDB: 2JFA). Using this template, the lowest energy conformation was computed for 14a–f (shown in Supplemental Figure 3). Only 14b preferentially bound to the small pocket between H3 and the corepressor (Figure 4f) while retaining hydrogen bonding to arginine-752 (Supplemental Figure 3b).
Nuclear Translocation of AR by AR-HDACi
Because the extent of AR nuclear localization is an important measure for antiandrogen compounds, we visualized and quantified the effect of the AR-HDACi conjugates on subcellular location of AR using yellow fluorescent protein tagged AR (YFP-AR).39–40 In the absence of ligands, AR remains primarily in the cytoplasm (DMSO vehicle, Figure 5a). The AR subcellular localization is not altered in the presence of normal HDACi (SAHA) as well. As expected, endogenous ligands (DHT, testosterone) induce strong nuclear localization of the AR (Figure 5a), a prerequisite for the initiation of the transcription of pro-growth genes which are harnessed by prostate cancers to achieve uncontrolled proliferation. Although classical antiandrogens such as bicalutamide are able to induce strong nuclear localization (Figures 5a and 5b) and DNA binding, they nevertheless block transcription of AR inducible genes by preventing recruitment of coactivators. This strong nuclear localization becomes a liability in some patients harboring a single amino acid substitution in the ligand binding domain which converts bicalutamide to an agonist.18 The more recently approved enzalutamide, although possessing AR binding affinity greater than bicalutamide, shows residual cytosolic AR localization (Figures 5a and 5b) attributed to the disruption of nuclear localization and impaired DNA binding.41 AR-HDACi with stronger AR binding affinity than bicalutamide or enzalutamide exhibit a range of AR nuclear localization both less than, equal to, or greater than enzalutamide (Figure 5b). Specifically, YFP-AR nuclear localization is induced to the same extend by both bicalutamide and 14d at either 1μM or 10μM (Supplemental Figure 4). This is consistent with the binding of the AR-HDACi to the AR ligand binding domain such that their methylene hydroxamate side-chain impedes helix-12 closure (Figure 4b) and subsequent assembly of transcriptional coregulators to different extents.
Figure 5.
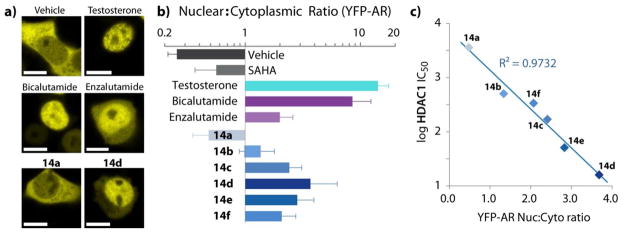
(a) Confocal images of YFP-AR translocation to the nucleus. YFP-AR was transfected into HEK-293T cells, dosed for 4 h and imaged live. Scale bars are 10 μm. (b) Ratio of YFP-AR fluorescence within the nucleus versus the cytoplasm. Values are the average of image pixel quantification from at least 4 cells ± SD. Testosterone was dosed at 1 μM, all other compounds at 10 μM. (c) HDAC1 inhibition activity versus YFP-AR nuclear to cytoplasmic localization ratio for aryl nilutamide conjugates.
Interestingly, among AR-HDACi conjugates with AR binding affinity stronger than bicalutamide or enzalutamide (14a–f) there is a strong linear correlation (R2 = 0.9732) between HDAC1 inhibition activity and the extent to which these conjugates induce nuclear localization of YFP-AR (Figure 5c). The observed correlation between AR binding affinity and HDAC1 inhibition could simply be due to the enhanced drug exposure to the cell nucleus localized HDAC1, the concomitant effect of AR-drug complex translocation into the nucleus. Alternatively, because the acetylation of histones flanking the AR response element promoter regions is critical for the assembly of AR transcriptional complexes,42 inhibiting HDAC1 activity (increasing histone acetylation) should promote AR complex formation, increasing both the extent and duration of AR-drug complex occupancy in the nucleus. In this scenario, the ratio of AR localized in the nucleus could be a function of HDAC1 activity. Cytosolic HDAC inhibition is unlikely to cause this observation, because HDAC6 knock down experiments have resulted in an opposite effect on AR nuclear localization.43
Indeed, it is exciting to find a quantitative correlation between HDAC inhibition and accumulation of AR in the nucleus. AR-dependent nuclear localization may contribute to improved inhibition of HDAC in the nucleus of AR containing cells, which we anticipate to result in cell-type selective antiproliferative effects.
Whole Cell Antiproliferative Activity
To investigate cell-type selectivity and potency, we evaluated the anti-proliferative activity of these conjugates in both AR-expressing LNCaP (hormone dependent prostate cancer) and AR-negative DU145 (hormone refractory, metastatic prostate cancer) (Table 2).
Table 2.
Whole cell anti-proliferative activity IC50 (μM) against prostate cancer cell lines.
| analogue | LNCaP (AR+) | DU-145 (AR−) |
|---|---|---|
| 14a | 14.4 ± 1.0 | >40b |
| 14b | 3.7 ± 0.9 | 14.1 ± 0.9 |
| 14c | 21.4 ± 10.3 | 7.4 ± 0.5 |
| 14d | 1.1 ± 0.4 | 1.0 ± 0.4 |
| 14e | 3.3 ± 1.3 | 10.5 ± 1.6 |
| 14f | 3.3 ± 1.0 | 30.5 ± 3.6 |
| 15a | 5.6 ± 0.4 | 35.9 ± 0.9 |
| 15b | 14.5 ± 1.4 | >40b |
| 15c | 10.7 ± 2.5 | >40b |
| 15d | 3.8 ± 1.5 | 13.2 ± 1.0 |
| 15e | 8.8 ± 1.6 | 11.8 ± 1.1 |
| 15f | 2.8 ± 0.2 | 5.0 ± 1.2 |
| SAHA | 1.1 ± 0.3 | 2.0 ± 0.8 |
| bicalutamide | >80b | >80b |
| enzalutamide | 42.6 ± 6.0 | >80b |
IC50 values are an average of at least two independent experiments, ± SD.
IC50 not determinable up to highest concentrations tested.
As expected, enzalutamide shows preferential toxicity to AR dependent LNCaP while SAHA does not show any pronounced AR dependency in its cytotoxicity to either cell lines. Gratifyingly, we observed that most conjugates (11 out of 13) are generally more cytotoxic against AR dependent LNCaP (Table 2 and Figure 6), as compared to the AR independent DU145. This strongly suggests that the secondary ability to bind AR has indeed conferred cell-type selectivity to these AR-HDACi conjugates. The most active compounds in each series (14d and 15d) are between 40–80-fold and 10–20-fold more active relative to the standard antiandrogens bicalutamide and enzalutamide respectively. The enhanced activity of HDACi (both SAHA and the new compounds presented here) over standard antiandrogens is retained in both the androgen-induced growth of LNCaP and the androgen-independent growth of DU145 (where bicalutamide has been previously shown to have no activity).41,44 It should be noted that reported LNCaP IC50 values for enzalutamide and bicalutamide are highly variable, and our results are at the high end of this range.44–46
Figure 6.

Box plot of all AR-HDACi conjugate anti-proliferative activity in LNCaP (AR+) and DU145 (AR−) cells.
A closer analysis of the cell growth inhibition data in Table 2 revealed that anti-proliferative activity against DU145 generally increases with the strength of the compounds’ HDAC1 inhibition activity (Supplemental Figure 5d). Against LNCaP, HDAC8 inhibition activity has the strongest correlation with inhibition of cell growth, especially among the alkyl nilutamide HDACi 15a–f (Supplemental Figure 5f). Compound 14c, the only drug with greater growth inhibition activity against DU145 relative to LNCaP is interestingly the weakest HDAC8 inhibitor of the entire series, a sensitivity that could be explored further.
Additional cell types (Supplemental Table 2) were screened with compounds 14d (the most potent compound against HDAC1, HDAC6, LNCaP and DU145) and 15d (best HDAC1 inhibitor with the alkyl cap group and more selective for LNCaP over DU145). The broad potency of 14d (attributed primarily to its strong HDAC activity) is similar to that of SAHA across other cancer cell lines, whereas 15d shows potency only against AR dependent cell line LNCaP. Excitingly, we found that against non-cancerous VERO cells, 14d is 10-fold less toxic than parent SAHA (Figure 7).
Figure 7.
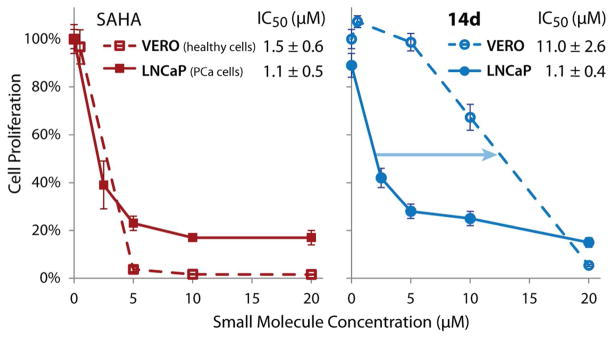
Dose response curves for SAHA and 14d in both LNCaP (AR+ prostate cancer) and VERO (healthy kidney cells).
Conclusion
Combining HDAC inhibition and antiandrogen therapy causes synergistic prostate cancer cell death in vitro (in AR expressing cells).47 This observation has provided the rationale for an ongoing phase I/II trial of LBH589 (HDACi panobinostat) combined with bicalutamide in castration-resistant PCa (ClinicalTrials.gov identifier: NCT00878436). However, given the prior inability of very promising preclinical data to translate into efficacy against PCa in patients, presumably due to lack of solid tumor accumulation of HDACi, the outcome of this effort remains uncertain. Moreover, two different drugs have inherently different adsorption, biodistribution, metabolism and excretion profiles. This can prohibit dosing strategies aimed at achieving synergies dependent on drugs being simultaneously present within a cancer cell. In contrast, combining both properties within a single molecule can circumvent difficult pharmacokinetics by enhancing tumor accumulation and promoting co-localization of both activities. The ability of these AR-HDACi conjugates to antagonize AR and inhibit HDAC could confer an additive or a synergistic antiproliferative activity to a single molecule.
In summary, we have shown, using multiple techniques, that the AR-HDACi conjugates described herein utilize HDACi activity for potency, with their secondary activity (binding the AR) causing cell type selectivity. AR promoted HDACi delivery is an attractive strategy for selective therapy of all forms of prostate cancer. This is premised on two main reasons – (i) the biological function of the AR ligand-conjugates are independent of the anti-androgen activity of the appended AR ligand, as orthogonal HDAC inhibition will induce cell death upon AR enhanced cellular accumulation, and (ii) the AR will allow for very effective targeting, especially of those CRPC driven by AR overexpression, the more common and aggressive phenotype that is resistant to hormone treatment.13 Other CRPC mutations, including those driven by the expression of AR splice variants missing LBD, which are still dependent on wild-type AR for their prostate tumor growth promoting effects,48–49 could be targeted by these AR-HDACi conjugates. Moreover, the membrane AR or testosterone sensing GPRC6A may also facilitate active uptake into prostate tumor tissue. Indeed, we have shown that antiandrogens appropriately linked to nanoparticles can facilitate cell uptake even in castration resistant DU145 PCa cells, which although they have a silenced AR, over express GPRC6A.24 Our ongoing efforts are on the investigations of the in vivo biodistribution, antitumor effects, and further SAR studies of the lead compounds.
METHODS
Chemicals
Bicalutamide and testosterone were a kind gift from Dr. Shafiq Khan (Clark Atlanta University, Atlanta, GA). Enzalutamide was purchased from Selleckchem. All other chemicals (including SAHA) were purchased from Sigma Aldrich. Anhydrous solvents and other reagents were purchased and used without further purification. For experiments, 10 mM stocks of the compounds were dissolved in DMSO and stored at −20°C.
HDAC Activity Assay
In vitro HDAC inhibition was assayed through a contract agreement with BPS Bioscience (www.bpsbioscience.com). All of the compounds were dissolved in DMSO. A series of dilutions of the compounds were prepared with 10% DMSO in HDAC assay buffer and 5 μl of the dilution was added to a 50 μl reaction so that the final concentration of DMSO is 1% in all of reactions. The enzymatic reactions were conducted in duplicate at 37 °C for 30 min in a 50 μl mixture containing HDAC assay buffer, 5 μg BSA, HDAC substrate, HDAC enzyme (human recombinant HDAC1, HDAC6 or HDAC8) and various concentrations of each compound. After enzymatic reactions, 50 μl of 2x HDAC Developer was added to each well and the plate was incubated at room temperature for an additional 15 min. Fluorescence intensity was measured at an excitation of 360 nm and an emission of 460 nm using a Biotek Synergy microplate reader.
HDAC Activity Data Analysis
The fluorescent intensity data were analyzed using the computer software, Graphpad Prism. In the absence of the compound, the fluorescent intensity (Ft) in each data set was defined as 100% activity. In the absence of HDAC, the fluorescent intensity (Fb) in each data set was defined as 0% activity. The percent activity in the presence of each compound was calculated according to the following equation: %activity = (F−Fb)/(Ft−Fb), where F= the fluorescent intensity in the presence of the compound. The values of % activity versus a series of compound concentrations were then plotted using non-linear regression analysis of Sigmoidal dose-response curve generated with the equation Y=B+(T−B)/1+10((LogEC50-X)×Hill Slope), where Y=percent activity, B=minimum percent activity, T=maximum percent activity, X= logarithm of compound and Hill Slope=slope factor or Hill coefficient. The IC50 value was determined by the concentration causing a half-maximal percent activity.
Western blot analysis
Total cellular proteins were prepared and analyzed by western blot as described previously.50 Briefly, cell lysates were mixed with Laemmli buffer. Individual samples (30–35 μg proteins) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis in 8 or 10% gels and transferred to polyvinylidene difluoride membranes (Millipore). After blocking with 5% fat-free milk in Tris-Buffered Saline Tween (TBST) for 1 h at room temperature, the membranes were incubated with appropriate dilutions of specific primary antibodies (1:250 dilution for acetylated α-tubulin antibody) overnight at 4°C. After washing, the blots were incubated with anti-rabbit or anti-mouse IgG-HRPs for 1 h. The blots were developed in enhanced chemiluminescence mixture (Thermo Fisher Scientific, Rockford, IL) and the density of specific protein bands were determined by QuantityOne image analysis software.
AR Ligand Binding Affinity
Radioligand binding (Eurofins Panlabs) was performed using rat androgen receptor and [3H]mibolerone (PanVera) in buffer (50 mM Tris-HCl, 0.8 M NaCl, 10% Glycerol, 2 mM Dithiothreitol, 0.1% BSA, 2% EtOH, pH 7.4). 78 ng of AR was incubated with 1.5 nM [3H]mibolerone for 4 h at 4 °C, then incubated with a hydroxyapatite slurry over 15 minutes and filtered. The filters are washed 3 times and counted to determine [3H]mibolerone specifically bound. Compounds were tested in a logarithmic concentration range from 10 nM to 100 μM to determine Ki and IC50 values.
SHBG Steroid-Binding Capacity Assay
A ligand saturation analysis method51 was used to determine the relative steroid binding capacity of SHBG to its natural ligands in the presence of the novel antiandrogenic compounds. Briefly, serum samples from a woman treated with ethinyl estradiol were stripped of their existing steroids with dextran-coated charcoal (DCC) prior to incubation with [3H]DHT (specific activity 119.2 Ci mmol−1; PerkinElmer) and the antiandrogen of interest. Samples were prepared and analyzed both in the presence or absence of 100 fold molar excess of unlabelled DHT to account for non-specific binding. Free steroids, unbound to serum SHBG were removed using DCC as a separation reagent. The remaining tritiated ligands bound to SHBG were measured by liquid scintillation analysis.
Luciferase assay cell lines and plasmids
The human embryonic kidney cell line, HEK 293T, was purchased from ATCC and cultured in DMEM with 10% fetal bovine serum (FBS). The pReceiverAR vector was purchased from GeneCopoeia. The reporter plasmid, pARLuc, was purchased from Signosis and contains firefly luciferase downstream of AR response elements. pCMXβGal was used to express β-galactosidase as an internal control, and to assess transfection efficiency.
Transfection and Luciferase Assay for AR activity
Cells were cultured in phenol red-free DMEM (MediaTech) with 10% charcoal dextran treated FBS (Atlanta Biologicals) to an approximate confluence of 60–70% in 48-well plates. Cells were then transiently transfected with 100 ng of DNA (20:40:40; pReceiverAR:pARLuc:pCMXβGal) per well as previously described.52 Following 8 h incubation at 37 °C, media was aspirated from cells, and the cells were then dosed with phenol-red free DMEM with 10% charcoal stripped FBS medium with 10 μM AR-HDACi conjugate. For antagonism assays, 200 pM testosterone was added to all wells in addition to antagonist ligand. Cell lysates were tested for luciferase and beta-galactosidase after 40 h incubation at 37 °C using a Berthold TriStar2 plate reader, as previously described.53 Data reported represent the average of three sets luciferase assays, each performed in triplicate, normalized to β-galactosidase activity.
Molecular Docking Analysis
In silico docking was performed using Autodock Vina 54 run through PyRx to manage the workflow and PyMol to visualize the results. Ligands were prepared by first generating a3D structure in ChemBioDraw3D, followed by processing with Autodock Tools 1.5.4 to assign Gasteiger charges, merging non-polar hydrogens and set torsional bonds. Docking runs were performed within a 25–30 Å cubic search space surrounding the binding pocketand output modes were ranked according to binding affinity (BA). Autodock Vina identified molecular conformations with the best fit and strongest BA (global minimums) by a stochastic algorithm exploring surfaces/pockets of the rigid macromolecule, through an iterative series of local optimizations (changing shape, bond angles, and position of the ligand), evaluating both intermolecular (hydrophobic interactions, repulsions, hydrogen bonding, etc.) and intramolecular (torsion, rotational torque) factors.
YFP-AR Transfection and Confocal Microscopy
HEK-293T cells were grown in DMEM with 10% FBS. Cells were transferred to Nunc® Lab-Tek™ II Chamber Slide™ System (prepared with collagen matrix) using DMEM with 10% charcoal stripped FBS (CSS), 24 h prior to transfection. Transfection mix was prepared by mixing Lipofectamine with YFP-AR55 plasmid DNA and Optimem (according to manufacturer’s instructions), and incubated with cells for 8 h. Afterward the media was changed back to DMEM (10% CSS), incubated overnight. A 20 min treatment with NucBlue® Live ReadyProbes™ Reagent (Hoechst) was performed, followed by dosing with controls and compounds for 4 h. Images were taken using a Zeiss LSM 700–405 Confocal Microscope (IBB Core Facilities, Georgia Institute of Technology, Atlanta GA).
Cell Culture and Cell Viability Assay
The human breast cancer cell line MCF-7 was a generous gift from Dr. Al Merrill’s laboratory (Georgia Institute of Technology, GA). MDA-MB-231, DU145 and VERO cells were obtained from ATCC. Cell lines are verified by ATCC and only used while passage numbers are low (<25). Cells were routinely cultured in phenol-red free RPMI (Invitrogen) with 10% fetal bovine serum (FBS) (Hycone) with antibiotics. All cell cultures were incubated at 37 °C under a 5% CO2 atmosphere. For all experiments, cells were grown in 96-well cell culture treated microtiter plates (Techno Plastic Products AG) with the appropriate ligand in duplicate for 72 h. MTS and MTT colorimetric tests (CellTiter 96 Aqueous One Solution and CellTiter 96 Non-Radioactive Cell Proliferation Assays, Promega) were employed to determine cell viability per manufacturer instructions. Logit plot analysis was used to determine the IC50 values for each compound.
Supplementary Material
Acknowledgments
We are grateful to A. C. Millena for assistance with cell culture work. We thank M. Diamond for the kind gift of YFP-AR plasmid. We appreciate G. Hammond for helpful discussions on SHBG binding.
Funding Sources
This work was financially supported by National Institutes of Health grant R01CA131217 (AKO); G12MD007590, 5P20MD002285 (SK); by the Georgia Research Alliance grant GRA.VL13.B11 (AKO) and by the Department of Defense (DOD) W8I-08-1-0077 (SK). BEG, MJA and MKR are thankful recipients of the GAANN predoctoral fellowship from the Georgia Tech Center for Drug Design, Development, and Delivery.
ABBREVIATIONS
- HDAC
Histone deacetylase
- HAT
histone acetyltransferase
- HDACi
histone deacetylase inhibitor
- AR
androgen receptor
- YFP-AR
yellow fluorescent protein tagged androgen receptor
- PCa
prostate cancer
- ZBG
zinc binding group
- SAR
structure-activity relationship
- SHBG
sex hormone binding globulin
- AR-HDACi
androgen receptor targeted histone deacetylase inhibitor
Footnotes
Supporting Information. Predicted ADME properties; trends in biological activities; YFP-AR confocal images; details of compound synthesis and characterization; 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry AS, Watson RWG, Lawler M, Hollywood D. The epigenome as a therapeutic target in prostate cancer. Nat Rev Urol. 2010;7:668–680. doi: 10.1038/nrurol.2010.185. [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 5.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 6.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 8.Brochier C, Dennis G, Rivieccio MA, McLaughlin K, Coppola G, Ratan RR, Langley B. Specific Acetylation of p53 by HDAC Inhibition Prevents DNA Damage-Induced Apoptosis in Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:8621–32. doi: 10.1523/JNEUROSCI.5214-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marzio G, Wagener C, Gutierrez MI, Cartwright P, Helin K, Giacca M. E2F family members are differentially regulated by reversible acetylation. J Biol Chem. 2000;275:10887–10892. doi: 10.1074/jbc.275.15.10887. [DOI] [PubMed] [Google Scholar]
- 10.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Medicinal Chemistry. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, Pili R, Zwiebel J, Scher H, Hussain M. Vorinostat in Advanced Prostate Cancer Patients Progressing on Prior Chemotherapy (National Cancer Institute Trial 6862) Trial Results and Interleukin-6 Analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer. 2009;115:5541–5549. doi: 10.1002/cncr.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS, Investigators A. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. New England Journal of Medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 13.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nature Medicine. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 14.Papatsoris AG, Karamouzis MV, Papavassiliou AG. Novel biological agents for the treatment of hormone-refractory prostate cancer (HRPC) Current Medicinal Chemistry. 2005;12:277–296. doi: 10.2174/0929867053363306. [DOI] [PubMed] [Google Scholar]
- 15.Linja MJ, Savinainen KJ, Saramaki OR, Tammela TLJ, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 16.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y, Grillot K, Bischoff ED, Cal L, Aparicio A, Dorow S, Arora V, Shao G, Qian J, Zhao H, Yang GB, Cao CY, Sensintaffar J, Wasielewska T, Herbert MR, Bonnefous C, Darimont B, Scher HI, Smith-Jones P, Klang M, Smith ND, De Stanchina E, Wu N, Ouerfelli O, Rix PJ, Heyman RA, Jung ME, Sawyers CL, Hager JH. ARN-509: A Novel Antiandrogen for Prostate Cancer Treatment. Cancer Res. 2012;72:1494–1503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones JO, An WF, Diamond MI. AR Inhibitors Identified by High-Throughput Microscopy Detection of Conformational Change and Subcellular Localization. ACS Chem Biol. 2009;4:199–208. doi: 10.1021/cb900024z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–991. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen PC, Patil V, Guerrant W, Green P, Oyelere AK. Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorganic & Medicinal Chemistry. 2008;16:4839–4853. doi: 10.1016/j.bmc.2008.03.050. [DOI] [PubMed] [Google Scholar]
- 20.Van Dort ME, Jung YW. Synthesis and structure-activity studies of side-chain derivatized arylhydantoins for investigation as androgen receptor radioligands. Bioorg Med Chem Lett. 2001;11:1045–1047. doi: 10.1016/s0960-894x(01)00146-9. [DOI] [PubMed] [Google Scholar]
- 21.Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, Wongvipat J. Structure-Activity Relationship for Thiohydantoin Androgen Receptor Antagonists for Castration-Resistant Prostate Cancer (CRPC) J Med Chem. 2010;53:2779–2796. doi: 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharifi N, Hamel E, Lill MA, Risbood P, Kane CT, Hossain MT, Jones A, Dalton JT, Farrar WL. A bifunctional colchicinoid that binds to the androgen receptor. Mol Cancer Ther. 2007;6:2328–2336. doi: 10.1158/1535-7163.MCT-07-0163. [DOI] [PubMed] [Google Scholar]
- 23.Cogan PS, Koch TH. Rational design and synthesis of androgen receptor-targeted nonsteroidal anti-androgen ligands for the tumor-specific delivery of a doxorubicin-formaldehyde conjugate. J Med Chem. 2003;46:5258–5270. doi: 10.1021/jm0303305. [DOI] [PubMed] [Google Scholar]
- 24.Dreaden EC, Gryder BE, Austin LA, Tene Defo BA, Hayden SC, Pi M, Quarles LD, Oyelere AK, El-Sayed MA. Antiandrogen Gold Nanoparticles Dual-Target and Overcome Treatment Resistance in Hormone-Insensitive Prostate Cancer Cells. Bioconjugate Chem. 2012;23:1507–1512. doi: 10.1021/bc300158k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Development of protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular & Cellular Proteomics. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. Journal of the American Chemical Society. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 27.Bock VD, Hiemstra H, van Maarseveen JH. Cu-I-catalyzed alkyne-azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur J Org Chem. 2006:51–68. [Google Scholar]
- 28.Drummond DC, Noble CO, Kirpotin DB, Guo ZX, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annual Review of Pharmacology and Toxicology. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 29.Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A Novel Class of Small Molecule Inhibitors of HDAC6. ACS Chem Biol. 2012;7:330–338. doi: 10.1021/cb200134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.All radioactive AR binding tests were performed by Ricerca, LLC.
- 31.Hur E, Pfaff SJ, Payne ES, Grøn H, Buehrer BM, Fletterick RJ. Recognition and Accommodation at the Androgen Receptor Coactivator Binding Interface. PLoS Biol. 2004;2:e274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakeling AE, Furr BJA, Glen AT, Hughes LR. Receptor binding and biological activity of steroidal and nonsteroidal antiandrogens. Journal of Steroid Biochemistry. 1981;15:355–359. doi: 10.1016/0022-4731(81)90297-1. [DOI] [PubMed] [Google Scholar]
- 33.Avvakumov GV, Cherkasov A, Muller YA, Hammond GL. Structural analyses of sex hormone-binding globulin reveal novel ligands and function. Molecular and Cellular Endocrinology. 2010;316:13–23. doi: 10.1016/j.mce.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Bohl CE, Nair VA, Mustafa SM, Hong SS, Miller DD, Dalton JT. Preclinical pharmacology of a nonsteroidal ligand for androgen receptor-mediated imaging of prostate cancer. J Pharmacol Exp Ther. 2006;317:402–408. doi: 10.1124/jpet.105.094334. [DOI] [PubMed] [Google Scholar]
- 35.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, Liu B, Geng G, Wu JH. Study of the impact of the T877A mutation on ligand-induced helix-12 positioning of the androgen receptor resulted in design and synthesis of novel antiandrogens. Proteins: Structure, Function, and Bioinformatics. 2010;78:623–637. doi: 10.1002/prot.22592. [DOI] [PubMed] [Google Scholar]
- 37.Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson JA, Pike ACW. Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J Biol Chem. 2007;282:10449–10455. doi: 10.1074/jbc.M611424200. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Zuercher WJ, Consler TG, Lambert MH, Miller AB, Orband-Miller LA, McKee DD, Willson TM, Nolte RT. X-ray Crystal Structures of the Estrogen-related Receptor-γ Ligand Binding Domain in Three Functional States Reveal the Molecular Basis of Small Molecule Regulation. J Biol Chem. 2006;281:37773–37781. doi: 10.1074/jbc.M608410200. [DOI] [PubMed] [Google Scholar]
- 39.Levine PM, Lee E, Greenfield A, Bonneau R, Logan SK, Garabedian MJ, Kirshenbaum K. Androgen Receptor Antagonism by Divalent Ethisterone Conjugates in Castrate-Resistant Prostate Cancer Cells. ACS Chem Biol. 2012;7:1693–1701. doi: 10.1021/cb300332w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen HC, Shanmugasundaram K, Simon NI, Cai CM, Wang HY, Chen S, Balk SP, Rigby AC. In Silico Discovery of Androgen Receptor Antagonists with Activity in Castration Resistant Prostate Cancer. Mol Endocrinol. 2012;26:1836–1846. doi: 10.1210/me.2012-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia L, Berman BP, Jariwala U, Yan X, Cogan JP, Walters A, Chen T, Buchanan G, Frenkel B, Coetzee GA. Genomic Androgen Receptor-Occupied Regions with Different Functions, Defined by Histone Acetylation, Coregulators and Transcriptional Capacity. PLoS ONE. 2008;3:e3645. doi: 10.1371/journal.pone.0003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, Wang Z. HDAC6 Regulates Androgen Receptor Hypersensitivity and Nuclear Localization via Modulating Hsp90 Acetylation in Castration-Resistant Prostate Cancer. Mol Endocrinol. 2009;23:1963–1972. doi: 10.1210/me.2009-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreis W, Budman DR, Calabro A. A reexamination of PSC 833 (Valspodar) as a cytotoxic agent and in combination with anticancer agents. Cancer Chemother Pharmacol. 2001;47:78–82. doi: 10.1007/s002800000191. [DOI] [PubMed] [Google Scholar]
- 45.Kuruma H, Matsumoto H, Shiota M, Bishop J, Lamoureux F, Thomas C, Briere D, Los G, Gleave M, Fanjul A, Zoubeidi A. A Novel Antiandrogen, Compound 30, Suppresses Castration-Resistant and MDV3100-Resistant Prostate Cancer Growth In Vitro and In Vivo. Mol Cancer Ther. 2013;12:567–576. doi: 10.1158/1535-7163.MCT-12-0798. [DOI] [PubMed] [Google Scholar]
- 46.Danquah M, Duke CB, Patil R, Miller DD, Mahato RI. Combination Therapy of Antiandrogen and XIAP Inhibitor for Treating Advanced Prostate Cancer. Pharm Res. 2012;29:2079–2091. doi: 10.1007/s11095-012-0737-1. [DOI] [PubMed] [Google Scholar]
- 47.Marrocco DL, Tilley WD, Bianco-Miotto T, Evdokiou A, Scher HI, Rifkind RA, Marks PA, Richon VM, Butler LM. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol Cancer Ther. 2007;6:51–60. doi: 10.1158/1535-7163.MCT-06-0144. [DOI] [PubMed] [Google Scholar]
- 48.Attar RM, Takimoto CH, Gottardis MM. Castration-Resistant Prostate Cancer: Locking Up the Molecular Escape Routes. Clin Cancer Res. 2009;15:3251–3255. doi: 10.1158/1078-0432.CCR-08-1171. [DOI] [PubMed] [Google Scholar]
- 49.Watson PA, Chen YNF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vo BT, Cody B, Cao Y, Khan SA. Differential role of SloanKettering Institute (Ski) protein in Nodal and transforming growth factor-beta (TGF-beta)-induced Smad signaling in prostate cancer cells. Carcinogenesis. 2012;33:2054–2064. doi: 10.1093/carcin/bgs252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammond GL, Lahteenmaki PLA. A versatile method for the determination of serum cortisol binding globulin and sex hormone binding globulin binding capacities. Clin Chim Acta. 1983;132:101–110. doi: 10.1016/0009-8981(83)90237-1. [DOI] [PubMed] [Google Scholar]
- 52.Taylor J, Rohatgi P, Spencer HT, Doyle D, Azizi B. Characterization of a molecular switch system that regulates gene expression in mammalian cells through a small molecule. BMC Biotechnology. 2010;10:15. doi: 10.1186/1472-6750-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gryder BE, Rood MK, Johnson KA, Patil V, Raftery ED, Yao L-PD, Rice M, Azizi B, Doyle D, Oyelere AK. Histone Deacetylase Inhibitors Equipped with Estrogen Receptor Modulation Activity. J Med Chem. 2013 doi: 10.1021/jm400467w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaufele F, Carbonell X, Guerbadot M, Borngraeber S, Chapman MS, Ma AAK, Miner JN, Diamond MI. The structural basis of androgen receptor activation: Intramolecular and intermolecular amino–carboxy interactions. Proc Natl Acad Sci U S A. 2005;102:9802–9807. doi: 10.1073/pnas.0408819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.