

Summary
Nine years ago we published a manuscript that suggested a specialized role for calcineurin-nuclear factor of activated T-cells (NFAT) signaling in regulating pathologic cardiac hypertrophy preferentially over physiologic growth, and in fact, the later response was associated with reduced calcineurin-NFAT activity. Since this time we and others have continued to uncover how this signaling effector pathway functions in the heart in regulating specific aspects of the growth response during disease and with exercise.
Keywords: Hypertrophy, signaling, calcineurin, transcription
Pathologic cardiac hypertrophy is associated with neuroendocrine dysregulation after myocardial infarction injury or in response to long-standing hypertension, as well as in response to a myriad of other disease causing stimuli, while physiologic hypertrophy of the heart occurs with intense exercise training 1,2. Although the heart enlarges under both pathological and physiological stimuli, pathologic hypertrophy causes negative remodeling of the ventricles and often extreme concentric or eccentric hypertrophy whereas exercise typically produces a mild 10-20% growth of the ventricles in a uniform manner 1,2. The molecular pathways that mediate these 2 responses, as well as the downstream effectors and associated adaptations of the heart, are different. In the broadest sense, physiologic cardiac hypertrophy appears to be proximally mediated by insulin-like growth factor-1 (IGF-1) signaling through phosphoinositide 3-kinase (PI3K) and Akt and mammalian target of rapamycin (mTOR). Although other minor effectors/pathways can also have specific role in regulating aspects of physiologic cardiac hypertrophy. For a more comprehensive review of physiologic cardiac hypertrophy and the role of PI3K-Akt and signaling effectors please see the recent review by Maillet and colleagues 1. In contrast to physiologic cardiac hypertrophy, a much larger array of specialized signaling pathways, such as calcineurin-NFAT, appear to underlie pathologic cardiac hypertrophy and negative remodeling of the ventricles (Figure 1) 1,2.
Figure 1. Calcineurin/NFAT signaling participates in pathological hypertrophy but not physiologic hypertrophy.
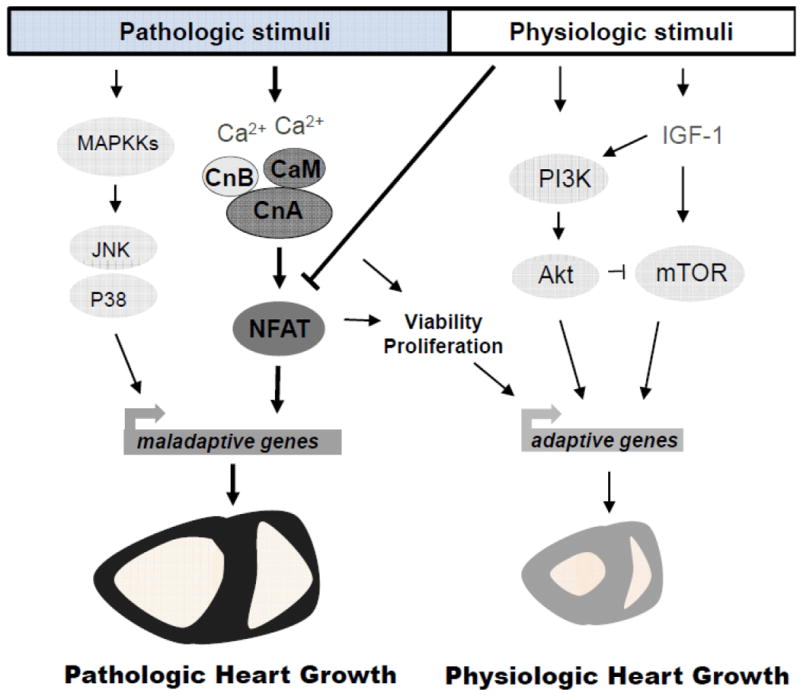
While physiologic cardiac hypertrophy can be induced by various stimuli such as insulin-like growth factor 1 (IGF-1), which activates phosphoinositide 3-kinase (PI3K), Akt and the mammalian target of rapamycin (mTOR) signaling effectors, pathologic hypertrophy can be triggered by the activation of the mitogen-activated protein kinases (MAPK) c-Jun N-terminal kinase (JNK) or p38 kinase and the Ca2+-calmodulin-activated phosphatase calcineurin. Sustained Ca2+ elevations leads to calcineurin activation as a heterotrimeric complex composed of calcineurin B (CnB), calmodulin (CaM) and the catalytic subunit calcineurin A (CnA). Calcineurin then dephosphorylates the nuclear factor of activated T-cells (NFAT) transcription factor in the cytosol allowing its translocation to the nucleus and induction of genes underlying pathologic cardiac hypertrophy. However, more recent genetic approaches in the mouse also show some adaptive or physiologic functions of calcineurin in permitting myocyte proliferation and protection from cell death. Finally, physiologic exercise stimulation down-regulates calcineurin/NFAT signaling, coincident with a lack of fetal gene induction despite mild hypertrophic growth of the heart.
That divergent signaling pathways can underlie pathologic versus physiologic cardiac hypertrophy was suggested by Wilkins et al. when they observed that exercise-induced growth occurred in the absence of induction of the fetal gene program classically associated with the development of pathological hypertrophy 3. The pathology-induced fetal gene program includes induction of mRNA for atrial natriuretic factor (ANF), b-type natriuretic peptide (BNP), skeletal α-actin, and β-myosin heavy chain (β-MHC) 1,2. Wilkins et al. showed that these pathologic marker genes were induced by transverse aortic constriction (TAC) in mice, but not after swimming exercise or growth hormone/IGF-1 injections 3. Indeed, Leinwand and colleagues also showed that exercise-induced physiologic remodeling of the heart was associated with downregulation of the pathologic fetal gene program and suppression of NFAT activity 4. These very simple observations suggested that physiologic growth of the myocardium utilizes specialized regulatory pathways and signaling circuits compared with many of the disease causing pathways that are associated with induction of the fetal gene program. For example, signaling through calcineurin-NFAT leads to induction of BNP and β-MHC expression in cardiac myocytes, which may be a direct effect since NFAT has been shown to bind the promoters of these 2 genes 5,6.
The Ca2+-dependent serine-threonine protein phosphatase calcineurin was identified as a central prohypertrophic signaling effector in the myocardium by us approximately 15 years ago 5. Calcineurin is the target of two widely used immunosuppressant drugs, cyclosporin A (CsA) and FK506, which exert their effect by directly inhibiting calcineurin through a complex with immunophilin/cyclophilin proteins 7,8. Calcineurin is an obligate dimer of a 57-61 kDa catalytic subunit (CnA) and a 19 kDa regulatory subunit (CnB) that becomes activated by sustained elevations in intracellular Ca2+ in association with direct binding of calmodulin 7,8. There are three genes that encode the CnA catalytic subunit (CnAα, β, and γ) and two genes that encode the CnB regulatory subunit (CnB1 and CnB2) 7,8. The most highly characterized and dedicated calcineurin targets are NFAT transcription factors, of which there are 4 individual genes, designated Nfatc1-c4 9. Once activated, calcineurin tightly binds and dephosphorylates conserved serine residues at the N-terminus of cytoplasmic NFAT transcription factors, permitting their translocation to the nucleus and activation of pathologic hypertrophic gene expression 7-9.
I will not attempt to review all the literature related to CsA and FK506 treatment of rodent models of cardiac hypertrophy (both prevention and regression), other than to state that the vast majority of these studies support the requirement of calcineurin in facilitating cardiac growth following pathologic stimuli 7,10. However, it is important to acknowledge that CsA and FK506 are probably not appropriate agents for treating hypertrophic heart disease given that much higher dosing is required that can lead to severe side effects 7. More definitively, we showed that cardiac-specific expression of the noncompetitive calcineurin inhibitory domain from A-kinase anchoring protein 79 (AKAP79) or Cabin-1 reduced myocardial hypertrophy after pathologic agonist infusion or TAC stimulation 11. Similarly, transgene-mediated overexpression of regulator of calcineurin 1 (RCAN1, previously known as MCIP1) also inhibited cardiac hypertrophy to diverse pathologic stimuli 12,13. Analogous to RCAN1 overexpression, transgene-mediated expression of a dominant negative mutant of calcineurin in the heart reduced pathologic cardiac hypertrophy in mice 14. Finally, CnAβ (Ppp3cb) gene-targeted mice, which had a 70% reduction in total cardiac calcineurin activity, were generated by us and shown to have blunted hypertrophy following 2 weeks of angiotensin II or isoproterenol infusion, as well as after TAC stimulation 15. With respect to gain-of-function studies, mice expressing an activated calcineurin mutant protein in the heart developed massive cardiac hypertrophy with negative ventricular remodeling 5, while mice lacking the calcineurin docking and inhibitory protein calsarcin-1 showed greater endogenous calcineurin activity and were sensitized to negative cardiac remodeling and greater disease with pressure overload stimulation 16. Collectively, these genetic approaches have established the role of calcineurin-NFAT signaling as an obligate regulator of the pathologic hypertrophic response.
More recently, we also generated mice with an inducible loss of all calcineurin activity in the heart, due to cardiac-specific deletion of the CnB1 (Ppp3r1) gene 17. While we were not able to assess hypertrophy in these mice after pathologic pressure overload stimulation because of acute lethality, the baseline phenotype actually suggested that calcineurin can have some adaptive and protective signaling functions in the heart. Specifically, cardiac-specific CnB1-loxP deleted mice showed reduced myocyte proliferation rates with fewer cells 17. These results are also consistent with greater myocyte apoptosis rates in the absence of CnB1 in the heart with acute pressure overload 17, as well as greater cell death and injury after ischemia-reperfusion injury in CnAβ deleted mice 18. Moreover, transgenic mice expressing an activated calcineurin mutant were protected from cell death after ischemia-reperfusion injury, despite the underlying pathologic hypertrophic phenotype 19. Thus, while calcineurin-NFAT signaling contributes to and is necessary for pathologic cardiac hypertrophy, this pathway also has some degree of physiologic importance in protecting from cell death and permitting myocyte proliferation.
A number of equally supportive studies have emerged with respect to NFAT and its role in the heart. Interestingly Nfatc1 gene-deleted mice die during embryogenesis form a defect in heart valve maturation 20,21. With respect to cardiac hypertrophy induced by pathologic stimuli, mice lacking either Nfatc2 or Nfatc3, but not Nfatc4, showed a significant reduction in the response 22,23. Moreover, mice with inhibited c-Jun N-terminal kinase (JNK) or p38α mitogen-activated protein kinase signaling in the heart, which directly resulted in greater NFAT activity (they are both important NFAT kinases that inhibit its activity) showed greater cardiac hypertrophy following pathologic stimulation 24,25. Finally, mice lacking Mkk4, which lies upstream of p38 and JNK signaling, showed increased NFAT activity and greater pathologic cardiac hypertrophy, but swimming-induced cardiac hypertrophy was unaffected 26.
The Wilkins et al paper was the first to suggest that calcineurin-NFAT signaling was more highly specialized in the adult heart for inducing cardiac hypertrophy and negative ventricular remodeling in response to pathologic insults 3. At that time, it was also one the first manuscripts describing a genetic tool allowing the direct in vivo assessment of the activity of a transcription factor 3,27-29. The transgenic line we generated contained an NFAT-dependent luciferase transcriptional reporter consisting of 9 multimerized NFAT binding sites upstream of a minimal promoter, which we showed specifically responded to calcineurin-NFAT signaling in vivo 3. For example, when this reporter transgenic line was crossed with mice containing the activated calcineurin transgene, NFAT activity was increase over 15-fold in a CsA-inhibitable manner 3. The NFAT reporter transgene was also significantly induced in the adult heart at essentially all stages of TAC stimulation, both early (2 days) and late when the hearts transitioned to failure (56 days). The reporter was also activated following MI injury to the heart, both the early compensatory phase and later when the hearts began to fail. However, this reporter was not stimulated in the adult heart following either swimming or voluntary wheel running-induced physiologic hypertrophy, and in fact, it was significantly repressed 3. This later result suggested that reductions in calcineurin-NFAT signaling were part of a more adaptive profile of physiologic hypertrophy of the myocardium to exercise stimulation. Consistent with these data, we also showed that IGF-1 or an activated Akt mutant, which are known regulators of adaptive or physiologic hypertrophy, did not induce the NFAT-luciferase reporter in cultured myocytes, but in fact likely repressed it 3. Finally, we also showed that CnAβ-/- mice, which are largely refractory to hypertrophy following pathologic stimulation as discussed earlier, hypertrophied normally following IGF-I-growth hormone injections over 2 weeks time, further indicating that calcineurin-NFAT signaling does not underlie adaptive or physiologic growth of the heart in vivo.
Since our report in 2004 others have used the NFAT-luciferase reporter transgenic mice and confirmed our results. For example, the reporter is significantly activated within the heart following pathologic TAC stimulation, even as early as 24 hrs 30,31, while Heineke and colleagues confirmed its induction in the heart following myocardial infarction injury 32. Facundo et al. showed that calcineurin-NFAT activity was also induced in the heart with pathologic pressure overload stimulation but in a manner dependent on β-O-linked N-acetylglucosamine modification 33. Mice with pathologic cardiac remodeling due to a leaky Ca2+ mutant ryanodine receptor also showed induction of NFAT-luciferase activity in the heart 34. With respect to physiologic hypertrophy, Oliveira et al. reported that exercise training in mice diminished NFATc3 translocation within cardiomyocytes 35. Moreover, De Windt and colleagues showed that Nfatc2-/- mice showed less pathologic remodeling and hypertrophy in response to disease associated stimuli, yet these mice hypertrophied normally in response to voluntary exercise training, collectively suggesting again that the calcineurin-NFAT signaling pathway does not underlie physiologic hypertrophy of the heart 23. Finally, the NFAT-luciferase reporter transgenic line was also used to assess calcineurin-NFAT activity in other tissues such as the skeletal muscle, vessels, pancreas and kidney 36-39.
In conclusion, the Wilkins et al report from 2004 in many ways defined a novel signaling paradigm related to calcineurin-NFAT, such that this pathway plays an important role in maladaptive growth of the adult heart following disease-causing stimuli 3. Physiologic growth stimulation even appears to downregulate calcineurin-NFAT signaling, as well as suppress expression of the classic fetal gene program in the heart that normally associates with pathologic remodeling. However, given the primacy of calcineurin-NFAT signaling, it is not surprising that complete deletion of this pathway in CnB1 null mice, or even partial deletion in CnAβ null mice, can predispose to developmental defects with less myocyte proliferation as well as enhanced susceptibility to cell death. Thus, while this signaling module preferentially regulates pathologic responses of the adult heart, it also has adaptive functions at baseline or during development that promotes both myocyte survival and the proper maturation of the heart with correct myocyte proliferation levels.
Acknowledgments
Source of funding:
National Institutes of Health and Howard Hughes Medical Institute.
This is a commentary on article Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94(1)
Footnotes
Disclosures:
None (no financial or other conflicts of interest)
References
- 1.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 4.Konhilas JP, Watson PA, Maass A, Boucek DM, Horn T, Stauffer BL, Luckey SW, Rosenberg P, Leinwand LA. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res. 2006;98:540–548. doi: 10.1161/01.RES.0000205766.97556.00. [DOI] [PubMed] [Google Scholar]
- 5.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai S, Kerppola TK. Opposing roles of FoxP1 and Nfat3 in transcriptional control of cardiomyocyte hypertrophy. Mol Cell Biol. 2011;31:3068–3080. doi: 10.1128/MCB.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 8.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 9.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 10.Heineke J, Ritter O. Cardiomyocyte calcineurin signaling in subcellular domains: from the sarcolemma to the nucleus and beyond. J Mol Cell Cardiol. 2012;52:62–73. doi: 10.1016/j.yjmcc.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 11.De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, Glascock BJ, Kimball TF, del Monte F, Hajjar RJ, Molkentin JD. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3322–3327. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill JA, Rothermel B, Yoo KD, Cabuay B, Demetroulis E, Weiss RM, Kutschke W, Bassel-Duby R, Williams RS. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J Biol Chem. 2002;277:10251–10255. doi: 10.1074/jbc.M110722200. [DOI] [PubMed] [Google Scholar]
- 13.Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3328–3333. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, Nagai R, Yazaki Y, Komuro I. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation. 2001;104:97–101. doi: 10.1161/01.cir.104.1.97. [DOI] [PubMed] [Google Scholar]
- 15.Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Impaired cardiac hypertrophic response in Calcineurin Abeta-deficient mice. Proc Natl Acad Sci USA. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frey N, Barrientos T, Shelton JM, Frank D, Rütten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, Richardson JA, Katus HA, Hill JA, Olson EN. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med. 2004;10:1336–1343. doi: 10.1038/nm1132. [DOI] [PubMed] [Google Scholar]
- 17.Maillet M, Davis J, Auger-Messier M, York A, Osinska H, Piquereau J, Lorenz JN, Robbins J, Ventura-Clapier R, Molkentin JD. Heart-specific deletion of CnB1 reveals multiple mechanisms whereby calcineurin regulates cardiac growth and function. J Biol Chem. 2010;285:6716–6724. doi: 10.1074/jbc.M109.056143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bueno OF, Lips DJ, Kaiser RA, Wilkins BJ, Dai YS, Glascock BJ, Klevitsky R, Hewett TE, Kimball TR, Aronow BJ, Doevendans PA, Molkentin JD. Calcineurin Abeta gene targeting predisposes the myocardium to acute ischemia-induced apoptosis and dysfunction. Circ Res. 2004;94:91–99. doi: 10.1161/01.RES.0000107197.99679.77. [DOI] [PubMed] [Google Scholar]
- 19.De Windt LJ, Lim HW, Taigen T, Wencker D, Condorelli G, Dorn GW, 2nd, Kitsis RN, Molkentin JD. Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo: An apoptosis-independent model of dilated heart failure. Circ Res. 2000;86:255–263. doi: 10.1161/01.res.86.3.255. [DOI] [PubMed] [Google Scholar]
- 20.de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 21.Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 22.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH, De Windt LJ. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 24.Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 2003;22:5079–5089. doi: 10.1093/emboj/cdg474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu W, Zi M, Jin J, Prehar S, Oceandy D, Kimura TE, Lei M, Neyses L, Weston AH, Cartwright EJ, Wang X. Cardiac-specific deletion of mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ Res. 2009;104:905–914. doi: 10.1161/CIRCRESAHA.108.188292. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt-Ullrich R, Mémet S, Lilienbaum A, Feuillard J, Raphaël M, Israel A. NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development. 1996;122:2117–2128. doi: 10.1242/dev.122.7.2117. [DOI] [PubMed] [Google Scholar]
- 28.Ciana P, Di Luccio G, Belcredito S, Pollio G, Vegeto E, Tatangelo L, Tiveron C, Maggi A. Engineering of a mouse for the in vivo profiling of estrogen receptor activity. Mol Endocrinol. 2001;15:1104–1113. doi: 10.1210/mend.15.7.0658. [DOI] [PubMed] [Google Scholar]
- 29.Naya FJ, Wu C, Richardson JA, Overbeek P, Olson EN. Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2-dependent transgene. Development. 1999;126:2045–2052. doi: 10.1242/dev.126.10.2045. [DOI] [PubMed] [Google Scholar]
- 30.Donaldson C, Eder S, Baker C, Aronovitz MJ, Weiss AD, Hall-Porter M, Wang F, Ackerman A, Karas RH, Molkentin JD, Patten RD. Estrogen attenuates left ventricular and cardiomyocyte hypertrophy by an estrogen receptor-dependent pathway that increases calcineurin degradation. Circ Res. 2009;104:265–275. doi: 10.1161/CIRCRESAHA.108.190397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finsen AV, Lunde IG, Sjaastad I, Østli EK, Lyngra M, Jarstadmarken HO, Hasic A, Nygård S, Wilcox-Adelman SA, Goetinck PF, Lyberg T, Skrbic B, Florholmen G, Tønnessen T, Louch WE, Djurovic S, Carlson CR, Christensen G. Syndecan-4 is essential for development of concentric myocardial hypertrophy via stretch-induced activation of the calcineurin-NFAT pathway. PLoS One. 2011;6:e28302. doi: 10.1371/journal.pone.0028302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H, Wollert KC. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci USA. 2005;102:1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol. 2012;302:H2122–H2130. doi: 10.1152/ajpheart.00775.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaguchi N, Chakraborty A, Pasek DA, Molkentin JD, Meissner G. Dysfunctional ryanodine receptor and cardiac hypertrophy: role of signaling molecules. Am J Physiol Heart Circ Physiol. 2011;300:H2187–H2195. doi: 10.1152/ajpheart.00719.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveira RS, Ferreira JC, Gomes ER, Paixão NA, Rolim NP, Medeiros A, Guatimosim S, Brum PC. Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol. 2009;587:3899–3910. doi: 10.1113/jphysiol.2009.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parsons SA, Millay DP, Wilkins BJ, Bueno OF, Tsika GL, Neilson JR, Liberatore CM, Yutzey KE, Crabtree GR, Tsika RW, Molkentin JD. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem. 2004;279:26192–26200. doi: 10.1074/jbc.M313800200. [DOI] [PubMed] [Google Scholar]
- 37.de Frutos S, Duling L, Alò D, Berry T, Jackson-Weaver O, Walker M, Kanagy N, González Bosc L. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H2382–2390. doi: 10.1152/ajpheart.00132.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gurda GT, Guo L, Lee SH, Molkentin JD, Williams JA. Cholecystokinin activates pancreatic calcineurin-NFAT signaling in vitro and in vivo. Mol Biol Cell. 2008;1:198–206. doi: 10.1091/mbc.E07-05-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Chang JH, Paik SY, Tang Y, Eisner W, Spurney RF. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol Endocrinol. 2011;8:1376–1386. doi: 10.1210/me.2011-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]