

Abstract
CCN family 2/connective tissue growth factor (CCN2/CTGF) promotes endochondral ossification. However, the role of CCN2 in the replacement of hypertrophic cartilage with bone is still unclear. The phenotype of Ccn2 null mice, having an expanded hypertrophic zone, indicates that the resorption of the cartilage extracellular matrix is impaired therein. Therefore, we analyzed the role of CCN2 in osteoclastogenesis because cartilage extracellular matrix is resorbed mainly by osteoclasts during endochondral ossification. Expression of the Ccn2 gene was upregulated in mouse macrophage cell line RAW264.7 on day 6 after treatment of glutathione S transferase (GST) fusion mouse receptor activator of NF‐κB ligand (GST‐RANKL), and a combination of recombinant CCN2 (rCCN2) and GST‐RANKL significantly enhanced tartrate‐resistant acid phosphatase (TRACP)–positive multinucleated cell formation compared with GST‐RANKL alone. Therefore, we suspected the involvement of CCN2 in cell‐cell fusion during osteoclastogenesis. To clarify the mechanism, we performed real‐time PCR analysis of gene expression, coimmunoprecipitation analysis, and solid‐phase binding assay of CCN2 and dendritic cell–specific transmembrane protein (DC‐STAMP), which is involved in cell‐cell fusion. The results showed that CCN2 induced and interacted with DC‐STAMP. Furthermore, GST‐RANKL–induced osteoclastogenesis was impaired in fetal liver cells from Ccn2 null mice, and the impaired osteoclast formation was rescued by the addition of exogenous rCCN2 or the forced expression of DC‐STAMP by a retroviral vector. These results suggest that CCN2 expressed during osteoclastogenesis promotes osteoclast formation via induction of and interaction with DC‐STAMP. © 2011 American Society for Bone and Mineral Research.
Keywords: OSTEOCLASTS, CCN FAMILY 2/CONNECTIVE TISSUE GROWTH FACTOR (CCN2/CTGF), RECEPTOR ACTIVATOR OF NF‐κB LIGAND (RANKL), DENDRITIC CELL–SPECIFIC TRANSMEMBRANE PROTEIN (DC‐STAMP)
Introduction
Bone undergoes a process termed remodeling, which involves the formation and resorption of bone tissues to provide maximal strength with minimal mass in the physiologic state. This bone remodeling needs to coordinate balances of function of bone‐forming osteoblasts and bone‐resorbing osteoclasts.1 Imbalances in bone remodeling are associated with most adult skeletal diseases, such as osteoporosis, periodontal disease, rheumatoid arthritis, and cancer bone metastasis.1 These skeletal diseases commonly are based on excess osteoclastic activity.1 Osteoclasts are multinucleated cells responsible for bone resorption and are derived from hematopoietic precursors of the monocyte/macrophage lineage.1, 2 Macrophage colony‐stimulating factor (M‐CSF) and receptor activator of nuclear factor‐κB ligand (RANKL) within the bone microenvironment are now known to be essential signaling molecules for the differentiation of these precursors into osteoclasts.1, 2 These factors are required to induce the expression of osteoclast marker genes, such as tartrate‐resistant acid phosphatase (Tracp) and cathepsin K. However, although M‐CSF and RANKL are critical for osteoclast formation, a number of other growth factors and cytokines in bone tissues are suggested to play an important role in regulating osteoclast differentiation.
Connective tissue growth factor (CTGF) is the second member of the CCN family of proteins (CCN2), and its structure is characterized by four distinct modules, that is, insulin‐like growth factor–binding protein‐like (IGFBP), von Willebrand factor type C (VWC), thrombospondin type 1 repeat (TSP1), and a carboxyl‐terminal cysteine knot (CT).3, 4 The CCN family consists of six distinct proteins (i.e. cysteine‐rich 61; Cyr61/CCN1, CTGF/CCN2, nephroblastoma overexpressed; Nov/CCN3, Wnt‐inducible secreted protein 1, 2, and 3; and Wisp1–3/CCN4–6 with similar structures as described earlier, except for Wisp2/CCN5, which lacks the CT module.3, 4 CCN family proteins are involved in a number of biological processes in development, tissue repair, and tumor suppression, but their exact functions are still unspecified. Previously, we reported that the gene expression of CCN2 was remarkably high in the prehypertrophic region of the growth plate in bones3 and that recombinant CCN2 (rCCN2) promoted the proliferation and differentiation of chondrocytes and osteoblasts in vitro5, 6 and maintained the integrity of the articular chondrocytes7 and tissues in vivo.8 In addition, rCCN2 also stimulated proliferation, migration, and tube formation of vascular endothelial cells in vitro and angiogenesis in vivo.9 These findings indicate that CCN2 not only promotes endochondral ossification by enhancing the proliferation and maturation of chondrocytes and osteoblasts and survival of endothelial cells but also maintains the articular cartilage tissues under physiologic conditions. In fact, an in vivo study revealed that Ccn2 null mice died soon after birth owing to respiratory distress as a result of severe skeletal abnormalities,10 and primary chondrocytes derived from the rib cage and osteoblasts from the calvaria of Ccn2 knockout mice exhibited impaired DNA synthesis and faulty extracellular matrix production.11, 12 As such, both the endochondral and intramembranous ossification is impaired in Ccn2 null mice.10, 11, 12 In particular, the most striking histologic feature of the Ccn2 null growth plate is an enlarged hypertrophic zone.10 This phenotype may be associated with impaired removal of the cartilage extracellular matrix (ECM) by chondroclasts/osteoclasts. Previously, we showed that the expression of CCN2 was upregulated in bone callouses during fracture repair.13 Furthermore, we recently reported that CCN2 induced the expression of vascular endothelial growth factor (VEGF) via upregulation of hypoxia‐inducible factor (HIF) 1α and interacted with bone morphogenetic protein 2 (BMP‐2).14, 15 These results suggest that CCN2 may control a network of growth factors during drastic bone remodeling events, such as endochondral ossification or bone fracture repair, by acting as a “signal conductor.” On the other hand, the osteolytic metastasis of a human breast cancer cell line, MDA231, was decreased by treatment with a CCN2‐neutralizing antibody.16 Moreover, downregulation of CCN2 in bone marrow cells by an antisense S‐oligodeoxynucleotide‐inhibited TRACP+ osteoclast‐like cell formation.16 Because CCN2 is highly expressed in MDA231 cells,17 bone destruction induced by MDA231 cells may occur owing to excess osteoclastic functions activated by CCN2. These findings indicate that CCN2 also contributes to osteolysis under a pathogenic condition. Furthermore, it was reported recently that CCN2 production induced by tumor necrosis factor α (TNF‐α) promoted articular damage by increased osteoclasts in rheumatoid arthritis patients.18 On the basis of these findings, we hypothesized that CCN2 regulates osteoclast differentiation or functions under both physiologic and pathogenetic conditions. To clarify the molecular mechanistic basis of this hypothesis, we examined the effect of CCN2 on osteoclast differentiation by using murine macrophage‐like cell line RAW264.7 and Ccn2 null osteoclast progenitors from fetal liver.
Materials and Methods
Materials
Alpha‐modified Eagle's medium (α‐MEM) and fetal bovine serum (FBS) were purchased from ICN Biomedicals (Aurora, OH, USA) and Cancera International (Rexcalale, Ontario, Canada), respectively. Plastic dishes and multiwell plates were obtained from Greiner Bio‐One (Frickenhausen, Germany). Anti‐glutathione S transferase (GST) and anti‐histidine (His) were purchased from Bethyl Laboratories (Montgomery, TX, USA), and anti‐hemagglutinin (anti‐HA), anti‐nuclear factor of activated Tc1 (NFATc1), and anti‐matrix metalloproteinase 9 (MMP‐9) were from Covance (Princeton, NJ, USA), Santa Cruz Biotechnology (Santa Cruz, CA, USA), and Triple Point Biologics, Inc. (Forest Grove, OR, USA), respectively. Anti‐CCN2 monoclonal IgG(8–86) and rabbit polyclonal anti‐CCN2 antiserum were prepared,19 and rabbit polyclonal anti‐CCN2 antibody was obtained from Santa Cruz Biotechnology. Recombinant CCN2 (rCCN2) was purified as described previously.5, 20 For immunoprecipitation–Western blot analysis, polyhistidine (His)–tagged rCCN2 was produced by Escherichia coli.15, 21
Construction and preparation of GST‐fused RANKL and DC‐STAMP
The extracellular domain of RANKL and full‐length dendritic cell–specific transmembrane protein (DC‐STAMP) cDNAs were obtained by using the gene‐specific primers listed in Table 1.22, 23 These cDNAs were inserted into the expression vector pGEX‐2TK and expressed in E. coli. The resulting products were purified by using glutathione‐Sepharose gels (Sigma, St Louis, MO, USA) and employed in experiments.22 For a control experiment, the parental vector was used to produce and purify the GST protein.
Table 1.
Sense (S) and Antisense (AS) Primers for Target Genes
| Gene | Accession no. | Primer sequence | Expected size (bp) |
|---|---|---|---|
| Cathepsin K | NM_007802 | (S) 5'‐ATATGTGGGCCAGGATGAAAGTT‐3' | 89 |
| (AS) 5'‐TCGTTCCCCACAGGAATCTCT‐3' | |||
| Trap | NM_007388 | (S) 5'‐CGACCATTGTTAGCCACATACG‐3' | 76 |
| (AS) 5'‐TCGTCCTGAAGATACTGCAGGTT‐3' | |||
| Ccn2 | NM_010217 | (S) 5'‐CCACCCGAGTTACCAATGAC‐3' | 168 |
| (AS) 5'‐GTGCAGCCAGAAAGCTCA‐3' | |||
| NFATc1 | NM_016791 | (S) 5'‐TGAGGCTGGTCTTCCGAGTT‐3' | 90 |
| (AS) 5'‐CGCTGGGAACACTCGATAGG‐3' | |||
| DC‐STAMP | NM_029422 | (S) 5'‐CTAGCTGGCTGGACTTCATCC‐3' | 305 |
| (AS) 5'‐TCATGCTGTCTAGGAGACCTC‐3' | |||
| GAPDH | NM_008084 | (S) 5'‐GCCAAAAGGGTCATCATCTC ‐3' | 215 |
| (AS) 5'‐GTCTTCTGGGTGGCAGTGAT‐3' | |||
| Oligonucleotides used for making of expression vectors | |||
| RANKL | pGEX2TK | (S) 5'‐ATACCAATTGCGGATCCTAACAGAATATCAG‐3' | |
| (AS) 5'‐ATACCAATTGGAAATGAGTCTCAGTCAATG‐3' | |||
| DC‐STAMP | pGEX2TK | (S) 5'‐GGCCGAATTCCGATGAGGCTCTGGACCTTGGGCACCAGTATT‐3' | |
| (AS) 5'‐ACGGAGATCTTTAATGATGATGATGATGAT | |||
| GATGTAGATCATCTTCATTTGCAGGGATTGTCTGCT‐3' | |||
| DC‐STAMP | pcDNA3.1 | (S) 5'‐GGCCGAATTCCATGAGGCTCTGGACCTTGGGCACCAGTATT‐3' | |
| (AS) 5'‐ACGGTCTAGAAGTAGATCATCTTCATTTGCAGGGATTGTCTGCT‐3' | |||
Cell culture and osteoclastogenesis
The murine macrophage‐like cell line RAW264.7 was routinely cultured in a 5% CO2 atmosphere at 37°C in α‐MEM supplemented with 10% FBS. Ccn2 null and wild‐type mice used in this study were described previously.10 Primary cultures of fetal liver cells from these mice on embryonic day 14.5 were isolated as described previously.24 All animal experiments in this study were conducted according to the Guidelines for Animal Research of the Okayama University and were approved by the Animal Committee. For osteoclastogenesis experiments, RAW264.7 cells were inoculated at a density of 1 × 104/cm2 into wells of a 96‐ or 12‐well multiwell plate and incubated in the presence or absence of GST‐RANKL for 7 days until typical multinucleated cells were observed. Fetal liver cells were cultured in a 5% CO2 atmosphere at 37°C in α‐MEM supplemented with 10% FBS, M‐CSF, and GST‐RANKL for 7 to 10 days until typical multinucleated cells were appeared.24
Tartrate‐resistant acid phosphatase (TRACP) staining
Cells were fixed with 3.5% paraformaldehyde in PBS at 4°C for 60 minutes. The cells then were treated with 0.2% Triton X‐100 in PBS at room temperature for 5 minutes and rinsed twice with PBS. Finally, the fixed cells were incubated with 0.01% naphthol AS–MX phosphate (Sigma) and 0.05% fast red violet LB salt (Sigma) in the presence of 50 mM of sodium tartrate and 90 mM of sodium acetate (pH 5.0) for TRACP staining and then rinsed 3 times with PBS.25
Transfection assays
The day prior to transfection with plasmids, RAW264.7 cells were inoculated at a density of 1 × 104/cm2 into the 6‐well multiwell plates (Greiner Bio‐One) containing α‐MEM with 10% FBS. Then this medium was replaced with Opti‐MEM (Invitrogen, Carlsbad, CA, USA) without any serum, and the cells were transiently transfected for 18 hours using FuGENE6 reagent (Roche, Basel, Switzerland). The ratio of DNA to FuGENE6 was 1:2 (volume/weight), with a DNA amount of 3 µg.26 On the day of stimulation, the medium was discarded, and fresh α‐MEM containing 10% FBS and GST‐RANKL was added. The cells were collected 2 days later and subjected to immunoprecipitation–Western blot analysis.
Retrovirus infection
Culture supernatants containing replication‐defective recombinant retroviruses were prepared by using Retrovirus Packaging Kit (Takara Shuzo, Tokyo, Japan) from 293T packaging cells with pMFG‐Dc‐stamp‐His. After mouse fetal liver cells were incubated with virus‐containing supernatants for 8 hours, supernatants were removed, and the infected cells were cultured with M‐CSF and GST‐RANKL for 7 to 10 days until typical multinucleated cells appeared.
Indirect immunofluorescence analysis
RAW264.7 cells were inoculated onto chamber slides (Nunc, Inc., Naperville, IL, USA), and the next day the cells were infected with a retroviral vector for the overexpression of His‐tagged DC‐STAMP and were cultured with GST‐RANKL. After 8 days, cultures were washed with PBS, fixed with 3.5% paraformaldehyde for 30 minutes at room temperature, and made permeable with 0.1% NP‐40 in PBS. Indirect immunofluorescence analysis was performed as described previously.27
Real‐time RT‐PCR analysis
Total RNA was isolated from RAW264.7 cells by using ISOGEN reagent (Nippon Gene, Tokyo, Japan). First‐strand cDNA was synthesized from 1 µg of total RNA with avian myeloblastosis virus (AMV)–derived reverse transcriptase (RT), and subsequent quantitative RT‐PCR was performed as described previously.8 The amplification condition was as follows: 95°C (5 seconds), 64°C (10 seconds), and 72°C (15 seconds) for 45 cycles by using a StepOne plus real‐time PCR system (Applied Biosystems, Carlsbad, CA, USA) or LightCycler (Roche). Primer sets used to detect various cDNAs are given in Table 1.
Western blot analysis
Proteins separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) were transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Little Chalfont, UK) by using a semidry transfer apparatus (ATTO, Tokyo, Japan). Western blot analysis was carried out essentially as described previously.28
Solid‐phase binding assay
A 96‐well multiwell plate was precoated with rCCN2 (1, 5, and 20 µg/mL) at 4°C overnight, and then GST‐fused DC‐STAMP with a His tag was added to the precoated plate. For competitive binding assay, recombinant DC‐STAMP was labeled with biotin by a using biotin labeling kit–NH2 (Dojindo Laboratories, Kumamoto, Japan). We measured optical absorbance (450 nm) representing the binding of DC‐STAMP to CCN2 by performing a colorimetrical assay using anti‐His antibody or streptoavidin.15, 21
Bone resorption assay and small interfering RNA transfection
RAW264.7 cells were inoculated at a density of 1 × 104/cm2 into a calcium phosphate apatite–coated 24‐well multiwell plate (OAAS‐24, Oscotec, Cheonan, Korea). The next day, the cells were treated with GST‐RANKL. After 3 days, these cells were transfected with a small interfering RNA (siRNA, 25 nM) targeted against mouse Ccn2 by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The siRNA was purchased from Greiner Bio‐One, and the nucleotide sequences were 5'‐CCAUGAUGCGAGCCAACUGtt‐3' (sense) and 5'‐CAGUUGGCUCGCAUCAUGGtt‐3' (antisense). Thereafter, the cells treated with siRNA were cultured with or without rCCN2. At 72 hours after the transfection, the medium was removed, and each well was washed with PBS. In order to detach the cells, a solution of 5% sodium hypochlorite was applied to the well for 5 minutes. To count the number of resorption pits, each well then was stained with 0.05% (v/v) toluidine blue for 5 minutes and washed with distilled water several times. The number of bone resorption pits was counted under a microscope.
Statistical analysis
Unless otherwise specified, all experiments were repeated at least twice, and similar results were obtained. Statistical analysis was performed by performing Student's t test.
Results
Upregulation of CCN2 during osteoclastogenesis
We purified recombinant mouse RANKL by using a Gst gene fusion system, as described previously.22 We investigated the effects of GST‐RANKL on osteoclastogenesis using mouse macrophage lineage cell line RAW264.7. As a result, GST‐RANKL treatment induced the formation of multinucleated cells positive for TRACP staining, whereas GST treatment did not (data not shown). Under these conditions, we examined the time course of the expression of Ccn2 and osteoclast differentiation marker genes during RAW264.7 cell differentiation. During osteoclast differentiation, the expression level of NFATc1, which is a critical transcriptional factor for osteoclasts, started to increase on day 1 and continued until day 7. On the other hand, the expression levels of Tracp, Cathepsin K, Ccn2, and Dc‐stamp mRNAs increased on days 6 and 7 (Fig. 1 A). In addition, CCN2 also was induced at the protein level in the osteoclast‐like cells time dependently (Fig. 1 B). These findings indicate that CCN2 was increased at both the mRNA and protein levels at the late stage of osteoclast differentiation, thus suggesting that CCN2 is regulated depending on osteoclast differentiation but not by stimulation of RANKL directly.
Figure 1.
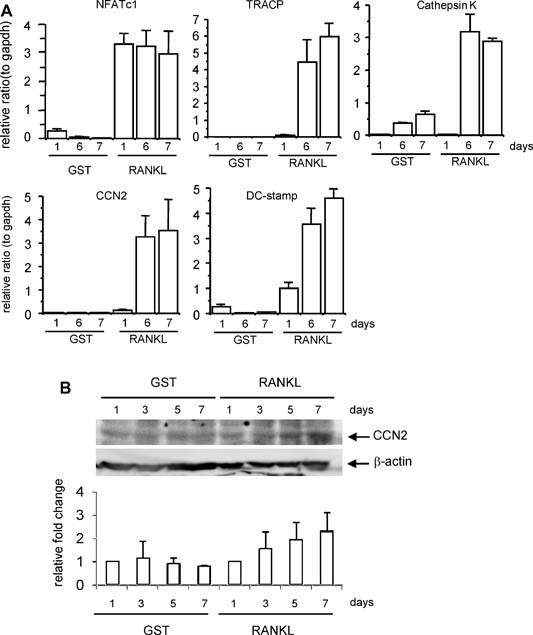
Expression and production of CCN2 induced in osteoclast‐like cells by GST‐RANKL. (A) The expressions of osteoclast marker genes in RAW264.7 cells on days 1, 6, and 7 after treatment with GST‐RANKL were determined by real‐time RT‐PCR. The expression of the NFATc1 gene was upregulated on day 1 after the addition of GST‐RANKL and the expressions of Tracp, Ccn2, Dc‐stamp, and cathepsin K genes were upregulated on days 6 and 7. Data are presented as mean and SD of two sets of independent cultures. (B) CCN2 protein production in RAW264.7 cells stimulated with GST‐RANKL was determined by Western blot analysis. The band density of CCN2 for the extract of the GST‐RANKL–treated cells was increased time dependently (upper panel). Signals for β‐actin were used to normalize the band densities (bottom panel). The lower graph represents the relative value of CCN2 normalized to the amount of β‐actin using densitometric analysis. Each column is standardized against the value of day 1 sample and all columns are presented as mean and SD of two individual experiments.
CCN2 synergistically promotes RANKL‐induced osteoclast differentiation
To test whether CCN2 exerted any effect on the differentiation of RAW264.7 cells, we next evaluated the formation of multinucleated TRACP+ cells on the treatment with CCN2 in the presence of GST‐RANKL in a time course. When RAW264.7 cells were treated with the combination of CCN2 and GST‐RANKL on day 2, the number of TRACP+ multinucleated cells was not increased compared with treatment with GST‐RANKL alone (Fig. 2 A). However, the number of TRACP+ cells with more than three nuclei started to increase on day 4 of treatment with the combination of CCN2 with GST‐RANKL, and on days 6 and 7, the number was significantly increased compared with the cells treated with GST‐RANKL alone (Fig. 2 A). On the other hand, the cells did not form these TRACP+ multinucleated cells when treated with CCN2 or GST in the absence of GST‐RANKL (Fig. 2 A). These findings suggest that CCN2 promotes RANKL‐initiated differentiation of RAW264.7 cells at the late stage of osteoclastogenesis but had no effect at the early stage. In addition, the results that CCN2 had no effect by itself may indicate that there is no cell surface molecular counterpart interacted with CCN2 on macrophages without RANKL treatment. Furthermore, we analyzed the protein level of NFATc1, which is a critical transcriptional factor in osteoclast differentiation, and MMP‐9, which plays an important role in bone resorption in RAW cells treated with a combination of CCN2 and RANKL. As shown in Fig. 2 B, both NFATc1 and MMP‐9 were increased further by treatment with a combination of CCN2 and GST‐RANKL than by the treatment with GST‐RANKL alone. To clarify in detail the role of CCN2 in the differentiation of RAW264.7 cells, we investigated the effect of the combination of CCN2 with GST‐RANKL on the mRNA expression of Dc‐stamp, representing functional cell surface molecules of osteoclasts, in cultured RAW264.7 cells by performing real‐time RT‐PCR analysis. As shown in Fig. 3, on day 1 after stimulating with a combination of CCN2 and GST‐RANKL or GST‐RANKL alone, gene expression levels of Dc‐stamp showed no difference. However, on day 4, gene expression level of Dc‐stamp was increased significantly in RAW264.7 cells stimulated by the combination of rCCN2 and GST‐RANKL compared with those treated with GST‐RANKL alone. Furthermore, on day 8, Dc‐stamp gene expression was upregulated significantly by treatment with a combination of CCN2 and GST‐RANKL. Interestingly, treatment with CCN2 alone significantly increased the expression of Dc‐stamp in RAW264.7 cells on days 4 and 8. These findings suggest that CCN2 induced the upregulation of DC‐STAMP, which then promoted osteoclastogenesis in collaboration with RANKL‐mediated signaling.
Figure 2.
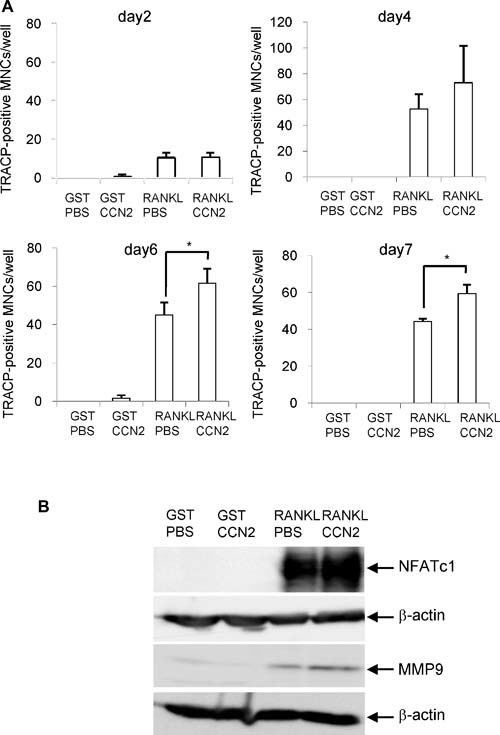
(A) Effect of CCN2 on osteoclast formation induced by GST‐RANKL. CCN2 increased TRACP+ multinucleated cell (MNC) formation from RAW264.7 cells. Cells were inoculated at a density of 1 × 104/cm2 in 12‐well multiwell plates, and the next day they were stimulated by GST‐RANKL or GST in the presence or absence of rCCN2 (50 ng/mL). After 3 days of treatment with these factors, the cells were refreshed with fresh α‐MEM containing 10% serum with these factors and cultured for 7 days. Differentiated RAW264.7 cells were defined and quantified as TRACP+ multinucleated cells (MNCs) on days 2, 4, 6, and 7. Data are the means ± SD of triplicate cultures. The asterisk indicates a significant difference from the sample RANKL/PBS (p < .05). (B) Effect of CCN2 on the production of NFATc1 and MMP‐9, which are osteoclast differentiation markers. RAW264.7 cells were treated with GST‐RANKL or GST in the presence or absence of rCCN2 for 6 days or 8 days. Then, cell lysate was collected and Western blot analysis was performed by using anti‐NFATc1 or anti‐MMP9 antibody. The amount of NFATc1 was increased in the cells treated with combination of CCN2 and GST‐RANKL, compared with that with GST‐RANKL alone for 6 days (upper panel). β‐Actin was used as an internal control (lower panel). Similarly, the amount of MMP‐9 also was increased in the cells treated with combination of CCN2 and GST‐RANKL for 8 days.
Figure 3.
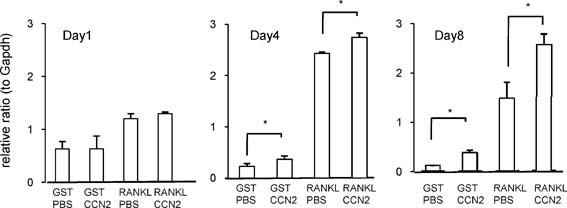
Effect of CCN2 on the expression of Dc‐stamp gene in RAW264.7 cells. RAW264.7 cells were inoculated on 12‐well multiwell plates or 35‐mm dishes and treated with GST‐RANKL or GST in the presence and absence of rCCN2 (50 ng/mL) on the next day after inoculation. After 3 days of treatment with these factors, the medium was replaced with fresh α‐MEM containing 10% serum with these factors and cultured for 7 days. Total RNA was isolated on days 1, 4, and 8, and real‐time RT‐PCR analysis was performed by using the primers described in Table 1. Data are presented as mean and SD of three sets of independent cultures. PCR conditions were described under “Materials and Methods.” The asterisk indicates a significant difference (p < .05).
CCN2 interacts with DC‐STAMP on the surfaces of osteoclast‐like cells
The expression of Dc‐stamp was upregulated by treatment of RAW264.7 cells with CCN2. Next, to examine whether or not CCN2 interacted with DC‐STAMP during the differentiation of RAW264.7 cells, we first generated a recombinant retrovirus encoding mouse Dc‐stamp tagged at its C‐terminus with 6× histidine (pMFG‐Dc‐stamp‐His) and infected RAW264.7 cells with this retrovirus. After infection, we examined the localization of DC‐STAMP and CCN2 in GST‐RANKL–induced osteoclast‐like RAW264.7 cells by indirect immunofluorescence analysis. As shown in Fig. 4 A, RAW264.7 cells overexpressing DC‐STAMP‐His differentiated into TRACP+ multinucleated giant cells in the presence of GST‐RANKL (phase and TRACP), and His‐tagged DC‐STAMP was detected in the cytoplasm and on the cell surfaces of the multinucleated giant cells (Fig. 4 A; anti‐His). Interestingly, CCN2 also was detected on the cell surface (Fig. 4 A; anti‐CCN2). By merging the images of the fluorescent signal from anti‐CCN2 with the anti‐His‐associated fluorescence, colocalization of the anti‐CCN2 and anti‐His signals on the cell surface was observed (yellow signals, Fig. 4 A). On the other hand, CCN2 was not detected in those cells in the absence of GST‐RANKL. These findings indicate that CCN2 is colocalized with DC‐STAMP on the multinucleated cells, thus suggesting that CCN2 interacts with DC‐STAMP. To test this hypothesis, we performed immunoprecipitation–Western blot analysis using a recombinant CCN2 tagged at its C‐terminus with 6× His. We newly generated a plasmid encoding mouse Dc‐stamp tagged at its C‐terminus with hemagglutinin (Dc‐stamp‐HA) for the purpose of detection of rCCN2 tagged with 6× His and transfected RAW264.7 cells with this plasmid. As shown in Fig. 4 B (upper panel), lysates of RAW cells transfected to express DC‐STAMP‐HA were prepared and incubated with or without rCCN2 tagged with 6× His and then immunoprecipitated with anti‐HA antibody. The immunoprecipitate then was probed with anti‐His antibody. As observed in Fig. 4 B (lower panel), DC‐STAMP‐HA was detected in the proteins immunoprecipitated with anti‐HA antibody from lysates of RAW cells that had been transfected with DC‐STAMP‐HA, but it was not detected in cells transfected with empty vector. Under the same conditions, CCN2‐His was distinctly detected only when the lysate of RAW cells expressing Dc‐stamp‐HA was used. These data demonstrate that CCN2 binds to DC‐STAMP in the cells. To confirm the direct interaction of CCN2 with DC‐STAMP, we next generated a recombinant mouse DC‐STAMP tagged at its N‐terminus with GST and at its C‐terminus with 6× His (Fig. 5 A). The integrity of the purified recombinant DC‐STAMP was confirmed by Coomassie Brillant Blue (CBB) staining and Western blotting using anti‐GST and anti‐His antibodies (Fig. 5 B). Using this protein, we performed a solid‐phase binding assay as described previously.15, 21 As shown in Fig. 5 C (panel a), recombinant DC‐STAMP bound directly to CCN2 in a dose‐dependent manner. Furthermore, to test whether CCN2 specifically interacted with DC‐STAMP or not, we performed a competitive binding assay (Fig. 5 C, panel b). The addition of 100‐fold unlabeled DC‐STAMP decreased the binding of biotin‐labeled DC‐STAMP to CCN2. These findings indicate that CCN2 can bind directly to DC‐STAMP both in the cell and in vitro.
Figure 4.
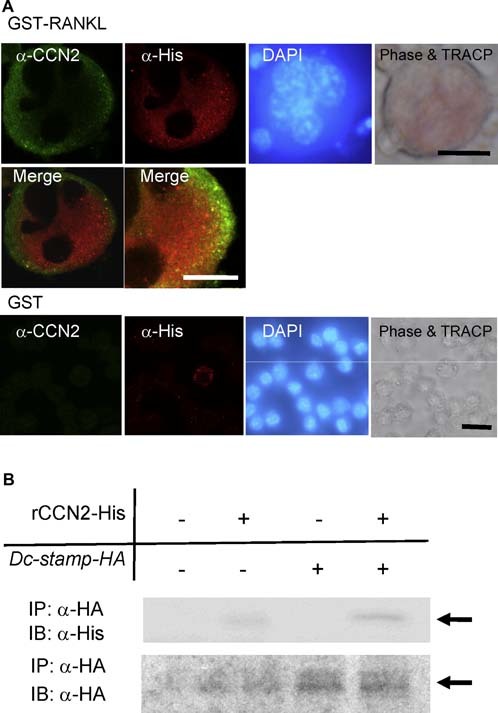
Colocalization and interaction of CCN2 with DC‐STAMP in osteoclast‐like cells. (A) Immunofluorescence analysis of CCN2 and DC‐STAMP expression on RAW264.7 cells treated with GST‐RANKL. RAW264.7 cells were inoculated at a density of 1 × 104 /cm2 into a 4‐well chamber slide, and the next day the medium was removed and infected with retroviruses, having pMGF‐Dc‐stamp‐His, containing 8 µg/mL of polybrene (hexadimethrine bromide). After 8 hours, the retrovirus solution was replaced with fresh α‐MEM containing 10% serum with GST‐RANKL or GST, and the cells were cultured for 7 days. Thereafter, RAW264.7 cells that had differentiated into multinucleated giant cells were analyzed by laser‐scanning confocal microscopy. The same field was viewed by phase‐contrast microscopy, nuclear staining with DAPI, and TRACP staining. CCN2 showed cell surface distribution (green), and DC‐STAMP showed cell surface plus cytoplasmic distribution (red). In the merged image, colocalization of the two molecules was observed, and the higher‐magnification image showed the colocalization of two molecules more clearly. On the other hand, CCN2 and DC‐STAMP was at very low levels in RAW cells treated with GST. The bar represents 25 µm and 10 µm (high‐magnification image). (B) Western blot analysis of CCN2 binding to DC‐STAMP immunoprecipitated with anti‐HA antibody. RAW264.7 cells were inoculated at a density of 1 × 105 /well into a 6‐well multiwell plate, and the next day the medium was replaced with serum‐free medium. Then the cells were transfected with a DC‐STAMP expression plasmid containing an HA tag by using FuGENE6. After 18 hours, the medium was replaced with fresh α‐MEM containing 10% serum with GST‐RANKL or GST, and the cells were cultured for 2 days. Thereafter, the cell lysates were collected and incubated without or with 1 µg rCCN2 tagged with His at 4°C for 2 hours. Then immunoprecipitaion was performed with Ezview Red anti‐HA affinity gel (Sigma) at 4°C. After 1 hour, precipitates were subjected to SDS‐PAGE and Western blotting with anti‐His antibody (upper panel). The arrow indicates the signal for CCN2. In the lower panel, the same membrane was reacted with anti‐HA antibody. The arrow indicates the signal for DC‐STAMP‐HA.
Figure 5.
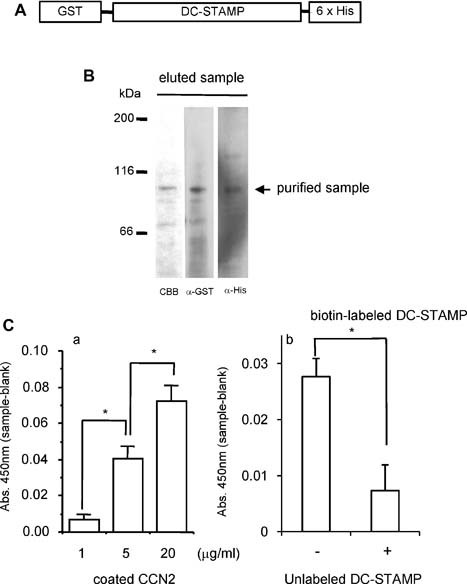
Solid‐phase binding assay of CCN2 to DC‐STAMP. (A) Schema represents recombinant DC‐STAMP protein carrying a GST tag at its N‐terminus and a 6× His tag at its C‐terminus. (B) Purification of recombinant DC‐STAMP by using a Gst gene fusion system. Gst‐fused mouse Dc‐stamp cDNA in a pGEX vector was expressed in E. coli, and the lysate was applied to glutathione‐Sepharose. The protein eluted with 20 mM reduced glutathione was separated by 7.5% SDS‐PAGE. Then the gel was stained with CBB, and Western blot analysis was performed by using anti‐GST or anti‐His antibody. The arrow indicates purified DC‐STAMP proteins. (C) Solid‐phase binding assay of CCN2 to GST‐fused DC‐STAMP. (a) To a 96‐well multiwell plate precoated with different concentrations of rCCN2 (1, 5, and 20 µg/mL), 6× His‐tagged DC‐STAMP (1 µg/mL) was added, and the plate then was incubated at 37°C for 2 hours. Total binding was determined by immunoreactivity to the His tag. Background binding was determined by immunoreactivity to the His‐tagged DC‐STAMP in the wells precoated with BSA at the same concentration used for each rCCN2 concentration. Specific binding was calculated by subtracting background binding from total binding. Data represent mean ± SD of triplicate samples. The asterisk indicates a significant difference (p < .01). (b) Competition assay for the binding of CCN2 to DC‐STAMP. Biotin‐labeled DC‐STAMP (0.5 µg/mL) in the presence or absence of 100‐fold unlabeled DC‐STAMP was incubated at 37°C for 2 hours in the wells precoated with rCCN2 at the concentration of 5 µg/mL. Bound proteins were determined by their immunoreactivity with anti‐biotin antibody. Data represent means ± SD of triplicate samples. The asterisk indicates a significant difference (p < .05).
Formation of TRACP+ multinucleated cells from fetal liver cells is impaired in Ccn2 null mice
Since we clarified that CCN2 promoted the expression and interacted with DC‐STAMP, we tested whether RANKL‐induced osteoclastogenesis requires CCN2 by examining fetal liver cells on embryonic day 14.5. We chose these cells because it was difficult to isolate bone marrow cells from Ccn2 null mice owing to the fact that these mice die soon after birth from respiratory distress as a result of severe skeletal abnormalities.10 In fact, as shown in previous data10 and in Fig. 6 A, the radius from Ccn2 mutants on embryonic day 18.5 showed a concave surface with the kink in the middle, and we previously found less alkaline phosphatase staining in cortical and trabecular bones of Ccn2 null mice. In addition, as shown in Fig. 6 A (right panel), TRACP+ cells along with the bone trabeculae of Ccn2 mutants were decreased compared with those of wild type mice. Taken together with previous findings,10 these data indicate that the balance of bone formation and bone resorption may be impaired in Ccn2 null mice. As expected, the fetal liver cells from wild‐type mice differentiated into TRACP+ multinucleated cells by treatment with GST‐RANKL and M‐CSF, whereas such differentiation was hardly induced in the cells from Ccn2 null embryos (Fig. 6 B). Indeed, the number of multinucleated TRACP+ cells obtained for the wild‐type embryonic liver was approximately eightfold higher than that for the Ccn2 null liver (Fig. 6 B). Consistent with these findings, the expression levels of osteoclast marker genes were decreased in fetal liver cells from Ccn2 null mice compared with their wild type counterparts (Fig. 6 C). These results indicate that the expression of Dc‐stamp was decreased in the cells without CCN2, which impaired the cell‐cell fusion, thus suggesting that CCN2 promotes osteoclast differentiation via regulation of the expression level of the Dc‐stamp gene.
Figure 6.
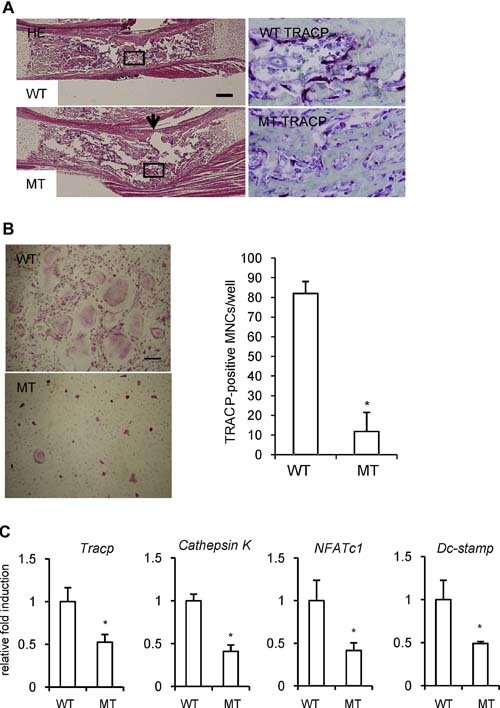
Impaired formation of TRACP+ multinucleated cells generated by M‐CSF‐ and RANKL‐treated fetal liver cells from Ccn2 null mice. (A) Hematoxylin‐eosin (H‐E) staining of radii from wild‐type and Ccn2 null mice (left panel) and TRACP staining in the box of H‐E staining (right panel) on embryonic day 18.5. The bar represents 200 µm. The number of TRACP+ cells was decreased in Ccn2 mutants compared with wild‐type mice. The arrow indicates the concave surface of the kink in Ccn2 mutants. (B) To investigate the effect of CCN2 on osteoclastogenesis, we isolated fetal liver cells from Ccn2 null mice on embryonic day 14.5 and treated them with M‐CSF (10 ng/mL) for 3 days. The M‐CSF–treated adherent cells were treated with M‐CSF and GST‐RANKL for 7 to 10 days, fixed, and stained for TRACP. (Left panel) The cells from wild‐type mice (WT) treated with M‐CSF and GST‐RANKL differentiated into TRACP+ multinucleated cells (MNCs), whereas the formation of TRACP+ MNCs from Ccn2 null mice (MT) was impaired. The bar represents 50 µm. (Right panel) The graph shows quantification of TRACP+ MNCs with more than three nuclei. Data are the means ± SDs (n = 4). The asterisk indicates a significant difference from WT (p < .01). (C) The expression of osteoclast marker genes was determined by real‐time RT‐PCR. Real‐time RT‐PCR analysis was performed as described under “Materials and Methods.” Data are presented as means and SDs for wild type (n = 3) and Ccn2 null (n = 4) mice. The asterisk indicates a significant difference from WT (p < .05).
Impaired osteoclastogenesis is rescued by rCCN2 or Dc‐stamp overexpression in Ccn2‐deficient cells
Next, to investigate whether impaired osteoclastogenesis of Ccn2‐deficient cells is rescued by the addition of rCCN2 or not, we enumerated TRACP+ Ccn2 null cells with more than three nuclei stimulated by RANKL and M‐CSF in the presence of rCCN2. As shown in Fig. 7 A, the number of TRACP+ multinucleated Ccn2 null cells was approximately threefold higher in the presence of rCCN2 than in the absence of rCCN2. In addition, to investigate whether the functional deficiency in Ccn2‐deficient cells as osteoclasts could be rescued by the addition of exogenous CCN2, we performed bone resorption assays. For this objective, Ccn2 null cells were not applicable because DC‐STAMP was downregulated as well. Therefore, we used RNA interference technology to silence transient CCN2 production to avoid the downregulation of Dc‐stamp on bone resorption. In fact, the gene expression of Dc‐stamp in RAW264.7 cells was not decreased by the mild and transient silencing of CCN2 production by siRNA technique (data not shown). As shown in Fig. 7 B, under these conditions, the number of bone resorption pits was decreased at a marginally significant level (p = .07). Finally, when the cells treated with the Ccn2 siRNA were stimulated by rCCN2, the number of bone resorption pits was increased significantly beyond control level (Fig. 7 B), suggesting a critical interaction between CCN2 and DC‐STAMP. As a further characterization, the effect of rCCN2 on the gene expression of Dc‐stamp in Ccn2 null cells was evaluated by performing real‐time RT‐PCR analysis. As shown in Fig. 7 C, the expression level of Dc‐stamp was upregulated in Ccn2 null cells treated with rCCN2, but the Tracp gene was not (data not shown). These findings suggest that CCN2 may regulate Dc‐stamp gene expression directly in osteoclast precursors. Furthermore, to confirm whether the upregulation of Dc‐stamp is involved in osteoclastogenesis, the number of TRACP+ multinucleated cells in Ccn2 null mice was counted when Dc‐stamp‐His was overexpressed using a retroviral vector. As shown in Fig. 7 D, we confirmed that DC‐STAMP‐His was overexpressed in Ccn2 null cells by a recombinant retrovirus pMFG‐Dc‐stamp‐His in the presence of M‐CSF and RANKL by performing immunocytochemical analysis, and the number of TRACP+ cells with more than three nuclei was increased after treatment with the recombinant virus compared with the control culture. These data indicate that impaired osteoclastogenesis of Ccn2‐deficient cells also was rescued by overexpression of the Dc‐stamp gene.
Figure 7.
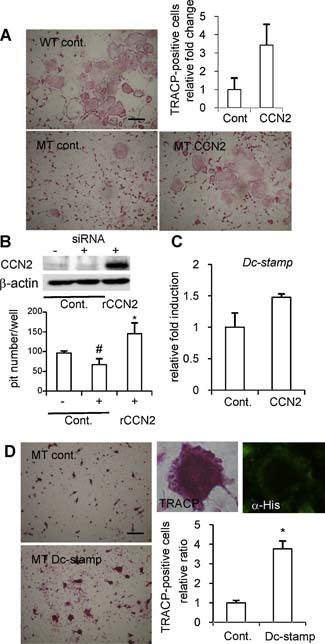
Effect of the addition of exogenous CCN2 or the forced expression of Dc‐stamp on rescue of impaired osteoclastogenesis in Ccn2‐deficient cells treated with M‐CSF and RANKL. (A) Rescue of osteoclastogenesis by the addition of exogenous CCN2. Fetal liver cells from Ccn2 null mice on embryonic day 14.5 were treated with M‐CSF and GST‐RANKL in the presence or absence of rCCN2 for 7 to 10 days, fixed, and stained for TRACP (left panel and lower right panel). The bar represents 50 µm. (Upper right panel) The graph shows quantification of TRACP+ multinucleated cells (MNCs) with more than three nuclei. Data are the means ± SDs of triplicate cultures. (B) Effect of exogenous CCN2 on the bone‐resorption activity in RAW264.7 cells treated with siRNA of Ccn2 (25 nM). (Upper panel) Western blot analysis by using an anti‐CCN2 antibody revealed that CCN2 production was decreased in siRNA‐treated cells. (Lower panel) RAW264.7 cells treated with siRNA of Ccn2 were cultured in an OAAS plate with or without rCCN2. After 7 days, cells were detached, and each well was stained with 0.05% toluidine blue for 5 minutes. The number of bone resorption pits in each well was calculated using a microscope. Data are the means ± SDs of three wells. The asterisk indicates a significant difference from siRNA‐treated cells (p < .05), whereas the number sign (#) indicates a marginally significant difference (p = .07). (C) Effect of exogenous CCN2 on the expression of the Dc‐stamp gene in Ccn2 null cells stimulated by M‐CSF and GST‐RANKL. Expression of Dc‐stamp gene was upregulated by the addition of CCN2 in Ccn2 mutants. Data are presented as mean and SD of two sets of independent cultures. (D) Rescue of osteoclastogenesis by Dc‐stamp overexpression. Ccn2 null fetal liver cells were cultured with M‐CSF and GST‐RANKL for 10 days after retroviral gene transfer of Dc‐stamp‐His. (Left) The formation of TRACP+ MNCs from Ccn2 null mice after Dc‐stamp overexpression was increased. The bar represents 50 µm. The graph shows quantification of TRACP+ MNCs with more than three nuclei (right). The asterisk indicates a significant difference from control culture (p < .05). TRACP staining or immunoreactivity to anti‐His antibody was detected in the same MNCs (upper right).
Discussion
In this study, we showed that RANKL‐induced osteoclastogenesis was positively regulated by CCN2. We hypothesize that CCN2 mediates another signaling pathway rather than RANKL signaling directly during osteoclast differentiation because gene expression of CCN2 was upregulated later than expression of NFATc1 regulated directly by RANKL signaling during osteoclastogenesis (Fig. 1 A). Moreover, we showed that CCN2 protein production was increased at the late of differentiation of RAW264.7 cells treated with RANKL (Fig. 1 B). Therefore, we hypothesized that CCN2 may be involved in the late stage of osteoclast differentiation. Because it was reported that although TRACP+ cells were found in the bone marrow of Ccn2 null mice, multinucleated osteoclasts were not seen,10 we investigated whether or not CCN2 was involved in cell fusion. Recently, a molecule involved in osteoclastic cell fusion was isolated.29 This molecule is a protein with putative seven‐transmembrane domains that was identified originally during gene‐expression screening of dendritic cells.29, 30 Therefore, this protein is termed dendritic cell‐specific transmembrane protein (DC‐STAMP).29, 30 In Dc‐stamp null mice, TRACP+ cells were present, but no multinucleated osteoclasts were identified in bone sections.29 In addition, it also was reported that overexpression of DC‐STAMP enhanced the formation of TRACP+ multinucleated cells from RANKL‐stimulated osteoclast precursors.23 These findings indicate that DC‐STAMP promotes the formation of multinucleated osteoclast‐like cells and thus suggests that this molecule has an essential role in cell‐cell fusion. It is well known that the expression of DC‐STAMP is strongly induced by interleukin 4 (IL‐4) or RANKL.31 In this study, we showed for the first time that the gene expression level of Dc‐stamp was significantly upregulated by stimulation of CCN2 alone (Fig. 3). Furthermore, we clarified that CCN2 directly interacted with DC‐STAMP (Figs. 4 and 5). These results suggest that CCN2 promotes cell‐cell fusion via upregulation of the expression of and interaction with DC‐STAMP. In fact, according to the results of our TRACP staining of bone sections from Ccn2 null mice, the number of TRACP+ multinucleated cells in those mice was decreased compared with that in their wild‐type littermates (Fig. 6). These findings support the hypothesis that CCN2 promotes osteoclast differentiation through the stimulation of cell‐cell fusion. To confirm this hypothesis, we initially tried to characterize in vitro cultures of bone marrow cells from Ccn2 null mice. However, because Ccn2 null mice showed neonatal lethality, it turned out to be difficult to isolate bone marrow cells from Ccn2 mutants.10 Therefore, we used fetal liver cells on embryonic day 14.5 from Ccn2 null mice instead.24 TRACP+ multinucleated osteoclasts were efficiently generated from mononuclear cells derived from wild‐type embryonic liver cells in the presence of M‐CSF and GST‐RANKL, whereas generation from fetal liver cells from Ccn2 null mice was much less (Fig. 6). Furthermore, we isolated total RNA from osteoclast‐like cells derived from Ccn2 null mice and investigated the expression of osteoclast marker genes by quantitative RT‐PCR. As a result, the expression levels of the osteoclast marker genes were decreased in osteoclast‐like cells from Ccn2 null mice compared with that in the wild‐type littermate cells (Fig. 6). Interestingly, in Ccn2 null fetal liver cells, although the number of TRACP+ cells with more than three nuclei was increased by the forced expression of Dc‐stamp in the presence of M‐CSF and RANKL, the effect of giant cell formation by the forced expression of Dc‐stamp seems to be less than the effect of the addition of exogenous CCN2 (Fig. 7 A, MT CCN2, and C, MT Dc‐stamp). Collectively, these findings suggest that CCN2 plays an important role in osteoclast differentiation via DC‐STAMP both through the upregulation of gene expression and through molecular interaction in an autocrine manner.
CCN1/Cyr61 (CCN1) and CCN3/Nov (CCN3) are other members of the CCN family of proteins.3, 4 CCN1 is upregulated in response to serum 1α,25‐dihydroxyvitamin D3 and cytokines, such as IL‐1β or TNF‐α,32 and CCN3 is expressed in a variety of tissues, including bone and hypertrophic cartilage, where it acts act as a downstream effecter of transforming growth factor β1 (TGF‐β1).33 Previous studies showed that both CCN1 and CCN3 inhibited osteoclastogenesis.33, 34 CCN1 and CCN3 bind to αvβ3 integrin,35 a molecule that plays an important role in osteoclastogenesis. One possibility is that these factors may act as an antagonist against αvβ3 integrin during osteoclastogenesis, but it is also possible that the effect of CCN1 or CCN3 therein is independent of integrins. On the other hand, it has been reported that CCN2 binds to integrins in a similar manner as CCN1 or CCN3.35 In this study, we showed that CCN2 was expressed in mature osteoclasts and promoted osteoclastogenesis through stimulating Dc‐stamp gene expression and interaction. These findings suggest that interaction not only with integrin but also with other factors, such as DC‐STAMP, plays an important role in the effect of CCN2 on osteoclastogenesis. It also should be noted that our recent study has uncovered possible interaction of CCN2 and RANK (Aoyama et al., unpublished data). Recently, it was reported that overexpression of CCN2 in osteoblasts under the control of the osteocalcin promoter impaired bone formation and led to osteopenia.36 That article suggested that overexpression of CCN2 caused a decrease in bone formation by antagonizing BMP, Wnt, and insulin‐like growth factor 1 (IGF‐1) signaling and activity. However, decreased bone volume is caused by an imbalance between bone formation and bone resorption. In fact, the osteoclast number was slightly increased in 1‐month‐old male Ccn2 transgenic mice, although the authors presented no apparent evidence of increased bone resorption. In addition, recently we showed that CCN2 combined with BMP‐2 inhibited the proliferation but not differentiation of chondrocytes.15 Therefore, overexpression of CCN2 under the control of the osteocalcin promoter may inhibit the proliferation of osteoblasts via this interaction with BMP‐2. In view of our current data, taken together with our previous data showing that CCN2 stimulates the proliferation and differentiation of cultured osteoblastic cells,6 CCN2 promotes both formation and resorption of bone tissues owing to stimulation not only of proliferation and differentiation of osteoblasts but also of formation of osteoclasts, which may cause imbalances in bone formation and resorption leading to osteopenia. However, further investigation obviously is needed to uncover the entire regulatory system that balances bone formation with resorption in vivo.
In conclusion, our study revealed that CCN2 not only promotes the expression of the Dc‐stamp gene, which plays an important role in cell‐cell fusion, but also interacts with this molecule to promote osteoclast differentiation. Further investigation is under way to clarify whether or not CCN2 is a functional ligand for DC‐STAMP.
Disclosures
All the authors state that they have no conflicts of interest.
Acknowledgements
This work was supported in part by grants from the programs Grants‐in‐Aid for Scientific Research (C) (to TN and SK) and (S) (to MT) from the Japan Society for the Promotion of Sciences, and Exploratory Research (to MT) of the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Drs Takako Hattori, Eriko Aoyama, and Mitsuhiro Hoshijima for their helpful suggestions and Ms Yuki Nonami and Yoko Tada for secretarial assistances.
References
- 1. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. [DOI] [PubMed] [Google Scholar]
- 2. Yavropoulou MP, Yovos JG. Osteoclastogenesis: current knowledge and future perspective. J Musculoskelet Neuronal Interact. 2008;8:204–216. [PubMed] [Google Scholar]
- 3. Takigawa M, Nakanishi T, Kubota S, Nishida T. Role of CTGF/Hcs24/ecogenin in skeletal growth control. J Cell Physiol. 2003;194:256–266. [DOI] [PubMed] [Google Scholar]
- 4. Kubota S, Takigawa M. Role of CCN2/CTGF/Hcs24 in bone growth. Int Rev Cytol. 2007;257:1–41. [DOI] [PubMed] [Google Scholar]
- 5. Nakanishi T, Nishida T, Shimo T, et al. Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte–specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology. 2000;141:264–273. [DOI] [PubMed] [Google Scholar]
- 6. Nishida T, Nakanishi T, Asano M, Shimo T, Takigawa M. Effects of CTGF/Hcs24, a hypertrophic chondrocyte‐specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J Cell Physiol. 2000;184:197–206. [DOI] [PubMed] [Google Scholar]
- 7. Nishida T, Kubota S, Nakanishi T, et al. CTGF/Hcs24, a hypertrophic chondrocyte‐specific gene product, stimulates proliferation and differentiation, but not hypertrophy of cultured articular chondrocytes. J Cell Physiol. 2002;192:55–63. [DOI] [PubMed] [Google Scholar]
- 8. Nishida T, Kubota S, Kojima S, et al. Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor). J Bone Miner Res. 2004;19:1308–1319. [DOI] [PubMed] [Google Scholar]
- 9. Shimo T, Nakanishi T, Nishida T, et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 1999;126:137–145. [DOI] [PubMed] [Google Scholar]
- 10. Ivkovic S, Yoon BS, Popoff SN, et al. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishida T, Kawaki H, Baxter RM, Deyoung RA, Takigawa M, Lyons KM. CCN2 (connective tissue growth factor) is essential for extracellular matrix production and integrin signaling in chondrocytes. J Cell Commun Signal. 2007;1:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawaki H, Kubota S, Suzuki A, et al. Functional requirement of CCN2 for intramembranous bone formation in embryonic mice. Biochem Biophys Res Commun. 2008;366:450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakata E, Nakanishi T, Kawai A, et al. Expression of connective tissue growth factor/hypertrophic chondrocyte‐specific gene product 24 (CTGF/Hcs24) during fracture healing. Bone. 2002;31:441–447. [DOI] [PubMed] [Google Scholar]
- 14. Nishida T, Kondo S, Maeda A, Kubota S, Lyons KM, Takigawa M. CCN family 2/connective tissue growth factor (CCN2/CTGF) regulates the expression of Vegf through Hif‐1α expression in a chondrocytic cell line, HCS‐2/8, under hypoxic condition. Bone. 2009;44:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maeda A, Nishida T, Aoyama E, Kubota S, Lyons KM, Takigawa M. CCN family 2/connective tissue growth factor modulates BMP signaling as a signal conductor, which action regulates the proliferation and differentiation of chondrocytes. J Biochem. 2009;145:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shimo T, Kubota S, Yoshioka N, et al. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J Bone Miner Res. 2006;21:1045–1059. [DOI] [PubMed] [Google Scholar]
- 17. Shimo T, Kubota S, Kondo S, et al. Connective tissue growth factor as a major angiogenic agent that is induced by hypoxia in a human breast cancer cell line. Cancer Lett. 2001;174:57–64. [DOI] [PubMed] [Google Scholar]
- 18. Nozawa K, Fujishiro M, Kawasaki M, et al. Connective tissue growth factor promotes articular damage by increased osteoclastogenesis in patients with rheumatoid arthritis. Arthritis Res Ther. 2009;11:R174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minato M, Kubota S, Kawaki H, et al. Module‐specific antibodies against human connective tissue growth factor: utility for structural and functional analysis of the factor as related to chondrocytes. J Biochem. 2004;135:347–354. [DOI] [PubMed] [Google Scholar]
- 20. Nishida T, Nakanishi T, Shimo T, et al. Demonstration of receptors specific for connective tissue growth factor on a human chondrocytic cell line (HCS‐2/8). Biochem Biophys Res Commun. 1998;247:905–909. [DOI] [PubMed] [Google Scholar]
- 21. Aoyama E, Hattori T, Hoshijima M, et al. N‐terminal domains of CCN family 2/connective tissue growth factor bind to aggrecan. Biochem J. 2009;420:413–420. [DOI] [PubMed] [Google Scholar]
- 22. Meiyanto E, Hoshijima M, Ogawa T, Ishida N, Takeya T. Osteoclast differentiation factor moduates cell cycle machinery and causes a delay in S phase progression in RAW264 cells. Biochem Biophys Res Commun. 2001;282:278–283. [DOI] [PubMed] [Google Scholar]
- 23. Kukita T, Wada N, Kukita A, et al. RANKL‐induced DC‐STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanazawa K, Kudo A. TRAF2 is essential for TNF‐α‐induced osteoclastogenesis. J Bone Miner Res. 2005;20:840–847. [DOI] [PubMed] [Google Scholar]
- 25. Hotokezaka H, Sakai E, Kanaoka K, et al. U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast‐like cells. J Biol Chem. 2002;277:47366–47372. [DOI] [PubMed] [Google Scholar]
- 26. Coste A, Dubourdeau M, Linas MD, et al. PPARγ promotes mannose receptor gene expression in murine macrophages and contributes to the induction of this receptor by IL‐13. Immunity. 2003;19:329–339. [DOI] [PubMed] [Google Scholar]
- 27. Kubota S, Hattori T, Shimo T, Nakanishi T, Takigawa M. Novel intracellular effects of human connective tissue growth factor expressed in Cos‐7 cells. FEBS Lett. 2000;474:58–62. [DOI] [PubMed] [Google Scholar]
- 28. Nishida T, Kubota S, Fukunaga T, et al. CTGF/Hcs24, hypertrophic chondrocyte‐specific gene product, interacts with perlecan in regulating the proliferation and differentiation of chondrocytes. J Cell Physiol. 2003;196:265–275. [DOI] [PubMed] [Google Scholar]
- 29. Yagi M, Miyamoto T, Sawatani Y, et al. DC‐STAMP is essential for cell‐cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hartgers FC, Vissers JL, Looman MW, et al. DC‐STAMP, a novel multimembrane‐spanning molecule preferentially expressed by dendritic cells. Eur J Immunol. 2000;30:3585–3590. [DOI] [PubMed] [Google Scholar]
- 31. Staege H, Brauchlin A, Schoedon G, Schaffner A. Two novel genes FIND and LIND differentially expressed in deactivated and Listeria‐infected human macrophages. Immunogenetics. 2001;53:105–113. [DOI] [PubMed] [Google Scholar]
- 32. Schütze N, Lechner A, Groll C, et al. The human analog of murine cystein rich protein 61 is a 1α, 25‐dihydroxyvitamin D3 responsive immediate early gene in human fetal osteoblasts: regulation by cytokines, growth factors, and serum. Endocrinology. 1998;139:1761–1770. [DOI] [PubMed] [Google Scholar]
- 33. Canalis E, Smerdel‐Ramoya A, Durant D, Economides AN, Beamer WG, Zanotti S. Nephroblastoma overexpressed (Nov) inactivation sensitizes osteoblasts to bone morphogenetic protein‐2, but nov is dispensable for skeletal homeostasis. Endocrinology. 2010;151:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crockett JC, Schütze N, Tosh D, et al. The matricellular protein cyr61 inhibits osteoclastogenesis by a mechanism independent of αvβ3 and αvβ5 . Endocrinology. 2007;148:5761–5768. [DOI] [PubMed] [Google Scholar]
- 35. Lau LF, Lam SCT. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res. 1999;248:44–57. [DOI] [PubMed] [Google Scholar]
- 36. Smerdel‐Ramoya A, Zanotti S, Stadmeyer L, Durant D, Canalis E. Skeletal overexpression of connective tissue growth factor impairs bone formation and causes osteopenia. Endocrinology. 2008;149:4374–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]