

Abstract
Elucidating gene regulatory network (GRN) from large scale experimental data remains a central challenge in systems biology. Recently, numerous techniques, particularly consensus driven approaches combining different algorithms, have become a potentially promising strategy to infer accurate GRNs. Here, we develop a novel consensus inference algorithm, TopkNet that can integrate multiple algorithms to infer GRNs. Comprehensive performance benchmarking on a cloud computing framework demonstrated that (i) a simple strategy to combine many algorithms does not always lead to performance improvement compared to the cost of consensus and (ii) TopkNet integrating only high-performance algorithms provide significant performance improvement compared to the best individual algorithms and community prediction. These results suggest that a priori determination of high-performance algorithms is a key to reconstruct an unknown regulatory network. Similarity among gene-expression datasets can be useful to determine potential optimal algorithms for reconstruction of unknown regulatory networks, i.e., if expression-data associated with known regulatory network is similar to that with unknown regulatory network, optimal algorithms determined for the known regulatory network can be repurposed to infer the unknown regulatory network. Based on this observation, we developed a quantitative measure of similarity among gene-expression datasets and demonstrated that, if similarity between the two expression datasets is high, TopkNet integrating algorithms that are optimal for known dataset perform well on the unknown dataset. The consensus framework, TopkNet, together with the similarity measure proposed in this study provides a powerful strategy towards harnessing the wisdom of the crowds in reconstruction of unknown regulatory networks.
Author Summary
Elucidating gene regulatory networks is crucial to understand disease mechanisms at the system level. A large number of algorithms have been developed to infer gene regulatory networks from gene-expression datasets. If you remember the success of IBM's Watson in ”Jeopardy!„ quiz show, the critical features of Watson were the use of very large numbers of heterogeneous algorithms generating various hypotheses and to select one of which as the answer. We took similar approach, “TopkNet”, to see if “Wisdom of Crowd” approach can be applied for network reconstruction. We discovered that “Wisdom of Crowd” is a powerful approach where integration of optimal algorithms for a given dataset can achieve better results than the best individual algorithm. However, such an analysis begs the question “How to choose optimal algorithms for a given dataset?” We found that similarity among gene-expression datasets is a key to select optimal algorithms, i.e., if dataset A for which optimal algorithms are known is similar to dataset B, the optimal algorithms for dataset A may be also optimal for dataset B. Thus, our “TopkNet” together with similarity measure among datasets can provide a powerful strategy towards harnessing “Wisdom of Crowd” in high-quality reconstruction of gene regulatory networks.
Introduction
Most genes do not exert their functions in isolation [1], but make their functions through regulations among them. Such regulatory interactions are in the same cell, between different cells, and even between different organs, forming large-scale gene regulatory networks (GRNs). The impact of genetic abnormality can spread through regulatory interactions in GRNs and alter the activity of other genes that do not have any genetic defects [2]. Analyses of GRNs are key to identify disease mechanisms and possible therapeutic targets for the future [1]. Therefore, reconstruction of accurate and comprehensive GRNs from genome-wide experimental data (e.g., gene expression data from DNA microarray experiments) is one of the fundamental challenges in systems biology [3], [4].
A plethora of algorithms have been developed to infer GRNs from gene expression data, i.e., mutual-information (MI) based algorithms [5]–[12], correlation-based algorithms [5], Bayesian networks (BNs) [13]–[17], regression-based algorithms [18]–[22], graphical gaussian model (ggm) [23], meta predictors that combine several different methods [24], [25], and several other approaches that were recently proposed [26]–[32], i.e., random forests based algorithm [26] (GENIE3) and two-way ANOVA based algorithm [27] (ANOVA). Each network-inference algorithm generates a confidence score for a link between two genes from expression data and assumes that a predicted link with higher confidence score is more reliable. Systematic and comparative assessment of the performance of these inference algorithms remains a major challenge in network reconstruction [33].
Several studies compared performances of the network-inference algorithms [7], [8], [9], [34]. Especially, the DREAM5 (Dialogue on Reverse Engineering Assessment and Methods) challenge evaluated performances of many and diverse network-inference algorithms (29 algorithms submitted by challenge participants and 6 commonly used “off-the-shelf” algorithms) by using benchmark dataset composed of large-scale Escherichia coli, Saccharomyces cerevisiae, and in silico regulatory networks and their corresponding expression datasets [35]. The evaluation demonstrated that no single individual algorithm performs optimally across all the three expression-datasets, i.e., GENIE3 and ANOVA perform optimally for E. coli dataset, while two algorithms based on regression techniques are optimal for in silico dataset. Further, algorithm-specific biases influence the recovery of different regulation patterns, i.e., MI and correlation based algorithms can recover feed-forward loops most reliably, while regression and BNs can more accurately recover linear cascades than MI and correlation based algorithms [35].
Above observations suggest that different network-inference algorithms have different strengths and weaknesses [33], . A natural corollary to the observations is that combining multiple network-inference algorithms may be a good strategy to infer an accurate and comprehensive GRN. Recently, Marbach et al. proposed a new network-inference algorithm, “Community Prediction”, by combining several network-inference algorithms that were submitted to DREAM5 challenge [35]. The Community Prediction combining 29 algorithms (“off-the-self” algorithms are not used) shows higher or at least comparable performance to the best among the 29 algorithms across all DREAM5 datasets. Further, performance of community prediction increases as the number of integrated algorithms increases. Thus, community prediction based on integration of many algorithms can be a robust approach to infer GRNs across diverse datasets and will provide a powerful framework to reconstruct unknown regulatory networks.
Analysis of DREAM5 results [35] reveal that algorithms complement each other in a context-specific manner and harnessing the combined strengths and weaknesses of diverse techniques can lead to high quality inference networks. Thus, it is important to analyze the anatomy of diversity and quantify it. This is particularly important to systematically evaluate the characteristics of individual techniques and leverage their diversity in finding an optimal combination set for a specific experimental data context. Recently, Marbach et al. showed that integration of algorithms with high-diversity outperform that with low-diversity [35]. However, their diversity analysis is qualitative and, to our knowledge, there is no measure to quantify algorithm diversity.
Analysis of small in silico datasets of the DREAM3 challenge demonstrated that integration of the best five algorithms outperforms integration of all algorithms submitted to the challenge [33]. Selection of optimal algorithms for a given expression data and integration of the selected optimal algorithms may be more powerful strategy to reconstruct accurate GRNs than using many algorithms. Development of a method to determine optimal algorithms is a key to reconstruct accurate and comprehensive GRNs, although it is difficult to identify beforehand optimal algorithms for reconstruction of an unknown regulatory network because of biological and experimental variations among expression datasets.
A measure to quantify similarity among gene-expression datasets could be a clue to determine the optimal algorithms for reconstruction of unknown regulatory networks. This is because, if expression-data associated with known regulatory network (e.g., the DREAM5 datasets) is similar to that with unknown regulatory network, optimal algorithms for data with known regulatory network could be also optimal for data with unknown regulatory network.
Motivated by the above observations and issues, this paper focuses on four strategies towards building a comprehensive network reconstruction platform –
A computational framework to integrate diverse inference algorithms.
Systematically assess the performance of the framework against the DREAM5 datasets composed of genome-wide transcriptional regulatory networks and their corresponding expression data from actual microarray experiments as well as in silico simulation.
Develop a measure to quantify diversity among inference techniques towards identifying optimal combination of algorithms which elucidate accurate GRNs.
Develop a measure to quantify similarity among expression datasets towards selecting optimal algorithms for reconstruction of unknown regulatory networks.
To investigate these possible strategies, we first develop a novel network-inference algorithm that can combine multiple network-inference algorithms. Second, to evaluate inference performances of the algorithms precisely, we used the DREAM5 datasets composed of E. coli and S. cerevisiae transcriptional regulatory networks and their corresponding expression data from large-scale microarray experiments, together with synthetic network and corresponding expression datasets (http://wiki.c2b2.columbia.edu/dream/index.php/D5c4). A cloud-based computing framework was developed on the Amazon Web Services (AWS) system to systematically benchmark the large data-sets and compute-intensive algorithms. Third, we define a mathematical function quantifying diversity between algorithm pairs to analyze the anatomy of diversity and its role in improving the performance of reverse engineering techniques. Finally, we present a similarity measure among expression-datasets and its potential to identify optimal algorithms for reconstruction of unknown regulatory networks.
Results
We developed a computational workflow for the combination of network-inference algorithms and systematic assessment of their performance. The workflow of our framework is composed of three steps (see Supplementary figure S1 and Materials and Methods for details):
Inference Methods: We obtained confidence score between gene pairs based on 29 algorithms submitted by DREAM5 participants and 6 commonly used “off the shelf” algorithms from Marbach et al [35]. Furthermore, we calculated confidence score between two genes based on other three algorithms, i.e., c3net [9], ggm [23], and mrnet [8] algorithms. We used, in total, 38 network-inference algorithms for the study.
Top k Net: A novel algorithm to generate predicted list of regulatory links by the network-inference algorithms that can combine multiple network-inference algorithms (in this case, 38 algorithms) (see Figure 1).
Performance Assessment: Comparative evaluation of the performance of TopkNet with that of the 38 network-inference algorithms and Community Prediction, benchmarked using the DREAM5 network-inference challenge dataset composed of the large synthetic data (number of genes = 1,643 and sample size = 805), large-scale E. coli and S. celevisiae networks (number of gene = 4,511 and 5,950, respectively), and their corresponding real microarray gene expression data (with sample size of 805 and 536, respectively). Table 1 summarizes the different data-sets employed in the performance assessment for this study. We used a cloud-computing infrastructure built on Amazon EC2 instances to infer GRNs from the large-scale DREAM5 dataset (see Materials and Methods for details).
Figure 1. Example of a prediction by TopkNet formed from five individual network predictions.

(a) Target Network. Circles and links are genes and regulatory links among genes, respectively. (b) The five lists are ranked according to the confidence levels of links, the most reliable prediction is at the top of the list and has the highest rank, i.e., Algorithm1 assigns the highest confidence level and the rank value of 1 to a link between nodes 1 and 2. The true link of the target network is highlighted in yellow. We regard links with rank of 1–7 as regulatory links inferred by the algorithms because the target network composed of 7 links. Red lines and blue dashed lines represent true positive and false negative links, respectively. (c) Five rank values for each link and the mean value among the five values. Green, red, orange, blue, and purple represent rank values from Algorithm1, Algorithm2, Algorithm3, Algorithm4, and Algorithm5, respectively. (d) Rank value of a link by TopkNet and that by Community Prediction. Top1Net and Top2Net regards 1st and 2nd highest value among five rank values for a link as the rank value of the link, respectively. Community Prediction calculates the mean value among five rank values for a link and regards the mean as the rank of the link. For example, rank of the links between genes 1 and 2 for Community Prediction is 7.4. This example illustrates how Top1Net can be more accurate than the other algorithms.
Table 1. The DREAM5 datasets used in this study.
| - | In silico1 | E. coli 2 | S. celevisiae 3 |
| Number of genes | 1,643 | 4,511 | 5,950 |
| Number of samples | 805 | 805 | 536 |
In silico Dream 5 dataset.
Dream 5 dataset from E.coli.
Dream5 dataset from S.cerevisiae.
Top k Net (the maximum value of k is the number of integrated algorithms, 38 in this case) is based on leveraging the diversity of the different techniques by combining the confidence of each gene pair interaction computed by the algorithms. TopkNet applied a bagging method, which was introduced by Breiman L [36], to combine confidence scores between each gene pair from multiple network inference algorithms. Top1Net assumes that two genes have a regulatory links between them, if at least one network-inference algorithm assigns high confidence level to the link between them, while Top38Net assumes that two genes have a regulatory link between them, only if all the 38 algorithms assign high confidence levels to the link between them (see below and Figure 1 for details). TopkNet with k = 2–37 and Community prediction (which takes the average of ranks assigned by different algorithms), are intermediates between Top1Net and Top38Net.
Figure 1 gives an illustrative example of Top k Net (for simplicity, in this illustrative example, we used 5 individual algorithms) on a sample target network (Figure 1A). As shown in Figure 1B, a network-inference algorithm assigns a confidence level to each link and links are ranked according to their confidence levels, i.e., a link with higher confidence level has higher rank value. For each link, 5 individual network-inference algorithms (represented by different colors) assigned 5 rank values to each link (Figure 1C). Among the five rank values of each link, Top k Net regards kth highest rank value as the rank value of the link (Figure 1D). For example, five rank values (1, 3.5, 10, 10.5, and 12) are assigned to the regulatory link between nodes 1 and 2 (Figure 1C). In this case, Top1Net and Top2Net regards 1 and 3.5 as the rank value of the link, respectively (see Figure 1D). As shown in color spectrums in Figure 1D, Top k Net algorithms reconstruct GRNs which include predicted connections from multiple algorithms.
Based on the observation that network-inference algorithms tend to assign high confidence levels to true-positive links [6], [34], Top1Net algorithm would infer the largest number of true-positive links among all algorithms, i.e., Top k Net algorithms would infer smaller number of true-positive links as the value of k increases and, at the same time, can avoid inferring false-positive links. Thus, in general, Top1Net would outperform other algorithms in terms of inference performances, as seen in the predicted network in Figure 1.
Comparative performance assessment
Network inference algorithms have increased following Moore's Law (doubling every two years) [33], [37]. Consequently, it has become increasing important to develop comprehensive performance benchmarking platforms to compare their relative strengths and weaknesses and leverage them to improve quality of inferred network. Two key components fundamental to performance assessment are representative metrics to quantify performance and standardized data sets on which to evaluate them.
In this section, we first outline these components employed in this study on the basis of which the performance of TopkNet is evaluated.
Benchmarking data sets
The performance of gene network reconstruction algorithms require benchmarking against various data sets representing network dynamics (for example, gene expression profiles) for which the underlying network is known. However, the ability to generate biologically plausible networks and validate them against experimental data remains a fundamental tenet in network reconstruction. In this respect, the DREAM initiative provides a community platform for the objective assessment of inference methods. The DREAM challenges provide a common framework on which to evaluate inference techniques against well-characterized data sets. In this study, we used large scale experimental data from the DREAM5 network inference challenge.
Performance metrics
True-positive rate, false-positive rate, recall, and precision are representative metrics to evaluate performances of network inference algorithms (see Materials and Methods for details). True-positive (false-positive) rate is, among all true (false) links, how many of them are with ranks beyond a threshold. Precision is, among all links with ranks beyond a threshold, how many of them are true links, while recall is, among all true links, how many of them are with ranks beyond a threshold. For example, in Figure 1, Top2Net for threshold of 3.5 shows recall = 6/7 (one missing) and precision = 6/7 (one mistake). By changing threshold gradually, we obtained a receiver operating characteristic (ROC) and Precision/recall (PR) curves that are graphical plots of true-positive rate vs. false-positive rate and precision vs. recall, respectively. These curves are straightforward visual representation of performances of network-inference algorithms.
Further, we calculated three representative metrics, i.e., AUC-PR (area under the PR curve), AUC-ROC (area under the ROC curve), and max f-score (f-score is harmonic mean of precision and recall) for these algorithms (see Materials and Methods for details). AUC-PR and AUC-ROC evaluate the average performances of network-inference algorithms, while max f-score evaluates the optimal performance of network-inference algorithms. A network-inference algorithm with higher inference performance would show higher AUC-PR, AUC-ROC, and max f-score. Moreover, DREAM5 also provides performance benchmarking package which computes an overall score (OS) across the entire dataset [35]. By using the package, we also calculated overall score to evaluate performance of TopkNet algorithms against community prediction and 38 algorithms (for performance of the individual 38 algorithms, see Supplementary table S1).
TopkNet performance on DREAM5 data set
To evaluate how TopkNet leverages diversity amongst the candidate algorithms to infer consensus network, we used the DREAM5 benchmarking data comprised of large scale synthetic and experimental gene regulatory networks for E.coli and S.cerevesiae as outlined in Table 1 and computed different performance metrics on them. As seen in the PR and ROC curves for in silico, E. coli, and S. cerevisiae datasets (Supplementary figures S2 and S3), Top6net shows constantly higher performance compared to community prediction. Other three performance metrics (AUC-PR, AUC-ROC, and Max f-score) of TopkNet with k = 5–8 are also higher than those of community prediction for all the three datasets (see Figures 2B–J). Thus, overall score of TopkNet with k = 5–8 is significantly higher than that of community prediction (see Figure 2B). However, the performance metrics of TopkNet is only comparable to the best individual algorithms and not significantly better. Community prediction also showed significantly lower performances than the best individual algorithm.
Figure 2. Performances of TopkNet and community prediction based on integration of the 38 network-inference algorithms.

Black squares and lines show performances of TopkNet algorithm. For example, values at k = 1 represent performances of Top1Net algorithm. Red and green lines represent performances of community prediction and those of the best algorithm, respectively. (A) Overall score. (B) AUC-PR for in silico dataset. (C) AUC-ROC for in silico dataset. (D) Max f-score for in silico dataset. (E) AUC-PR for E. coli dataset. (F) AUC-ROC for E. coli dataset. (G) Max f-score for E. coli dataset. (H) AUC-PR for S. cerevisiae dataset. (I) AUC-ROC for S. cerevisiae dataset. (J) Max f-score for S. cerevisiae dataset.
These results indicate that, while TopkNet would provide better strategy to integrate multiple algorithms than community prediction, such a strategy does not always significant increase in performance compared to the cost of integration. As seen in this section, the overall score of the best individual algorithm (40.279) is comparable to that of TopkNet with k = 5–7 (40.110–41.251) and is much higher than that of community prediction and TopkNet with k = 1 (30.228 and 10.432, respectively). This is because, for the DREAM5 datasets, several low-performance algorithms assign high confidence scores to many false-positive links and such false links could decrease the performance of TopkNet (especially, with k = 1) and community prediction algorithms.
Thus, by integrating only high-performance algorithms that tend to assign high confidence score to true-positive link, TopkNet (especially, with k = 1) and community prediction may show much higher performances than the best individual algorithms. To investigate this issue, we evaluate TopkNet (and community prediction) based on integration of 10 optimal algorithms (algorithms within top 10 highest AUC-PR) for each of the in silico, E. coli. and S. cerevisiae datasets. As seen in Supplementary figures S4 and S5, PR and ROC curves of Top1Net are constantly over those of the best individual algorithm and community prediction for in silico and E. coli datasets, although, for S. cerevisiae, PR-curve of the best individual algorithm slightly over that of Top1Net. Other three metrics (AUC-PR, AUC-ROC, and Max f-score) of TopkNet with low k (k = 1 for in silico and E. coli and k = 2 for S. cerevisiae) are significantly higher or at least comparable to those of the best individual algorithm and community prediction (see Figures 3B–J). Therefore, the overall score of TopkNet with k = 1 and 2 (74.935 and 73.261, respectively) are significantly higher than that of the best individual algorithm (40.279) and community prediction (56.158) (see Figure 3A). These results highlight that integration of multiple high-performance algorithms by Top1Net or Top2Net consistently reconstructs the most accurate GRNs for different datasets.
Figure 3. Performances of TopkNet and Community prediction based on integration of top 10 highest-performance algorithms.

Black squares and lines show performances of TopkNet algorithm. For example, values at k = 1 represent performances of Top1Net algorithm. Red and green lines represent performances of community prediction and those of the best algorithm, respectively. (A) Overall score. (B) AUC-PR for in silico dataset. (C) AUC-ROC for in silico dataset. (D) Max f-score for in silico dataset. (E) AUC-PR for E. coli dataset. (F) AUC-ROC for E. coli dataset. (G) Max f-score for E. coli dataset. (H) AUC-PR for S. cerevisiae dataset. (I) AUC-ROC for S. cerevisiae dataset. (J) Max f-score for S. cerevisiae dataset.
As demonstrated in this section, selection of optimal algorithms for a given expression data and Top1Net, Top2Net, and community prediction based on integration of the selected optimal algorithms could be a powerful approach to reconstruct high-quality GRNs. However, currently, to our knowledge, there is no method to determine beforehand optimal algorithms for expression data associated with an unknown regulatory network. Development of a method to determine optimal algorithms is a key to reconstruct unknown regulatory networks (We investigate this issue in the next section).
Selection of optimal algorithm pairs to infer GRNs based on algorithm diversity
Different network-inference algorithms employ different and often complementary techniques to infer gene regulatory interactions from an expression dataset. Therefore, a consensus driven approach, which leverages diversity in network-inference algorithms, can infer more accurate and comprehensive GRNs than a single network-inference algorithm. However, as demonstrated in this study, a simple strategy of increasing the number of algorithms may not always yield significant performance gains compared to the cost of consensus, i.e., the computation cost (CPU time and memory usage).
It is pertinent to analyze the anatomy of diversity between different algorithms in a theoretical framework to answer the questions of -
To what extent, then, are the algorithms different from each other?
Does bringing diversity of the algorithms into community prediction improve the quality of inferred networks?
For the purposes, Marbach et al. conducted principal component analysis (PCA) on confidence scores from 35 network-inference algorithms [35]. They mapped 35 algorithms onto 2nd and 3rd principal components and grouped the algorithms into four clusters by visual inspection. The analysis demonstrated that integration of three algorithms from different clusters shows higher performance than that from the same cluster. It indicates that the diversity signature of the selected algorithms, and not just the number of algorithms, plays an important role in the performance of the network reconstruction techniques.
However, their algorithm diversity is qualitative and, to our knowledge, there is no quantitative measure for algorithm diversity. In order to quantify diversity among the individual algorithms employed in this study, we developed two quantitative measures of diversity which calculates distance between algorithms pairs on the basis of confidence scores of regulatory interactions inferred by the algorithms. One is based on simple Euclidean distance (EUC distance) and the other is based on EUC distance on 2nd and 3rd components from PCA analysis (PCA distance) (see Materials and Methods for details). In Figure 4, we provide a toy model to explain how diversity among network-inference algorithms is defined.
Figure 4. A toy example to calculate diversity among algorithms.

(A) Confidence scores from algorithms. Confidence score of a link between two genes were generated by each of three algorithms. In this case, each algorithm has 6 confidence scores for 6 links. Note that the three algorithms in this example are algorithms to infer non-directional algorithms and make symmetrical matrices of confidence scores, i.e., confidence score of link from gene1 to gene 2 is same as that from gene2 to gene1. Thus, for simplicity, upper triangles of confidence score matrices are not shown in the figure. (B) Diversity among algorithms based on Euclidean distances. In this example, each of three algorithms has a vector of 6 confidence scores for 6 links between two genes. Euclidean distance between two vectors of confidence scores from two algorithms is calculated and is defined as diversity between the two algorithms. (C) Diversity among algorithms based on 2nd and 3rd components of PCA analysis. In this example, PCA analysis is conducted on three vectors of 6 confidence scores from three network-inference algorithms and the three algorithms are mapped on to 2nd and 3rd principal components (see left panel of C). Euclidean distance between two algorithms is calculated by using the 2nd and 3rd principal components and is defined as diversity between the two algorithms.
By using the diversity measures, we calculated distance among 10 optimal algorithms for each of the DREAM5 datasets to examine whether bringing quantified algorithm diversity into Top1Net (and Community prediction) improves the performances of network reconstruction. Based on the calculated distances, we defined high-diversity pairs as top 10% of algorithm pairs with highest distance, while low-diversity pairs are defined as bottom 10% of algorithm pairs with lowest distance. In this study, we have 45 algorithm pairs among 10 optimal algorithms and thus top 5 algorithm pairs with highest distance are high-diversity pairs, while bottom 5 algorithm pairs with lowest distance are low-diversity pairs.
Next, we evaluated the performances of Top1Net (or community prediction) based on integration of high-diversity pairs and those of low-diversity pair. As seen in Figures 5B–J and Supplementary figures S6B–J, AUC-PR, AUC-ROC, and max f-score of high-diversity pairs by EUC distance are higher or at least comparable to those of low-diversity pairs by EUC distance across all datasets. Especially, for in silico and E. coli datasets, AUC-PR and Max f-score of high-diversity pairs by EUC distance are significantly higher than that of low-diversity pairs by EUC distance. Thus, the overall score of high-diversity pairs is also significantly higher than that of low-diversity pair (P<0.05) (see Figure 5A and Supplementary figure S6A). The performances of high-diversity pairs by PCA distance are also higher or at least comparable to those of low-diversity pairs by PCA distance (see Supplementary figures S7 and S8). Furthermore, median value of the overall score of high-diversity pairs (47.725 and 50.250 by Top1Net, for EUC and PCA distances, respectively) are much higher than that by the best individual algorithms (40.279) and that by community prediction that integrates 38 network-inference algorithms (30.228). In summary, these results indicate that -,
Figure 5. Performances of Top1Net based on integration of high- or low-diversity algorithm pairs by EUC distance.

H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * and ** represent P<0.05 and P<0.01, by the Wilcoxon rank sum test.
Even for the same number of algorithms (in this case, two algorithms are integrated), the quantitative diversity to selected pairs can improve the performance of the consensus methods (TopkNet and community prediction).
Quantitative diversity-guided consensus can reduce the cost of consensus (only 2 algorithms integration instead of 38 algorithms integration in this case) without compromising the quality of the inferred network as shown in this study where the inference performance of high diversity pair is much higher than that of 38 algorithms combination.
Selection of optimal algorithms based on similarity among expression datasets towards reliable reconstruction of regulatory networks
Top1Net or Top2Net based on integration of highest-performance algorithms consistently reconstruct the most accurate GRNs, as demonstrated in the previous section (see Figure 3). However, as Marbach et al. mentioned, “Given the biological variation among organisms and the experimental variation among gene-expression datasets, it is difficult to determine beforehand which methods will perform optimally for reconstruction an unknown regulatory network” [35], and, to our knowledge, there is no method to select the optimal network-inference algorithms. Development of a method to select optimal network-inference algorithms for each of the expression datasets remains a major challenge in network reconstruction.
A measure to quantify similarity among expression datasets can be a key to select optimal network-inference algorithms for each of the datasets, because, if similarity between expression-data associated with known regulatory network (e.g., DREAM5 datasets) and that with unknown regulatory network is high, optimal algorithms for the known dataset can be repurposed to infer regulatory network from unknown dataset. Driven by this observation, we developed a similarity measure among gene-expression datasets based on algorithm diversity proposed in previous section.
First, we briefly explain the overview of the procedure to calculate similarity among expression datasets (see Figure 6 and Materials and Methods for the details). The procedure is composed of 4 steps. (1) The expression datasets were split into a dataset for which optimal algorithms are unknown (e.g, Data1 in Figure 6A) and datasets for which optimal algorithms are known (e.g., Data2 and Data3 in Figure 6A). (2) For each of the datasets, confidence scores of links were calculated by network-inference algorithms. In the example shown in Figure 6B, each of 5 algorithms calculates 6 confidence scores for 6 links. (3) By using the confidence scores calculated in the step (2), diversity among algorithms was calculated based on a distance measure proposed in the previous section (EUC and PCA based distances, see Figure 6C and Materials and Methods for details), for each of the datasets. In the example shown in Figure 6C, we have 10 algorithm pairs among 5 algorithms and thus, as shown in matrices in the figure, we have 10 distances between two algorithms for each of the three datasets. (4) By using algorithm diversity calculated in the step (3), we calculated correlation coefficient of the algorithm distances between two datasets (see Figure 6D). In terms of algorithm diversity, the correlation coefficient is regarded as similarity measure between the two datasets. In the example shown in Figure 6D, Data1 is more similar to Data2 than Data3. Thus, optimal algorithms for Data2 are better fit than those for Data3 to infer GRN from Data1.
Figure 6. Overview of a method to calculate similarity between two expression datasets.

(A) Datasets. Expression datasets were split into a dataset for which optimal algorithms are unknown (e.g., Data1) and datasets for which optimal algorithms are known (e.g., Data2 and Data3). (B) Confidence scores of links between two genes. For each of datasets, confidence scores from each of algorithms (e.g., algorithms, A1, A2, A3, A4, and A5) were calculated. (C) Diversity among algorithms. By using confidence scores calculated in (B), diversity among algorithms were calculated for each of three datasets. In this example, we examined five algorithms and thus, for each of the datasets, we have a vector of 10 distances between two algorithms. (D) Similarity between two expression datasets. Correlation coefficient between the vector of algorithm distances from Data1 and that from Data2 was calculated. The calculated correlation coefficient is defined as similarity between Data1 and Data2. In the example in this figure, Data1 is more similar to Data2 than Data3. Thus, optimal algorithms for Data2 could perform better than those for Data3 to infer GRN from Data1.
Next, to evaluate whether dataset similarity can be used to govern optimal selection of inference algorithms, we calculated the similarity among the DREAM5 gene-expression datasets and compared the performance of the algorithms across the datasets. As seen in Figure 7 and Table 2, correlation between S. cerevisiae and E. coli datasets (Spearman's correlation coefficient ρ = 0.99) is much higher than that between E. coli and in silico (ρ = 0.87 and 0.81 by EUC and PCA distances, respectively) and that between S. cerevisiae and in silico (ρ = 0.83). In terms of algorithm diversity, similarity between E. coli and S. cerevisiae datasets is much higher than that between E. coli and in silico and that between S. cerevisiae and in silico.
Figure 7. Similarity among gene-expression datasets based on algorithm diversity.

The scatter plots show correlation of algorithm distance between two gene-expression datasets. Each of points in scatter plots represents each of algorithm pairs. Because we have 703 algorithm pairs among 38 algorithms, 703 points are in each of the figures. Vertical axis represents (EUC or PCA) distance between two algorithms for one gene-expression dataset, while horizontal axis represents that for the other gene-expression dataset. (A) Scatter plots of EUC distance for in silico and E. coli datasets. (B) Scatter plots of EUC distance for in silico and S. cerevisiae dataset. (C) Scatter plots of EUC distance for E. coli and S. cerevisiae datasets (D) Scatter plots of PCA distance for in silico and E. coli datasets. (E) Scatter plots of PCA distance for in silico and S. cerevisiae datasets. (F) Scatter plots of PCA distance for E. coli and S. cerevisiae datasets.
Table 2. Correlation coefficient of algorithm distances and that of performance metrics across the DREAM5 gene-expression datasets.
| Dataset 1 | Dataset 2 | EUC distance1 | PCA distance2 |
| In silico 3 | E.coli 4 | 0.87 | 0.81 |
| In silico | S.cerevisiae 5 | 0.83 | 0.83 |
| E.coli | S.cerevisiae | 0.99 | 0.99 |
Spearman's correlation coefficient of algorithm distance (EUC distance) between Dataset 1 and Dataset 2.
Spearman's correlation coefficient of algorithm distance (PCA distance) between Dataset 1 and Dataset 2.
In silico Dream 5 dataset.
Dream 5 dataset from E.coli.
Dream5 dataset from S.cerevisiae.
Further correlation of algorithm performances between dataset pair with high similarity (e.g., E. coli and S. cerevisiae pair) is higher than that between dataset pair with low similarity (e.g., in silico and E. coli pair and in silico and S. cerevisiae pair) (see Supplementary figure S7 and Supplementary Table S2). These results indicate that, for dataset pair with high similarity, optimal network-inference algorithms for one dataset also tend to be optimal for the other dataset.
From above observations (observations in Figure 7, Supplementary figure S9, Table 2, and Supplementary table S2), we hypothesized that, if similarity between the two expression-datasets is high, integration of algorithms that are optimal for one dataset could perform well on the other dataset. To examine this issue in more detail, we integrated algorithms that are optimal for S. cerevisia dataset (algorithms with 10 highest AUC-PR values on the dataset) and those for the in silico dataset and evaluated their performance of these two integrations against E. coli dataset.
As seen in Figures 8A, B, and C, against the E.coli dataset, performances (AUC-PR, AUC-ROC, and max f-score) of optimal integration from S. cerevisiae dataset (green lines) are generally higher than those from in silico dataset (red lines). Further, against the S. cerevisiae dataset, we evaluate performances of optimal-algorithm integration from E. coli dataset and that for in silico dataset and found that optimal integration from E. coli dataset (green lines) generally outperform that from in silico dataset (red lines) (see Figures 8D, E, and F). Because similarity between S. cerevisiae and E. coli datasets are much higher than that between E. coli and in silico datasets and that between S. cerevisiae and in silico datasets (see Figure 7 and Table 2), these results support the above hypothesis.
Figure 8. Optimal algorithm selection based on similarity among expression datasets and its potential to improve network-inference accuracy.

Red lines show performance of TopkNet integrating algorithms that are optimal for a dataset with high-similarity, while green lines show that with low-similarity. Blue lines show performance of TopkNet integrating top 10 highest-performance algorithms. Dashed lines in red, green, and blue represent performance of community prediction integrating algorithms that are optimal for a dataset with high-similarity, that with low-similarity, and top 10 highest-performance algorithms, respectively. (A) AUC-PR for E. coli dataset. (B) AUC-ROC for E. coli dataset. (C) Max f-score for E. coli dataset. (D) AUC-PR for S. cerevisiae dataset. (E) AUC-ROC for S. cerevisiae dataset. (F) Max f-score for S. cerevisiae dataset.
Further, as shown in Figure 8, performance of Topknet integrating optimal algorithms from a dataset with high-similarity (green lines) is comparable to that integrating top 10 highest-performance algorithms (blue lines). Thus, data-similarity based optimal algorithm selection together with TopkNet (or community prediction) based integration of the selected optimal algorithms can be a powerful strategy to reconstruct unknown regulatory network.
Discussion
With an increasing corpus of inference algorithms, leveraging their diverse and sometimes complementary approaches in a community consensus can be a promising strategy for reconstruction of gene regulatory networks from large scale experimental data. A computational platform to systematically analyze, assess and leverage these diverse techniques is essential for the successful application of reverse engineering in biomedical research.
This study presents a reverse engineering framework which can flexibly integrate multiple inference algorithms, based on Top k Net - a novel technique for building a consensus network based on the algorithms. It is pertinent to note here that the consensus framework based on Top k Net can be flexibly extended to include various types of network-inference algorithms.
Comparative evaluation on the DREAM5 datasets showed that, although TopkNet based on 38-algorithm integration shows lower or at most comparable performance to the best individual algorithms, Top1Net based on integration of top 10 highest performance algorithms significantly outperforms the best individual algorithm as well as community prediction. The results demonstrated that (i) a simple strategy to combine many algorithms does not always lead to performance improvement compared to the cost of consensus and (ii) selection of high-performance algorithms for a given expression dataset and Top1Net based on integration of the selected high-performance algorithms could be a powerful strategy for reliable reverse engineering.
Why does Top1net algorithm integrating 10 optimal algorithms perform quite well and outperform the best individual method? This is because 10 optimal algorithms tend to assign high-confidence scores to true-positive links and Top1net method can recover many true-positive links that are with the highest confidence scores from 10 optimal algorithms. Furthermore, 10 optimal algorithms are based on different techniques (e.g., mutual information, regression, and other statistical techniques) and Top1net can leverage diversity from the optimal algorithms. For example, the optimal algorithms based on mutual-information and regression techniques can accurately recover true positive links in feed-forward loops and linear cascade modules, respectively [35], while Top1net could integrate the algorithms and accurately recover both feed-forward loops and linear cascade module in a GRN. Therefore, Top1net shows higher inference performance than the best individual algorithms.
Why, then, Top1net outperforms community prediction and Topknet with higher k? Community prediction and Topknet with larger k recover links with lower confidence scores than Top1net, i.e., community prediction uses mean among confidence scores from 10 optimal algorithms and Topknet uses kth highest confidence score from the algorithms. Links with lower confidence scores from optimal algorithms are more likely to be false-positive links and thus Top1net shows higher inference performance than community prediction and Topknet with higher k.
A key to reconstruct accurate GRNs is development of a method to determine optimal algorithms for a given expression dataset associated with unknown regulatory network. As mentioned in results, if similarity between expression-data associated with known regulatory network (i.e., DREAM5 datasets) and that with an unknown regulatory network is high, optimal algorithms for data with known regulatory network may be also optimal for reconstruction of the unknown regulatory network.
Based on this observation, we developed a measure to quantify similarity among the expression datasets based on algorithm diversity and demonstrated that, if similarity between the two expression-datasets is high, integration of algorithms that are optimal for one dataset could perform well on the other dataset. Thus, the similarity measure proposed in this study can be a good clue to identify optimal algorithms for reliable reconstruction of an unknown regulatory network.
The consensus framework outlined in this paper, TopkNet, together with analysis of similarity among expression datasets, provide a powerful platform towards harnessing the wisdom of the crowds approach in reconstruction of large scale gene regulatory networks.
Materials and Methods
DREAM5 datasets
We used the DREAM5 datasets (http://wiki.c2b2.columbia.edu/dream/index.php/D5c4) to evaluate performance of network-inference algorithms. The DREAM5 dataset composed of an in-silico network (1,643 genes), the real transcriptional regulatory network of E. coli (4,511 genes), that of S. celecisiae (5,950 genes), and corresponding expression dataset (805, 805, and 536 samples for the in-silico, E. coli, and S. celevisiae networks, respectively). The expression dataset of E. coli and that of S. celevisae are composed of hundreds of experiments, i.e., genetic, drug, and environmental perturbations. The in-silico network is generated by extracting a subnetwork composed of 1,643 genes from the E. coli transcriptional network. The expression datasets of the in-silico network was simulated by software GeneNetWeaver version 2.0 [38]. For the DREAM5 datasets, in the same manner to Marbach et al. [35], we used the links with the top 100,000 highest confidence scores by each network-inference algorithm to evaluate performance of the algorithm.
To evaluate performance of inference algorithms for the DREAM5 datasets, DREAM organizers provide a matlab software (http://wiki.c2b2.columbia.edu/dream/index.php/D5c4). The software calculates 4 metrics for each network, i.e., AUC-PR, AUC-ROC, AUC-PR p-value, and AUC-ROC p-value. AUC-PR (AUC-ROC) p-value is the probability that a given or greater AUC-PR (AUC-ROC) is obtained by random scoring of links. Furthermore, the software calculates an overall score that was used to evaluate the overall performance of the algorithms for all three networks (the large synthetic network, large real E. coli, and S. celevisiae GRNs) of the DREAM5 network inference challenge. The overall score (OS) is defines as OS = 0.5(p 1+p 2), where p 1 and p 2 are the mean of the log-transformed AUC-PR p-values and that of the log-transformed AUC-ROC p-values taken over the three networks of the DREAM5 challenge, respectively.
Confidence score of regulatory links from 38 network-inference algorithms
We obtained confidence scores between two genes by 35 algorithms (29 algorithms are from DREAM5 participants and 6 algorithms are commonly used “off-the shelf” algorithms) from supplementary file of Marbach et al. [35]. For c3net, ggm, and mrnet algorithms, we calculated confidence scores of regulatory link by using GeneNet package [39], c3net R package [9], [10], and minet R package [40], respectively. Because Marbach et al. used links with top 100,000 highest confidence scores from each of 35 algorithms for analyses [35], we used top 100,000 links from c3net, ggm, and mrnet for analyses in this study.
Metrics to evaluate performance of inference algorithms
For a given threshold value of confidence level, network-inference algorithms predict whether a pair of genes have regulatory link or not. A pair of genes with a predicted link is considered as a true positive (TP) if the link is present in the underlying synthetic network, while the pair is a false positive (FP) if the synthetic network does not have the link. Similarly, a pair of genes without a predicted link is considered as a true negative (TN) or false negative (FN) depending on whether the link exists or not in the underlying synthetic network, respectively. By using the values of TP, FP, TN, and FN, we can calculate several metrics to evaluate performances of network-inference algorithms.
One representative metric is precision/recall curve where the precision (p) and recall (r) are defined as 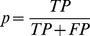 and
and 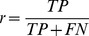 , respectively. By using many threshold values, we obtained a precision/recall curve that is a graphical plot of the precision vs. the recall and is a straight forward visual representation of performances of network-inference algorithms. The area under the precision/recall curve (AUC-PR) is a summary metric of precision/recall curve and measures the average accuracy of network-inference algorithms. Another representative metric is ROC curve that is a graphical plot of the true-positive rate vs. the false-positive rate. The area under the ROC curve (AUC-ROC) also represents the average inference performance of algorithms. On the other hand, max f-score [41] evaluates optimum performance of network-inference algorithms where f-score is defined as harmonic mean of the precision and recall (
, respectively. By using many threshold values, we obtained a precision/recall curve that is a graphical plot of the precision vs. the recall and is a straight forward visual representation of performances of network-inference algorithms. The area under the precision/recall curve (AUC-PR) is a summary metric of precision/recall curve and measures the average accuracy of network-inference algorithms. Another representative metric is ROC curve that is a graphical plot of the true-positive rate vs. the false-positive rate. The area under the ROC curve (AUC-ROC) also represents the average inference performance of algorithms. On the other hand, max f-score [41] evaluates optimum performance of network-inference algorithms where f-score is defined as harmonic mean of the precision and recall (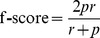 ). As predictions of network-inference algorithms become more accurate, the value of AUC-PR, AUC-ROC, and max f-score becomes higher. We used AUC-PR, AUC-ROC, and max f-score for performance evaluation. To obtain these three metrics, we used package provided by the DREAM5 team [35] (PR curve, ROC curve, AUC-PR, AUC-ROC, and overall score) and perl script provided by Küffner et al. (max f-score) [27].
). As predictions of network-inference algorithms become more accurate, the value of AUC-PR, AUC-ROC, and max f-score becomes higher. We used AUC-PR, AUC-ROC, and max f-score for performance evaluation. To obtain these three metrics, we used package provided by the DREAM5 team [35] (PR curve, ROC curve, AUC-PR, AUC-ROC, and overall score) and perl script provided by Küffner et al. (max f-score) [27].
Distances among network-inference algorithms
By using confidence scores among genes by network-inference algorithms, we calculated, D
EUC(X,Y), the simple Euclidean distance between two network-inference algorithms (EUC distance) X and Y for expression datasets with given number of genes and given sample size. Before giving a definition for D
EUC(X,Y), let us first define some notations. Let n be number of genes in the expression dataset and CS(i, j, X) be confidence value between genes i and j by algorithm X on the expression dataset. G = {(1,2), (2,3), … (i,j) …(n-1,n)} represents the list of all possible combinations of two genes for n genes. We defined the EUC distance between the two algorithms as 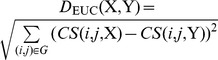 .
.
Further, we calculated, D
PCA(X,Y), the distance between two network-inference (X and Y) on 2nd and 3rd principal components (PCA distance) from PCA analysis on confidence scores of 38 algorithms. Let PC
2(X) and PC
3(X) be the 2nd and 3rd components of X, respectively. We defined the PCA distance between two algorithms as 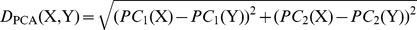 . For the PCA analysis, we used R code, pricomp2.R, obtained from http://aoki2.si.gunma-u.ac.jp/R/src/princomp2.R.
. For the PCA analysis, we used R code, pricomp2.R, obtained from http://aoki2.si.gunma-u.ac.jp/R/src/princomp2.R.
Similarity between two expression datasets based on algorithm diversity
By using distances among algorithms, we calculated, S(da1,da2), similarity between two expression datasets da1 and da2. Before giving definition of S(da1,da2), let us first define some notation. Let k and A = {a1, a2, …, ai, …, ak} be the number of algorithms and the list of the algorithms, respectively. AC = {(a1,a2),(a2,a3),…,(ai -1,ai),…, (ak -1,ak)} represents all possible combinations of two algorithms among k algorithms (k(k-1)/2 algorithm combinations). For example, in this study, we examined 38 algorithms and have 38*37/2 = 703 algorithm combinations. D(ai,aj,da1) represents distances between two algorithms a1 and a2 for da1. D da1{AC} = {D(a1,a2,da1), D(a2,a3,da1), …, D(ai -1,ai,da1), … D(ak -1,ak,da1)} represents a vector of k(k-1)/2 algorithm distances for da1 (in this study, we have a vector of 703 algorithm distances for each of DREAM5 datasets). We defined S(da1,da2) as Spearman's correlation coefficient between two vectors, D da1{AC} and D da2{AC}.
Cloud computing infrastructure on Amazon EC2 to infer GRNs from the large-scale DREAM5 expression datasets
To infer GRNs from the large-scale expression data of DREAM5 (expression data of E.coli and S. cerevisiae), we built a cloud computing infrastructure on Amazon EC2 “High-memory double” instances (34.2 GB memory and 4 virtual cores with 3.25 EC2 Compute Units each) with Redhad linux and R version 2.15.0 [42]. We placed all the input data on the ephemeral storage disk (850 GB) of the Amazon EC2 instances and TopkNet output results (e.g., a listing of confidence scores between genes) to files on the storage disk.
Supporting Information
The work flow of the experimental framework of this study. Expression datasets were obtained from the DREAM5 challenge web page (http://wiki.c2b2.columbia.edu/dream/index.php/The_DREAM_Project). Inferred network from the expression datasets by a network-inference algorithm is compared to the networks of the DREAM5 challenge (Step (iii)). See Materials and Methods for details.
(TIF)
PR curves of Top k Net and community prediction based on integration of the 38 individual algorithms. (A) PR curves for in silico datasets. (B) PR curves for E. coli dataset. (C) PR curves for S. cerevisiae dataset. Vertical and horizontal axes represent precision and recall, respectively.
(TIF)
ROC curves of Top k Net and community prediction based on integration of the 38 individual algorithms. (A) ROC curves for in silico datasets. (B) ROC curves for E. coli dataset. (C) ROC curves for S. cerevisiae dataset. Vertical and horizontal axes represent true-positive and false-positive rate, respectively.
(TIF)
PR curves of Top k Net and community prediction based on integration of the top 10 highest-performance algorithms. (A) PR curves for in silico datasets. (B) PR curves for E. coli dataset. (C) PR curves for S. cerevisiae dataset. Vertical and horizontal axes represent precision and recall, respectively.
(TIF)
ROC curves of Top k Net and community prediction based on integration of the top 10 highest-performance algorithms. (A) ROC curves for in silico datasets. (B) ROC curves for E. coli dataset. (C) ROC curves for S. cerevisiae dataset. Vertical and horizontal axes represent true-positive and false-positive rate, respectively.
(TIF)
Performances of community prediction based on integration of high- or low-diversity algorithm pairs by EUC distance. H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * and ** represent P<0.05 and P<0.01, by the Wilcoxon rank sum test.
(TIF)
Performances of Top1Net based on integration of high- or low- diversity algorithm pairs by PCA distance. H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * represents P<0.05 by the Wilcoxon rank sum test.
(TIF)
Performances of community prediction based on integration of high- or low- diversity algorithm pairs by PCA distance. H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * and ** represent P<0.05 and P<0.01, by the Wilcoxon rank sum test.
(TIF)
Comparison of algorithm performances across gene-expression datasets. The scatter plots show correlation of algorithm performance between two gene-expression datasets. Vertical axis represents algorithm performance for one gene-expression dataset, while horizontal axis represents that for the other gene-expression dataset. (A) Scatter plots of AUC-PR for in silico and E. coli datasets. (B) Scatter plots of AUC-PR for in silico and S. cerevisiae datasets. (C) Scatter plots of AUC-PR for E. coli and S. cerevisiae datasets. (D) Scatter plots of AUC-ROC for in silico and E. coli datasets. (E) Scatter plots of AUC-ROC for in silico and S. cerevisiae datasets. (F) Scatter plots of AUC-ROC for E. coli dataset and S. cerevisiae datasets. (G) Scatter plots of max f-score for in silico and E. coli datasets. (H) Scatter plots of max f-score for in silico and S. cerevisiae datasets. (I) Scatter plots of max f-score for E. coli and S. cerevisiae datasets.
(TIF)
Performances of the 38 individual algorithms. The table shows overall score, AUC-PR, and AUC-ROC of the 38 algorithms.
(XLS)
Correlation coefficient of performance metrics across the DREAM5 gene-expression datasets. The table shows Spearman's correlation coefficient of performance metrics across the DREAM5 gene expression datasets.
(DOC)
Acknowledgments
The authors thank Matsuoka Y, Kang H, Fujita K, Lopes T, and Shoemaker J for their useful comments and discussion.
Funding Statement
This work is, in part, supported by funding from the HD-Physiology Project of the Japan Society for the Promotion of Science (JSPS) to the Okinawa Institute of Science and Technology (OIST). Additional support is from a Canon Foundation Grant, the International Strategic Collaborative Research Program (BBSRC-JST) of the Japan Science and Technology Agency (JST), the Exploratory Research for Advanced Technology (ERATO) programme of JST to the Systems Biology Institute (SBI), a strategic cooperation partnership between the Luxembourg Centre for Systems Biomedicine and SBI, and from Toxicogenemics program of Ministry of Health, Labour and Welfare. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vidal M, Cusick ME, Barabasi AL (2011) Interactome networks and human disease. Cell 144: 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barabasi AL, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nature Review Genetics 12: 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bansal M, Belcastro V, Ambesi-Impiombato A, Bernardo DD (2007) How to infer gene networks from expression profiles. Molecular Systems Biology 3: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smet RD, Marchal K (2010) Advantages and limitations of current network inference methods. Nature Reviews Microbiology 8: 717–729. [DOI] [PubMed] [Google Scholar]
- 5. Butte A, Kohane I (2000) Mutual information relevance networks: functional genomic clustering using pair wise entropy measurements. Pacific Symposium on Biocomputing 5: 418–429. [DOI] [PubMed] [Google Scholar]
- 6. Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, et al. (2006) ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7: S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, et al. (2007) Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biology 5: e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyer PE, Kontos K, Lafitte F, Bontempi G (2007) Information-theoretic inference of large transcriptional regulatory networks. EURASIP Journal on Bioinformatics and Systems Biology 2007: 79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Altay G, Emmert-Streib F (2010a) Inferring the conservative causal core of gene regulatory networks. BMC Systems Biology 4: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Altay G, Emmert-Streib F (2011a) Structural influence of gene networks on their inference: analysis of C3NET. Biology Direct 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Altay G, Asim M, Markowetz F, Neal DE (2011b) Differential C3NET reveals disease networks of direct physical interactions. BMC Bioinformatics 12: 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mani S, Cooper GF (2004) Causal discovery using a bayesian local causal discovery algorithm. In: Proceedings of the World Congress on Medical Informatics 2004; 7–11 September 2004; San Francisco, California, United States. Medinfo 2004. Available: http://ebooks.iospress.nl/publication/21082. Accessed 15 June 2013.
- 13. Friedman N, Linial M, Nachman I, Pe'er D (2000) Using Bayesian networks to analyze expression data. Journal Computational Biology 7: 601–620. [DOI] [PubMed] [Google Scholar]
- 14. Yu J, Smith VA, Wang PP, Hartemink AJ, Jarvis ED (2004) Advances to bayesian network inference for generating causal networks from observational biological data. Bioinformatics 20: 3594–3603. [DOI] [PubMed] [Google Scholar]
- 15.Tsamardinos I, Aliferis CF, Statnikov A (2003) Time and sample efficient discovery of Markov blankets and direct causal relations. In: Proceedings of the Ninth ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 2003; 24–27 August; Washington, DC, United States. KDD 2003. Available: http://dl.acm.org/citation.cfm?id=956838. Accessed 15 June 2013.
- 16. Aliferis CF, Statnikov A, Tsamardinos I, Mani S, Koutsoukos XD (2010) Local causal and Markov blanket induction for causal discovery and feature selection for classification part I: algorithm and empirical evaluation. Journal of Machine Learning Research 11: 171–234. [Google Scholar]
- 17. Statnikov A, Aliferis C (2010) Analysis and computational dissection of molecular signature multiplicity. PLoS Computional Biology 6: e1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haury AC, Mordelet F, Vera-Licona P, Vert JP (2012) TIGRESS: Trustful Inference of Gene REgulation using Stability Selection. BMC Systems Biology 6: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan M, Lin Y (2006) Model selection and estimation in regression with grouped variables. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 68: 49–67. [Google Scholar]
- 20. Lèbre S, Becq J, Devaux F, Stumpf MPH, Lelandais G (2010) Statistical inference of the time-varying structure of gene-regulation networks. BMC Systems Biology 4: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meinshausen N, Bühlmann P (2010) Stability selection. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 72: 417–473. [Google Scholar]
- 22. van Someren EP, Vaes BL, Steegenga WT, Sijbers AM, Dechering KJ, et al. (2006) Least absolute regression network analysis of the murine osteoblast differentiation network. Bioinformatics 22: 477–484. [DOI] [PubMed] [Google Scholar]
- 23. Schafer J, Strimmer K (2005) An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics 21: 754–764. [DOI] [PubMed] [Google Scholar]
- 24. Greenfield A, Madar A, Ostrer H, Bonneau R (2010) DREAM4: Combining genetic and dynamic information to identify biological networks and dynamical models. PLoS ONE 5: e13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watkinson J, Liang K-C, Wang X, Zheng T, Anastassiou D (2009) Inference of regulatory gene interactions from expression data using three-way mutual information. Annals of the New York Academy of Sciences 1158: 302–313. [DOI] [PubMed] [Google Scholar]
- 26. Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P (2010) Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5: e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Küffner R, Petri T, Tavakkolkhah P, Windhager L, Zimmer R (2012) Inferring Gene Regulatory Networks by ANOVA. Bioinformatics 28: 1376–1382. [DOI] [PubMed] [Google Scholar]
- 28. Karlebach G, Shamir R (2012) Constructing logical models of gene regulatory networks by integrating transcription factor-DNA interactions with expression data: an entropy-based approach. Journal of Computational Biology 19: 30–41. [DOI] [PubMed] [Google Scholar]
- 29. Yeung KY, Bumgarner RE, Raftery AE (2005) Bayesian model averaging: development of an improved multi-class, gene selection and classification tool for microarray data. Bioinformatics 21: 2394–2402. [DOI] [PubMed] [Google Scholar]
- 30. Yip KY, Alexander RP, Yan K-K, Gerstein M (2010) Improved reconstruction of in silico gene regulatory networks by integrating knockout and perturbation data. PLoS ONE 5: e8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sîrbu A, Ruskin HJ, Crane M (2011) Stages of gene regulatory network inference: the evolutionary algorithm role. In: Evolutionary Algorithms, Kita E, editor. InTech. 521–546.
- 32. Song MJ, Lewis CK, Lance ER, Chesler EJ, Yordanova RK, et al. (2009) Reconstructing generalized logical networks of transcriptional regulation in mouse brain from temporal gene expression data. EURASIP Journal on Bioinformatics and Systems Biology 2009: 545176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, et al. (2010) Revealing strengths and weakness of methods for gene network inference. Proceedings of National Academy of Science USA 107: 6286–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altay G, Emmert-Streib F (2010b) Revealing differences in gene network inference algorithms on the network level by ensemble methods. Bioinformatics 26: 1738–1744. [DOI] [PubMed] [Google Scholar]
- 35. Marbach D, Costello JC, Küffner R, Vega NM, Prill RJ, et al. (2012) Wisdom of crowds for robust gene network inference. Nature Methods 9: 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Breiman L (1996) Bagging Predictors. Machine Learning 24: 123–140. [Google Scholar]
- 37. Stolovitzky G, Monroe D, Califano A (2007) Dialogue on reverse-engineering assessment and methods: The dream of high-throughput pathway inference. Annals of the New York Academy of Sciences 1115: 1–22. [DOI] [PubMed] [Google Scholar]
- 38. Schaffter T, Marbach D, Floreano D (2011) GeneNetWeaver: In silico benchmark generation and performance profiling of network inference methods. Bioinformatics 27: 2263–2270. [DOI] [PubMed] [Google Scholar]
- 39. Schafer J, Opgen-Rhein R (2006) Reverse engineering genetic networks using the GeneNet package. R News 6: 50–53. [Google Scholar]
- 40. Meyer PE, Lafitte F, Bontempi G (2008) minet: A R/Bioconductor package for inferring large transcriptional network using mutual information. BMC Bioinformatics 9: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rijsbergen CJV (1979) Information Retrieval. London: Butterworth-Heinemann. [Google Scholar]
- 42.R Development Core Team R: A language and environment for statistical computing, (R Foundation for Statistical Computing, Vienna, 2012)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The work flow of the experimental framework of this study. Expression datasets were obtained from the DREAM5 challenge web page (http://wiki.c2b2.columbia.edu/dream/index.php/The_DREAM_Project). Inferred network from the expression datasets by a network-inference algorithm is compared to the networks of the DREAM5 challenge (Step (iii)). See Materials and Methods for details.
(TIF)
PR curves of Top k Net and community prediction based on integration of the 38 individual algorithms. (A) PR curves for in silico datasets. (B) PR curves for E. coli dataset. (C) PR curves for S. cerevisiae dataset. Vertical and horizontal axes represent precision and recall, respectively.
(TIF)
ROC curves of Top k Net and community prediction based on integration of the 38 individual algorithms. (A) ROC curves for in silico datasets. (B) ROC curves for E. coli dataset. (C) ROC curves for S. cerevisiae dataset. Vertical and horizontal axes represent true-positive and false-positive rate, respectively.
(TIF)
PR curves of Top k Net and community prediction based on integration of the top 10 highest-performance algorithms. (A) PR curves for in silico datasets. (B) PR curves for E. coli dataset. (C) PR curves for S. cerevisiae dataset. Vertical and horizontal axes represent precision and recall, respectively.
(TIF)
ROC curves of Top k Net and community prediction based on integration of the top 10 highest-performance algorithms. (A) ROC curves for in silico datasets. (B) ROC curves for E. coli dataset. (C) ROC curves for S. cerevisiae dataset. Vertical and horizontal axes represent true-positive and false-positive rate, respectively.
(TIF)
Performances of community prediction based on integration of high- or low-diversity algorithm pairs by EUC distance. H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * and ** represent P<0.05 and P<0.01, by the Wilcoxon rank sum test.
(TIF)
Performances of Top1Net based on integration of high- or low- diversity algorithm pairs by PCA distance. H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * represents P<0.05 by the Wilcoxon rank sum test.
(TIF)
Performances of community prediction based on integration of high- or low- diversity algorithm pairs by PCA distance. H and L represent high-diversity and low-diversity algorithm pairs, respectively. (A) Box-plots of overall score. (B) Box-plots of AUC-PR for in silico dataset. (C) Box-plots of AUC-ROC for in silico dataset. (D) Box-plots of Max f-score for in silico dataset. (E) Box-plots of AUC-PR for E. coli dataset. (F) Box-plots of AUC-ROC for E. coli dataset. (G) Box-plots of Max f-score for E. coli dataset. (H) Box-plots of AUC-PR for S. cerevisiae dataset. (I) Box-plots of AUC-ROC for S. cerevisiae dataset. (J) Box-plots of max f-score for S. cerevisiae dataset. * and ** represent P<0.05 and P<0.01, by the Wilcoxon rank sum test.
(TIF)
Comparison of algorithm performances across gene-expression datasets. The scatter plots show correlation of algorithm performance between two gene-expression datasets. Vertical axis represents algorithm performance for one gene-expression dataset, while horizontal axis represents that for the other gene-expression dataset. (A) Scatter plots of AUC-PR for in silico and E. coli datasets. (B) Scatter plots of AUC-PR for in silico and S. cerevisiae datasets. (C) Scatter plots of AUC-PR for E. coli and S. cerevisiae datasets. (D) Scatter plots of AUC-ROC for in silico and E. coli datasets. (E) Scatter plots of AUC-ROC for in silico and S. cerevisiae datasets. (F) Scatter plots of AUC-ROC for E. coli dataset and S. cerevisiae datasets. (G) Scatter plots of max f-score for in silico and E. coli datasets. (H) Scatter plots of max f-score for in silico and S. cerevisiae datasets. (I) Scatter plots of max f-score for E. coli and S. cerevisiae datasets.
(TIF)
Performances of the 38 individual algorithms. The table shows overall score, AUC-PR, and AUC-ROC of the 38 algorithms.
(XLS)
Correlation coefficient of performance metrics across the DREAM5 gene-expression datasets. The table shows Spearman's correlation coefficient of performance metrics across the DREAM5 gene expression datasets.
(DOC)