

Abstract
The phylogenetic position of turtles within the vertebrate tree of life remains controversial. Conflicting conclusions from different studies are likely a consequence of systematic error in the tree construction process, rather than random error from small amounts of data. Using genomic data, we evaluate the phylogenetic position of turtles with both conventional concatenated data analysis and a “genes as characters” approach. Two datasets were constructed, one with seven species (human, opossum, zebra finch, chicken, green anole, Chinese pond turtle, and western clawed frog) and 4584 orthologous genes, and the second with four additional species (soft-shelled turtle, Nile crocodile, royal python, and tuatara) but only 1638 genes. Our concatenated data analysis strongly supported turtle as the sister-group to archosaurs (the archosaur hypothesis), similar to several recent genomic data based studies using similar methods. When using genes as characters and gene trees as character-state trees with equal weighting for each gene, however, our parsimony analysis suggested that turtles are possibly sister-group to diapsids, archosaurs, or lepidosaurs. None of these resolutions were strongly supported by bootstraps. Furthermore, our incongruence analysis clearly demonstrated that there is a large amount of inconsistency among genes and most of the conflict relates to the placement of turtles. We conclude that the uncertain placement of turtles is a reflection of the true state of nature. Concatenated data analysis of large and heterogeneous datasets likely suffers from systematic error and over-estimates of confidence as a consequence of a large number of characters. Using genes as characters offers an alternative for phylogenomic analysis. It has potential to reduce systematic error, such as data heterogeneity and long-branch attraction, and it can also avoid problems associated with computation time and model selection. Finally, treating genes as characters provides a convenient method for examining gene and genome evolution.
Introduction
Despite numerous recent attempts, the phylogenetic position of turtles remains controversial [1]–[6]. Traditionally, turtles were placed as the sister-group to A) all other living amniotes [7], or B) diapsids, but with various affiliations to different extinct groups [8]–[11] (see [12] for a review). These views are primarily based on morphological and fossil data. Most recent studies, however, favor a more nested position for turtles within diapsids. In addition to morphological data [13], [14], molecular data overwhelmingly support this placement. Several potential positions have been proposed, including turtles being the sister-group to C) lepidosaurs [2], [15], D) archosaurs [5], [6], [16]–[20], E) crocodilians [1], [21]–[23], and F) birds [24]. The alternative placements of turtles in the current phylogeny of living tetrapods are shown in Figure 1.
Figure 1. Alternative placements of turtles in the current phylogeny of living tetrapods.
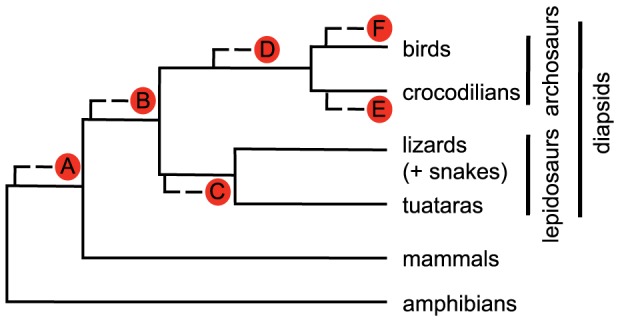
Conflicting conclusions are likely a consequence of systematic error rather than sampling error in the tree construction process, because most recent studies utilized substantial amounts of data. For example, Tzika et al. used transcriptome data of 4,689 genes [1], Lyson et al. used present/absent data of 186 microRNA families [2], and Wang et al. used 1,113 coding genes [6]. Conventional sequence data analysis uses nucleotide sites (or amino-acid residues) as characters. When data from a large number of genes are available, a supermatrix approach is often preferentially used (concatenated data analysis; e.g. [25]–[28]). Several well-recognized systematic errors are associated with this approach. Data heterogeneity is probably the most pronounced error in large dataset analysis, such as in phylogenomics [29], [30]. Additionally, when divergence is deep, multiple ‘hits’ in DNA substitution often lead to long-branch attraction [1], [31]. Furthermore, with a large and heterogeneous dataset, a realistic substitution model becomes elusive. Fitting different parts of the data with different sets of parameters may over-parameterize the model [32]. Several recent studies suggested that conventional concatenated sequence analysis might be problematic for phylogenetics with large amounts of data [30], [33].
A potentially better alternative in the age of phylogenomics is to use genes as characters, haplotypes as character states, and gene trees as character-state trees. Each gene represents one ordered multi-state character, and parsimony principle can be applied to reconstruct the species phylogeny (Figure 2). The idea of “locus (gene) as character” was first discussed in the isozyme community [34]. Doyle (1992) further explicitly proposed using a gene tree as one single species tree character [35]. Conceptually, genes are the unit of inheritance and function in Mendelian genetics, and arguably are a better choice than nucleotide sites (or amino-acid residues) as characters in evolutionary biology. Theoretically, this approach is capable of mitigating systematic error associated with supermatrix analysis using nucleotide sites as characters (e.g., data heterogeneity). The recent development of DNA sequencing technology makes massive amounts of sequence data readily available, which in turn makes the genes as characters approach a practical alternative. With the rapid development of phylogenomics, advantages associated with the genes as characters approach should be explored.
Figure 2. The procedural flowchart of using genes as characters in phylogenomic analysis.
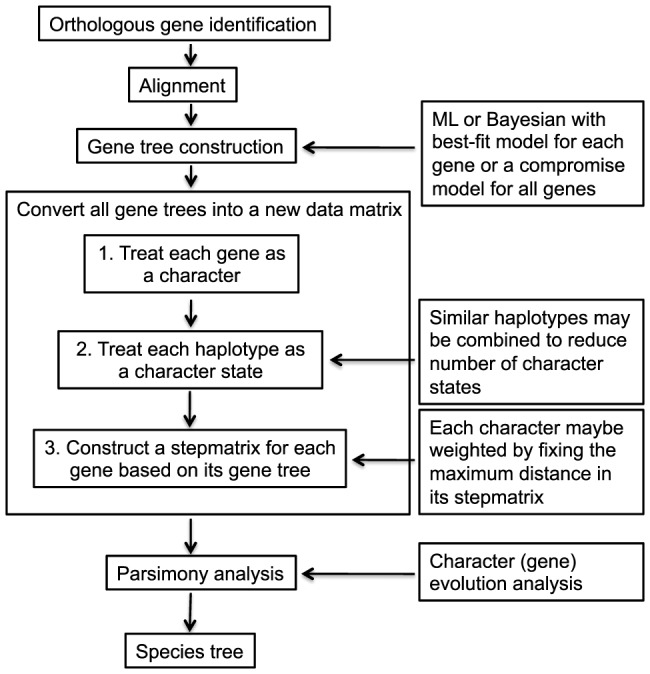
In this study, we evaluate the phylogenetic position of turtles within the vertebrate tree of life using the genes as characters approach with genomic data. We first sequenced a transcriptome of a turtle species and retrieved genomic data of ten other amniotes from other sources. Both concatenated data analysis and genes as characters approach were then applied and compared. Finally, utilization of phylogenetic information to understand genome evolution, including gene modular evolution and gene function association, was explored.
Results
Data acquisition
We first acquired high quality transcriptome data of a turtle species (Chinese pond turtle, Mauremys reevesii) by RNA sequencing. A total of 35,399,774 reads were generated through Illumina sequencing. After de novo assembly, 61,810 transcripts were produced with an N50 length of 1,571 base pairs (bps) and an average length of 1,072 bps. Sequence lengths ranged from 295 bps to 19,374 bps. Sequence information and the length distribution of transcripts are presented in Table S1 and Figure S1. Transcriptomic or genomic data of another ten vertebrate species were acquired from online databases, including green anole (Anolis carolinensis), Nile crocodile (Crocodylus niloticus), chicken (Gallus gallus), human (Homo sapiens), opossum (Monodelphis domestica), soft-shelled turtle (Pelodiscus sinensis), royal python (Python regius), zebra finch (Taeniopygia guttata), tuatara (Sphenodon punctatus), and western clawed frog (Xenopus tropicalis).
Putative orthologous genes were identified using the program HaMSTR (Hidden Markov Model based Search for Orthologs using Reciprocity; [36]) and the Amniota Orthology Dataset. Based on the six species in the Amniota Orthology Dataset, 5,587 putatively orthologous proteins and 5,584 coding genes of M. reevesii were initially identified. After removing the dubious orthologs based on the individual gene tree (see below), 4,584 genes remained for both amino-acid and nucleotide data for these seven species. When we added the transcriptome data of the other four species, Pelodiscus sinensis, Crocodylus niloticus, Python regius, and Sphenodon punctatus, the number of orthologous genes sharply decreased to 1,638 in the final amino-acid and nucleotide data. Considering the levels of details of the acquired data, we constructed two datasets for phylogenetic analysis. Seven species (human, opossum, zebra finch, chicken, green anole, Chinese pond turtle, and western clawed frog) had many more orthologous genes than the other species, and formed a 7-species dataset. The second dataset, the 11-species dataset, had more taxa but fewer genes. The transcriptome raw data of M. reevesii are deposited at the NCBI Sequence Read Archive (accession number SRA098741), and the putative orthologous gene sequence data of M. reevesii are deposited at GenBank (accession number GANJ01000000).
Phylogenetic inferences
Phylogenetic trees were constructed based on both amino-acid and coding region nucleotide sequences. Alignments were conducted for each individual gene. The western clawed frog served as outgroup in all phylogenetic analyses.
We first analyzed each gene separately to obtain gene trees using maximum likelihood (ML) method. We noticed that some gene trees produced by nucleotide sequences had unusually long branches, and careful inspection of the data suggested that low quality alignments, non-orthologous genes, or potential assembling errors might have contributed to the long branches. Consequently, gene trees with the longest branch length greater than 1.5, which included 1004 genes for the 7-species dataset and 642 genes for the 11-species dataset, were excluded from subsequent analyses.
Concatenated data analysis
All amino-acid and nucleotide sequences were pooled together, respectively. The data were analyzed using maximum parsimony (MP), ML, and Bayesian inference methods. The concatenated 7-species amino-acid dataset included 2,630,903 amino-acid residuals. Both the ML and MP tree strongly supported turtles being the sister-group to archosaurs (the archosaur hypothesis; Figure 3 A–B). The concatenated nucleotide sequence was 7,843,295 bp long. For parsimony analysis, due to computation constraints, the dataset was reduced to 3,824,484 bps by removing all constant sites. The ML and MP analyses produced the same topology as that of amino-acid sequences (Figure 3 C–D).
Figure 3. The phylogenetic hypotheses derived from the 7-species data.

Amino-acid and nucleotide sequences were analyzed by maximum parsimony (MP) and maximum likelihood (ML) methods, respectively. Numbers near the nodes are bootstrap values.
The concatenated 11-species amino-acid dataset included 808,806 amino-acid residuals, and both the ML and MP trees again placed turtles as sister-group to archosaurs (Figure 4 A–B). The concatenated nucleotide sequences included 2,431,766 nucleotide sites. All ML, MP, and Bayesian trees supported turtles being the sister-group to archosaurs (Figure 4 C–E). They had however, different placement for tuatara; while ML and Bayesian trees placed tuatara as the sister-group to squamates ( = lizards+snakes), MP tree placed tuatara as the basal clade to the (archodaurs+turtles) clade (Figure 4 C). Removing the hyper-variable 3rd codon position, the MP analysis resulted in tuatara being the sister-group to squamates (Figure 4 F). In all cases of concatenated analysis, all nodes received high bootstrap support ( = 100) or Bayesian posterior probability support ( = 99 or 100).
Figure 4. The phylogenetic hypotheses derived from the 11-species data.

Amino-acid and nucleotide sequences were analyzed by maximum parsimony (MP) and maximum likelihood (ML) methods, respectively. A Bayesian tree from nucleotide sequences and a MP tree from the 1st and 2nd codon position sequences are also presented. Numbers near the nodes are bootstrap values or Bayesian posterior probabilities (E).
Genes as characters method
We used two methods to summarize the information from gene trees into species trees. First, we used a simple count method, similar to the gene-support frequency measurement [30]. The numbers of genes that support various hypotheses were counted and compared. The hypothesis supported by the highest number of genes was preferred. Second, we performed a more elaborated parsimony analysis following the arguments of Doyle [35]. Each gene was treated as a single ordered multi-state character. Haplotypes of a gene were treated as character states and the gene trees were treated as the character-state trees. Step matrices were used to describe the character-state trees. We weighted all characters (genes) equally. The maximum pairwise distance in each step matrix was set to ten and all other steps were proportionally scaled. Finally, a parsimony analysis was performed. The procedure is summarized in Figure 2.
For the 7-species data, 4,584 individual proteins (∼574 amino-acids per protein) and coding genes (∼1,711 base pair per gene) were analyzed to reconstruct gene trees. Among them, 1,868 proteins (41%) and 2,117 coding genes (46%) supported the archosaur hypothesis. In contrast, only 811 proteins (18%) and 798 coding genes (17%) supported the lepidosaur hypothesis. Surprisingly, there were 535 (12%) proteins and 460 (10%) coding genes that supported the diapsid hypothesis (Table 1). For the 11-species data, 1,638 individual proteins and coding genes were analyzed to reconstruct gene trees. Among them, 80 proteins (5%) and 222 coding genes (14%) supported the archosaur hypothesis, 80 proteins (5%) and 121 coding genes (7%) supported the lepidosaur hypothesis, 22 proteins (1%) and 90 coding genes (5%) supported the crocodilian hypothesis, and 17 proteins (1%) and 47 coding genes (3%) supported the bird hypothesis (Table 1). Generally, the archosaur hypothesis received support from the largest number of genes. Noticeably, however, both the archosaur hypothesis and the lepidosaur hypothesis received support from the same number of proteins in the 11-species data. The diapsid hypothesis also received substantial support from a large number of genes (10–12%). In addition, amino-acid sequences were less informative than nucleotide sequences, and a larger portion of the gene trees from amino-acid sequences included polytomies.
Table 1. A simple tally of genes that support alternative hypotheses.
| Hypothesis | Tree topology | Number of genes (nucleotides) | Number of proteins (amino-acid) |
| 7-species dataset | |||
| Archosaur Hypothesis | ((ZF,Ch),PT) | 2117 (46%) | 1868 (41%) |
| (((ZF,Ch),PT)An) | 1502 | 1327 | |
| (((((ZF,Ch),PT),An),(Hu,Op)),Fr) | 1281 | 1149 | |
| Lepidosaur Hypothesis | (PT,An) | 798 (17%) | 811 (18%) |
| ((PT,An),(ZF,Ch)) | 618 | 597 | |
| ((((PT,An),(ZF,Ch)),(Hu,Op)),Fr) | 500 | 501 | |
| Diapsid Hypothesis | ((ZF,Ch,An),PT) | 460 (10%) | 535 (12%) |
| (((((ZF,Ch),An),PT),(Hu,Op)),Fr) | 379 | 450 | |
| Other topologies | 1209 (26%) | 1370 (30%) | |
| 11-species dataset | |||
| Archosaur Hypothesis | (((ZF,Ch),Cr),(PT,ST)) | 222 (14%) | 80 (5%) |
| ((((ZF,Ch),Cr),(PT,ST)),((An,Py),Tu)) | 106 | 32 | |
| Lepidosaur Hypothesis | (((An,Py),Tu),(PT,ST)) | 121 (7%) | 80 (5%) |
| ((((An,Py),Tu),(PT,ST)),((ZF,Ch),Cr)) | 32 | 21 | |
| Crocodilian Hypothesis | ((Cr,(PT,ST)),(ZF,Ch)) | 90 (5%) | 22 (1%) |
| Bird Hypothesis | (((ZF,Ch),(PT,ST)),Cr) | 47 (3%) | 17 (1%) |
| Other topologies | 1158 (71%) | 1439 (88%) |
Hu = Human (Homo sapiens), Fr = Western Clawed Frog (Xenopus tropicalis), ZF = Zebra Finch (Taeniopygia guttata), Ch = Chicken (Gallus gallus), An = Green Anole (Anolis carolinensis), PT = Chinese Pond Turtle (Mauremys reevesii), ST = Soft-shelled Turtle (Pelodiscus sinensis), Py = Royal Python (Python regius), Tu = Tuatara (Sphenodon punctatus), Op = Opossum (Monodelphis domestica), Cr = Nile Crocodile (Crocodylus niloticus).
For example, the clade ((ZF,Ch),PT) appears on 2117 gene trees without regarding other relationships. The 7-species dataset includes 4,584 putatively orthologous proteins and coding genes; the 11-species dataset includes 1,638 putatively orthologous proteins and coding genes.
Using each individual gene tree as a character-state tree, parsimony analyses produced very different results. The 7-species amino-acid dataset placed turtles as sister-group to the (birds+lizards) clade ( = diapsids; Figure 5 A), while the nucleotide dataset placed turtles as sister-group to lizards ( = lepidosaurs; Figure 5 B). Furthermore, the 11-species amino-acid dataset suggested turtles being the sister-group to the (birds+crocodilians) clade ( = archosaurs; Figure 5 C), while the nucleotide dataset suggested turtles being the sister-group to the (squamates+tuatara) clade ( = lepidosaurs; Figure 5 D). Noticeably, none of the above solutions received particularly high bootstrap support; the values of related nodes varied from 73 to 91.
Figure 5. The phylogenetic hypotheses derived from using genes as characters.

Individual gene tree was first estimated and used as character-state tree. Each gene was then treated as a character and a parsimony analysis was used to construct the species trees. Amino-acid and nucleotide sequences were analyzed separately. Numbers near the nodes are bootstrap values. Note the different placements of turtles and the low bootstrap values for the associated nodes.
Incongruence analysis
We examined the incongruence among genes with two methods. A SplitsTree analysis [37] was used to identify where, on the species tree, most incongruence occurred among the gene trees. A Phylcon analysis [38] was used to detect different levels of support of various genes to a particular topology.
The SplitTree analysis clearly pointed most uncertainty to the placement of turtles in the networks (Figure 6). With the 7-species data set, the uncertainty is located at the node that includes turtle, birds, and lizards (Figure 6A), although the nucleotide data further extended the uncertainty to include mammals (Figure 6 B). For the 11-species data, the uncertainty was one node, among turtles, birds and crocodilians (Figure 6 C–D). Figure 6 presents results with a threshold value of 0.15, which most clearly demonstrates the main contradictory splits.
Figure 6. Results of the SplitTree analysis.

Loops or boxes indicate the location of incongruence on the networks. The analysis examined four datasets, and all suggested strong incongruence regarding the phylogenetic position of turtles.
The Phylcon analysis revealed a substantial amount of incongruence among genes (Figure 7). For the 7-species data, the archosaur hypothesis (H1) was clearly better supported by a larger number of genes (lighter colored genes) than the lepidosaur hypothesis (H2) and the diapsid hypothesis (H3). For the 11-species data, the archosaur hypothesis (H1) and the crocodile hypothesis (H3) were slightly better supported than the lepidosaur hypothesis (H2) and the bird hypothesis (H4). The most noticeable feature of these heatmaps, however, was the amount of conflict among genes; even for the best supported topologies by the concatenated data (the archosaur hypothesis), a large number of genes strongly rejected them (dark colored genes), particularly in the 11-species data.
Figure 7. Heatmaps from the Phylcon analysis.

Each vertical line represents a gene and different colors represent different p values from the AU tests. A small p value (dark green) indicates that a gene rejects a topology. Topologies examined: 7-species data: 1. (((((ZF, Ch), PT),An),(Hu, Op)), Fr); 2. ((((ZF, Ch),(PT, An)),(Hu, Op)), Fr); 3. (((((ZF, Ch),An), PT),(Hu, Op)), Fr). 11-species data: 1. (((Hu, Op),((((ZF, Ch), Cr),(PT, ST)),((An,Py),Tu))),Fr); 2. (((Hu, Op),(((ZF, Ch), Cr),((PT, ST),((An,Py),Tu)))),Fr); 3. (((Hu, Op),(((ZF, Ch),(Cr,(PT, ST))),((An,Py),Tu))),Fr); 4. (((Hu, Op),((((ZF, Ch),(PT, ST)), Cr),((An,Py),Tu))),Fr). Hu = Human (Homo sapiens), Fr = Western Clawed Frog (Xenopus tropicalis), ZF = Zebra Finch (Taeniopygia guttata), Ch = Chicken (Gallus gallus), An = Green Anole (Anolis carolinensis), PT = Chinese Pond Turtle (Mauremys reevesii), ST = Soft-shelled Turtle (Pelodiscus sinensis), Py = Royal Python (Python regius), Tu = Tuatara (Sphenodon punctatus), Op = Opossum (Monodelphis domestica), Cr = Nile Crocodile (Crocodylus niloticus).
Gene evolution
Within a phylogenetic framework, we explored two aspects of gene evolution: the impact of genes experienced positive selection on phylogenetic inferences and gene modular evolution.
Positive selection
We used a maximum likelihood approach and the branch-site models [39] and detected signature of positive selection for 272 genes (out of 4,461) in the 7-species data and 31 genes (out of 957) in the 11-species data. We further performed a Chi-square test to examine any association between genes under selection and two primary alternative hypotheses, the archosaur and the lepidosaur hypotheses. For the 7-species dataset, 143 of the 272 genes supported the archosaur hypothesis and 37 of them support the lepidosaur hypothesis. The Chi-square test indicated that the proportion of the positively selected genes that support the archosaur hypothesis was significantly higher than expected and/or genes that support the lepidosaur hypothesis was significantly lower than expected (P = 0.0331; Table 2). Therefore, positive selection clearly had significant influence on the final tree topology.
Table 2. Chi-square test of the relationship between genes under positive selection and alternative hypotheses.
| Numbers genes support a clade | ((ZF, Ch), PT) | (PT, An) |
| Under positive selection | 143 (130.66) | 37 (49.34) |
| Neutral | 1917 (1929.35) | 741 (728.66) |
| Total | 2060 | 778 |
| Chi-square value | 4.54 | |
| P | 0.0331 |
ZF = Zebra Finch (Taeniopygia guttata), Ch = Chicken (Gallus gallus), PT = Chinese Pond Turtle (Mauremys reevesii), An = Green Anole (Anolis carolinensis).
Numbers in parentheses are expected numbers of genes under random distribution. Significantly more positively selected genes support the archosaur hypothesis.
Three genes (RUFY3, ENSG00000018189, GO: developmental protein; NUP37, ENSG00000075188, GO: Cell division, carbohydrate metabolic process and glucose transport; COG1, ENSG00000166685, GO: Golgi transport complex) were detected positive selection signal in both the 7-species and 11-species data. The function of these genes mainly involved biological regulation and cellular process. How these genes may have contributed to the evolution of turtles is worth exploring.
Gene function
To assess gene modular evolution, we tested whether genes supporting a particular topology were over-represented in a particular function category or metabolic pathway. The Gene Ontology (GO) annotation and the Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to classify gene function. At least 22 GO categories were over-represented among genes that support the archosaur hypothesis or the lepidosaur hypothesis (Table 3). For example, 61 genes that support the archosaur hypothesis were in the functional category of ‘organophosphate metabolic process’, which was higher than expected (49 under a random distribution). Similarly, 76 genes that support the lepidosaur hypothesis were in the functional category of ‘cell activation’, which was higher than expected (61 under a random distribution). In general, it appeared that genes associated with metabolic process and binding function were likely to support the archosaur hypothesis, while genes associated with fertilization, digestion, and development were more likely to support the lepidosaur hypothesis (Table 3).
Table 3. Chi-square tests of the relationship between genes functions and phylogenetic hypotheses.
| GO/KEGG category | A+ | A− | O+ | O− | X2 | P |
| A. Test of the Archosaur Hypothesis [((Zebra Finch, Chicken), Chinese Pond Turtle)] | ||||||
| Biological Process | ||||||
| organophosphate metabolic process (GO:0019637) | 61 (49) | 1975 (1987) | 46 (58) | 2342 (2030) | 5.3 | 0.0210 |
| hormone metabolic process (GO:0042445) | 49 (39) | 1987 (1997) | 35 (45) | 2353 (2343) | 5.2 | 0.0223 |
| macromolecule metabolic process (GO:0043170) | 1180 (1148) | 856 (888) | 1314 (1346) | 1074 (1042) | 3.8 | 0.0500 |
| primary metabolic process (GO:0044238) | 1419 (1385) | 617 (651) | 1590 (1624) | 798 (764) | 4.9 | 0.0269 |
| Cellular Component | ||||||
| organelle lumen (GO:0043233) | 682 (648) | 1354 (1388) | 726 (760) | 1662 (1628) | 4.8 | 0.0276 |
| extracellular matrix (GO:0031012) | 77 (65) | 1959 (1971) | 64 (76) | 2324 (2312) | 4.3 | 0.0376 |
| extracellular matrix part (GO:0044420) | 37 (25) | 1999 (2011) | 18 (30) | 2370 (2358) | 10.1 | 0.0015 |
| Molecular Function | ||||||
| nucleotide binding (GO:0000166) | 409 (381) | 1627 (1655) | 419 (447) | 1969 (1941) | 4.7 | 0.0307 |
| hormone binding (GO:0042562) | 25 (18) | 2011 (2018) | 15 (22) | 2373 (2366) | 4.4 | 0.0357 |
| cofactor binding (GO:0048037) | 72 (60) | 1964 (1976) | 59 (71) | 2329 (2317) | 4.3 | 0.0372 |
| KEGG pathway | ||||||
| inositol phosphate metabolism (map00562) | 28 (18) | 318 (328) | 10 (20) | 375 (365) | 11.2 | 0.0008 |
| phosphatidylinositol signaling system (map04070) | 31 (22) | 315 (324) | 15 (24) | 370 (361) | 7.9 | 0.0049 |
| arginine and proline metabolism (map00330) | 24 (18) | 322 (328) | 14 (20) | 371 (365) | 4.0 | 0.0448 |
| B. Test of the Lepidosaur Hypothesis [(Pond Turtle, Green Anole)] | ||||||
| Biological Process | ||||||
| cell activation (GO:0001775) | 76 (61) | 701 (716) | 274 (289) | 3373 (3358) | 4.5 | 0.0334 |
| translational initiation (GO:0006413) | 17 (10) | 760 (767) | 40 (47) | 3607 (3600) | 6.0 | 0.0143 |
| cellular component disassembly (GO:0022411) | 45 (34) | 732 (743) | 149 (160) | 3498 (3487) | 4.4 | 0.0350 |
| fertilization (GO:0009566) | 14 (8) | 763 (769) | 33 (39) | 3614 (3608) | 4.9 | 0.0268 |
| pollination (GO:0009856) | 16 (10) | 761 (767) | 40 (46) | 3607 (3601) | 4.7 | 0.0293 |
| system process (GO:0003008) | 156 (135) | 621 (642) | 610 (631) | 3037 (3016) | 5.0 | 0.0250 |
| digestion (GO:0007586) | 13 (6) | 764 (771) | 23 (30) | 3624 (3617) | 8.6 | 0.0033 |
| stem cell maintenance (GO:0019827) | 14 (8) | 763 (769) | 34 (40) | 3613 (3607) | 4.5 | 0.0336 |
| regulation of multi-organism process (GO:0043900) | 15 (9) | 762 (768) | 34 (40) | 3613 (3607) | 5.8 | 0.0158 |
| Cellular Component | ||||||
| cell projection (GO:0042995) | 130 (110) | 647 (667) | 499 (519) | 3148 (3128) | 4.9 | 0.0271 |
| membrane part (GO:0044425) | 329 (296) | 448 (481) | 1355 (1388) | 2292 (2259) | 7.3 | 0.0068 |
| Molecular Function | ||||||
| protein complex scaffold (GO:0032947) | 12 (5.6) | 765 (771) | 20 (26) | 3627 (3621) | 8.8 | 0.0029 |
| KEGG pathway | ||||||
| glycerolipid metabolism (map00561) | 14 (7) | 119 (126) | 25 (32) | 573 (566) | 8.7 | 0.0032 |
| glycerophospholipid metabolism (map00564) | 13 (7) | 120 (126) | 28 (34) | 570 (564) | 5.3 | 0.0210 |
Only significant results are presented. A: the archosaur hypothesis; L: the lepidosaur hypothesis; O: other hypotheses. +: number of genes which have a particular GO category or KEGG pathway; −: number of genes which do not have the GO category or KEGG pathway. Numbers in parentheses are expected numbers of genes under random distribution. For example, of the 2117 genes that support the Archosaur hypothesis, 2036 genes have the GO category of “biological process”; among them, 61 have the GO category of “organophosphate metabolic process” and 1975 genes do not have the term.
We detected five KEGG pathways that had biased representation. Genes involving the phosphatidylinositol signaling system, inositol phosphate and arginine and proline metabolism were more likely to support the archosaur hypothesis while genes involved with glycerolipid and glycerophospholipid metabolism were more likely to support the lepidosaur hypothesis (Table 3).
Discussion
Turtles as sister-group to Archosauria?
Using genes as characters, our analysis suggested that turtles are possibly the sister-group to diapsids, archosaurs, or lepidosaurs (Figure 5). Furthermore, considering the large amount of data, none of these solutions received a particularly high bootstrap support (77–91). This is clearly different from our concatenated data analysis, which supports the archosaur hypothesis with high bootstrap support ( = 100). Several recent genomic data based studies, such as Shaffer et al. [5] and Wang et al. [6], also used a concatenated approach and supported the archosaur hypothesis with high bootstrap support ( = 100). Rather than providing a clear answer, the genes as characters approach maintains the uncertainty about the phylogenetic position of turtles.
The observed level of gene incongruence suggests that the apparent solid support for the archosaur hypothesis is an artifact of concatenated analysis. Most studies supporting the archosaur hypothesis used a conventional concatenated analysis [5], [6], [19], [20], and problems associated with this approach in a phylogenomic context are well recognized [30], [33]. With a large amount of data, a putatively wrong phylogeny may be strongly supported with 100% bootstrap probability, and a topology can also be well supported even without support from any single gene [30], [33]. The high bootstrap values represent an over-estimate of confidence as consequence of large numbers of characters, and therefore, they are not appropriate measure of confidence for large concatenated datasets and should be abandoned in such cases [30]. Furthermore, our incongruence analysis clearly demonstrated that there is a large amount of inconsistency among genes and most of the conflict is surrounding the placement of turtles (Figure 6 & 7). The relatively low bootstrap values on our trees from genes as characters approach (Figure 5) are likely a reflection of the true state of nature, including conflict among gene trees and the uncertainty of the species tree. Different data treatment may favor one hypothesis over another, but they will unlikely provide any solution with statistical confidence.
Studies using other types of data and analyses reached different conclusions, and these studies cannot be discounted. For example, Lyson et al. [2] used microRNA data and supported the lepidosaur hypothesis. These data do not suffer the same problem as the concatenated sequence analysis and are proven to be reliable for phylogenetic information [40]. Hill's [12] intensive sampling of taxa, both fossil and extent, and morphological characters also resulted in turtles being the sister-group of lepidosaurs. All these conflicting results suggest that a solid conclusion of a turtle-archosaur affinity is pre-mature. This “turtle problem” is likely one of these cases that a solid phylogenetic solution is difficult, if not impossible, to achieve for several reasons, such as rapid speciation/divergence or ancient lineages, which have been extensively discussed by Delsuc et al. [41], Philippe et al. [42], and others.
Genes as characters in phylogenomics
Using genes as characters offers an alternative to concatenated analysis for large genomic data. The debate between consensus tree versus total evidence approaches has been at the centre of modern phylogenetics for decades, and benefits of analyzing each dataset separately are well-recognized (see review [41], [43]). In the era of phylogenomics, there are additional advantages of treating each gene as an independent character. First, using genes as characters likely reduces systematic error. With the massive amount of data, random error in phylogenomics becomes smaller, but systematic error, such as data heterogeneity, is often amplified [29], [41], [44]. Both theoretical exploration and empirical case studies have demonstrated that concatenated analysis may produce misleading results with high bootstrap supports [30], [33], and analyzing genes separately is capable of reducing these errors [29], [30], [44]. Using genes as characters will also reduce the long-branch attraction problem. The ultimate cause of the problem is that there are only four alternative states (A, C, G, T) for any nucleotide sites, and multiple hits are unavoidable when divergence is old. Consequently, the high homoplasy leads to long-branch attraction [31], [45]. With haplotypes as character states, the potential number of states has no limit, and the chance of ‘multiple hits’ at a haplotype level is remote. Second, the genes as characters approach provides a convenient way to detect incongruence and to apply weighting schemes. With genomic data, data quantity is no longer an issue; conveniently determining incongruence and properly weighting the high quality data are becoming not only possible but also necessary [30], [46]. We weighted all genes equally in this study; this scheme was aimed to scale back the impact of highly variable genes, which produce many informative characters in a concatenated analysis but are often considered less reliable [47]. Several indices have recently been proposed to measure gene reliability, such as gene stationarity [46], gene informativeness [48], and internode certainty [30]. They can potentially be used to establish weighting schemes. Third, using gene as the unit of evolutionary studies makes better biological sense. What constitutes a character in evolutionary biology is a contentious issue [49], but gene is arguably the unit of inheritance and the unit of function. These traits make it a better choice as a unit of evolution than nucleotide site or amino-acid residue. One major criticism of analyzing each dataset (gene) separately is the loss of information when converting datasets into gene trees (see [43] for a review). With all branch length information being preserved in the conversion, however, this loss is minimum. Perhaps more importantly, a minimum loss of information is significantly outweighed by the aforementioned benefits with large genomic data.
At the operational level, the genes as characters approach also offers several advantages. First, selecting a best-fit model for each individual gene is relatively easy, and it is difficult to fit a realistic model for complex and heterogeneous data [41], [47]. Second, this approach can largely avoid the problems associated with computation time. Existing models and algorithms are challenged when applied to genome-wide combined phylogenetic analysis [32], [33]. Parsimony analysis, however, is computationally inexpensive, and has potential to handle large dataset. Lastly, with large dataset, errors are unavoidable. Our weighting scheme can effectively minimize the impact of several types of errors. During our analysis, we found some gene trees having one or more unusually long branches, and they were likely results from low quality alignments, non-orthologous genes, or potential assembling errors. Our weighting scheme is capable of making these gene trees almost uninformative by scaling their branches, and effectively weeds out these errors.
Although consensus tree and supertree approaches have been widely implemented [37], [41], [43], [50], exploring their use with genomic data is just beginning [51]. With the gene incongruence problem being at the center of phylogenomic analysis [30], [41], [44], [52], separated analysis of individual gene appears to be a necessary first step. How to summarize the individual gene trees into species tree, however, still need more investigation [51], [52]. Several approaches have been proposed, including consensus trees, minimizing deep coalescent events, and likelihood or Bayesian based probabilistic estimates of species tree methods [52]. More recently, Liu et al. [51] proposed a new coalescence based approach and compared various nonparametric and parametric methods. Salichos and Rokas [30], on the other hand, emphasized on selecting more reliable genes for phylogenetic reconstruction. We subscribe to a Popperian view on hypotheses regarding historical events. History is unknown, and the best-corroborated hypothesis represents the best guess with the available data [41], [53]. In this regard, parsimony analysis provides a computationally easy and philosophically sound approach in phylogenomics.
From phylo- to genomics
Not only genomic data can improve the phylogenetic resolution of the tree of life, phylogenetic analysis can make significant contributions to the understanding of genomics as well. Phylogenies have always been the foundation for comparative studies of many aspects of genomes, such as genome size [54]. Additionally, phylogenetic analysis has a unique opportunity to contribute to genomics. Eisen (1998) early on suggested phylogenetic analysis as a tool to improve functional predictions for uncharacterized genes in a genome [55]. Recently, Lee et al. went further and suggested that genes or gene clusters that changed at a particular node could be used to derive or test hypotheses regarding the biological processes that are potentially responsible for the more interesting organismal level evolution [56]. In this regard, genes that are in conflict with the species tree may be the most interesting. Other aspects, such as modular evolution of genomes as in the case of the study of the origin of life [57], can also be explored in a phylogenetic context.
We have found clear evidence that the evolution of turtles was accompanied by positive selection on many genes (Table 2). On one hand, these genes may cloud the evolutionary history of turtles and make phylogenetic reconstruction difficult. In the case of turtles, genes that have experienced positive selection have a higher tendency to support the archosaur hypothesis (Table 2), which may contribute to the confusion regarding the phylogenetic position of turtles. On the other hand, they may offer important clues with respect to how turtles evolved, as Lee et al. suggested [56]. Along this line, detailed analysis with more genomic data may reveal genes that are responsible for the novel turtle body plan, which is a hot topic in modern evolutionary biology [58].
We also found clear evidence for modular evolution (Table 3). Genes in the same function category tend to support the same hypothesis. Although our study on turtles is not as pronounced as prokaryote to eukaryote dichotomy [57], it is not surprising that genes evolve in modular fashion. With the rapid accumulation of genomic data, genome wide evaluation of gene interactions and functional modules is an attractive direction for future research [59].
The genes as characters approach offers convenient tools for evaluating character evolution. In our case, we classified the genes into “supporting the archosaur hypothesis”, “supporting the lepidosaur hypothesis” and others with simplified nodes. In a more complex situation, genes can potentially be classified using several well-characterized indices, such as consistency index (CI) or retention index (RI). For example, a gene (character) has a CI greater than 0.9 on a particular topology (hypothesis) may be considered as “supporting the hypothesis”.
Conclusions
Using genes as characters, our analysis of 4500+ genes suggested that the phylogenetic placement of turtles within vertebrates remains uncertain. Gene heterogeneity and hence conflicting information from various genes is likely the cause of this uncertainty. Although different data treatment may point to one way or another, a confident solution may be elusive.
The genes as characters approach may lessen systematic error associated with phylogenomic analysis. It may also offer convenience in substitution model selection and computation. Perhaps more importantly, it provides us with an opportunity to simultaneously understand the evolutionary history of individual genes as well as the whole genome. Recent advances in genomics will undoubtedly expedite the final resolution of the tree of life. In return, phylogenetics has much to offer for a better understanding of genomics.
Materials and Methods
Data acquisition
We first acquired high quality transcriptome data of a turtle species by RNA sequencing. Two adult Chinese pond turtles (M. reevesii) were purchased from a pet store at Chengdu, China. The specimens were euthanized by an overdose injection of sodium pentobarbital, and dissected immediately after death. RNA was separately extracted from brain, muscle, liver and heart tissues and mixed in approximately equal quantities. A cDNA library was constructed and subsequently sequenced on an Illumina HiSeq2000 platform; both were carried out by BGI Inc (Shenzhen, China). A total of 5G of raw read data were obtained.
A de novo assembly was performed. The raw reads were first cleaned by filtering out the adapter sequences. Reads with more than 5% unknown base calls, low quality reads (<Q20), and possible human and Escherichia coli contamination were also discarded. We used a multiple K-mer length and coverage cutoff value approach for assembly [60]. Thirty raw assemblies were produced using the program Velvet and OASES [61], [62] with five K-mer lengths (21, 31, 41, 51, and 61) and six coverage cutoff values (2, 3, 6, 10, 15, and 20). Sequence overlaps and redundancies were then eliminated to produce the final assembly using the programs CD-HIT-EST and CAP3 [63], [64]. All reads were mapped back to the final assembly to revisit all potential assembling errors using program SOAP2 [65].
Transcriptomic or genomic data of another ten vertebrate species were acquired from online databases. We first acquired genomic data of six vertebrate species from the Amniota Orthology Dataset (http://www.deep-phylogeny.org/hamstr/download/datasets/hmmer3/), which include green anole (Anolis carolinensis), chicken (Gallus gallus), human (Homo sapiens), opossum (Monodelphis domestica), zebra finch (Taeniopygia guttata), and western clawed frog (Xenopus tropicalis). Additionally, we downloaded transcriptomic data of the soft-shelled turtle (Pelodiscus sinensis), Nile crocodile (Crocodylus niloticus), royal python (Python regius), and tuatara (Sphenodon punctatus) [1], [66]–[68]. After adding the Chinese pond turtle, our dataset included a total of 11 species, and had at least two representatives of each major lineage of living amniotes (with the exception of a single crocodilian). The western clawed frog served as outgroup for phylogenetic analysis.
Putative orthologous genes were identified using the program HaMSTR (Hidden Markov Model based Search for Orthologs using Reciprocity; [36]) and the Amniota Orthology Dataset. For each core ortholog in the Amniota Orthology Dataset, HaMSTR searches a set of “unigenes” and identifies the best putative orthologs. The default parameters were used in all searches.
Phylogenetic analysis
Phylogenetic trees were constructed based on both amino-acid and nucleotide sequences. Coding regions of the cDNA sequences were extracted and the UTRs were removed using local BlastX based on the amino-acid sequences. During this process, a small portion of coding sequences received low hit values of BlastX (e-value cut-off of 1e-5), and genes associated with these sequences were excluded from the subsequent analyses. Sequences of 1st and 2nd codon positions were also extracted for subsequent analyses.
Two datasets were constructed for phylogenetic analysis based on the levels of detail of the acquired data. Seven species (human, opossum, zebra finch, chicken, green anole, Chinese pond turtle, and western clawed frog) had many more orthologous genes than the other species, and formed a 7-species dataset. The second dataset, the 11-species dataset, had more taxa but fewer genes. These two datasets would allow us to test the effect of sampling, more genes or more taxa, and the 11-species dataset would also provide opportunity to test the crocodilian hypothesis.
Alignment
For amino-acid sequences, a newly developed fast and high-quality protein alignment method, Clustal Omega [69], was employed with its default parameters. All nucleotide sequences were aligned using a perl script with guidance from the amino-acid sequence alignment. Alignments of a random sample of genes were also manually checked to ensure the performance of the program. Poorly aligned columns and gap-rich regions were removed using TrimAl 1.2 [70] with the heuristic automated method.
Gene tree construction
We first analyzed each gene separately to obtain gene trees. Gene trees were constructed using maximum likelihood (ML) method with RAxML7.3.2 [71]. The JTT-F+G model for amino-acid sequences and GTR+G model for nucleotide sequences were used for all genes as a compromise to simplify the analysis process. Gene trees with unusually long branches (>1.5), which might have caused by low quality alignments, non-orthologous genes, or potential assembling errors, were excluded from subsequent analyses.
Concatenated data analysis
After removing dubious orthologs, the remaining amino-acid and nucleotide sequences were pooled together, respectively. The 7-species data (both amino-acid and nucleotide sequences) were analyzed using maximum parsimony (MP) and ML methods. For the 11-species data, the amino-acid sequences were analyzed using MP and ML methods, and the nucleotide sequences were analyzed using MP, ML and Bayesian inference. A separate MP analysis was also applied to the 11-species nucleotide data after removing all 3rd codon position sequences.
The parsimony analysis was carried out using PAUP 4.10b [72]. Searching parameters included TBR branch swapping and 100 random addition replicates. A non-parametric bootstrap [73] with 1000 replicates was used to assess nodal support. For the large 7-species concatenated dataset, the data exceeded the memory limit of our PC (8G) and subsequently, all constant sites were removed prior to tree search. This would not impact the searching process but might have a small impact on bootstrap values [74].
Likelihood analysis was performed using RAxML. The JTT-F+G model for amino-acid sequences was selected as the best-fit model by a perl script (ProteinModelSelection.pl) implemented in the program. We used the GTR+G model for nucleotide sequences, which was the most commonly used DNA substitution model in RAxML. Nodal support was assessed by a non-parametric bootstrap with 100 replicates for amino-acid sequences and 1000 replicates for nucleotide sequences.
Bayesian inference was performed using PhyloBayes 3.3f with the CAT model [75], [76]. The model is a site-specific infinite mixture model, which is particularly suitable for phylogenomic studies with a large number of genes. We only applied this analysis to nucleotides of the 11-species dataset, because the 7-species dataset was too large to run this program (memory limited). We also removed all constant sites to improve the mixing as recommended by the authors. CAT-GTR model was used and two independent chains were executed. Trees were sampled every 10 cycles, and the first 20% of sampled trees were discarded as burn-in. The maximum difference and minimum effective size between the two independent runs were used to test the convergence; when the maximum discrepancy was <0.3 and minimum effective size was >50, the results were considered acceptable. The automatic stopping rule was employed; searches automatically stopped when the two chains reached convergence.
Genes as characters approach
Following the arguments of Doyle [35], each gene was treated as a single character. We used two methods to summarize the information from gene trees into species trees. First, we used a simple count method. The numbers of genes that support various hypotheses were counted and compared. The hypothesis supported by the highest number of genes was preferred. Second, we performed a more elaborated parsimony analysis following a systematic procedure (Figure 2).
Each gene was treated as a single ordered multi-state character. Ordered multi-state characters have high information content in their transformation series and are often considered “ideal” characters [77]. We assumed that these genes evolved largely independently to satisfy the independent character assumption of parsimony analysis.
Haplotypes of a gene were treated as character states and the gene trees were treated as the character-state trees. Step matrices were used to describe the character-state trees. The pairwise distance between two haplotypes on a gene tree represents the cost in tree-length units of the corresponding transformations on the species tree.
We weighted all characters (genes) equally. The maximum pairwise distance in each step matrix was set to ten and all other steps were proportionally scaled. Two digits after the decimal points were kept in the step matrices in order to force the use of floating-point evaluation of tree lengths rather than integer lengths. An in-house perl script was used to batch convert the ML gene trees into step matrix formats.
Finally, parsimony analyses were performed in PAUP. Heuristic search was employed with TBR branch swapping and 100 random addition replicates. Bootstrap values were estimated with 1000 replicates. The procedure is summarized in a flowchart and presented in Figure 2.
Detecting incongruence
It is well recognized that data heterogeneity is a major problem in phylogenomic analysis [29], [30]. Different genes may have experienced different evolutionary histories and support different topologies. We examined the incongruence among genes with two methods.
SplitsTree analysis
We used a SplitsTree analysis to identify where, on the species tree, most incongruence occurred among the gene trees. All gene trees produced from separated analyses of genes were fed into the program SplitsTree 4 [37]. Consensus networks were constructed from those gene trees with split threshold values ranging from 0.15 to 0.30. The hyper-dimensional boxes (loops) in the networks represent areas with incompatible splits. The degree of denseness of boxes in a network reflects the intensity of contradictory evidence for grouping certain taxa, and the length of an edge is determined by the weight assigned to it [37]. In contrast, a network without boxes represents complete congruence.
Phylcon analysis
This analysis was used to detect different levels of support of various genes to a particular topology. Three rooted topologies of the 7-species data and four rooted topologies of the 11-species data were examined, which represented the primary alternative hypotheses. RAxML was used to compute the per-site log likelihood values for these topologies, with JTT-F+G (amino-acid) or GTR+G (nucleotide) substitution model. An AU test [78] was performed with CONSEL [79] based on these likelihood values. For any gene, a small P value (e.g., <0.05) for a topology indicates that the topology is significantly worse than the best tree, hence the gene rejects the topology. The P values for each individual gene and each topology were converted to heatmaps using the Phylcon package in R [38], which could be easily visualized.
Exploring gene (character) evolution
Within a phylogenetic framework, we explored two aspects of gene evolution. It is well known that positive selection has a major impact on the evolutionary history of genes and may mislead phylogenetic construction [80], [81]. In addition, genes may evolve in a modular fashion. Genes with a similar function may co-evolve and support a particular tree topology [57].
Test of positive selection
We employed a maximum likelihood approach [39] to test for positive selection using the branch-site models implemented in the program CODEML [82]. This test detects positive selection on a particular tree branch and requires unrooted trees. Individual gene trees from RAxML were converted to unrooted trees. The turtle lineage was defined as foreground (ω2), and the other lineages were defined as background branches (ω1). The initial kappa and omega were set to 2.0 and 0.4, respectively. A Chi-square test was then conducted for each gene to determine whether ω2 was significantly higher than ω1 and ω0 (average ω across the entire tree), in which case the gene might have experienced positive selection. We conducted this part of the analysis for both the 7-species data and the 11-species data.
After identifying genes with positive selection, we performed a Chi-square test to examine any association between genes under selection and two primary alternative hypotheses, the archosaur and the lepidosaur hypotheses. This analysis was only applied to the 7-species data, which included a large number of genes and had high statistical power.
Gene functions and metabolic pathways
To assess gene modular evolution, we tested whether genes supporting a particular topology were over-represented in a particular function category or metabolic pathway. The Gene Ontology (GO) annotation and the Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to classify gene function and we performed both analyses using BLAST2GO with an E cut-off value of 1e-6 [83]. We then conducted Chi-square tests to detect any relationship between gene functions and the archosaur and lepidosaur hypotheses. Only GO categories that had more than five genes were included in the Chi-square tests. This analysis was only applied to the 7-species data for high statistical power.
Ethics Statement
All animal collection and utility protocols were approved by the Chengdu Institute of Biology Animal Use Ethics Committee.
Supporting Information
Length distribution of transcripts of the pond turtle ( Mauremys reevesii ).
(EPS)
Summary of the transcriptome data for the pond turtle (Mauremys reevesii).
(DOCX)
Acknowledgments
We would like to thank Dr. Z. Yang for his advice on phylogenomic analysis and the use of PAML. Thanks also to Drs. A. Stamatakis and E. Susko for their help with model selection in RAxML and the use of Phylcon. Dr. J. P. Bogart, M. Fu, and T. F. Mitterboeck provided valuable comments on an early version of this manuscript.
Funding Statement
This work is supported by the Sichuan Provincial Government (China) (grant # Y1D3011) and a NSERC (Canada) Discovery grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Tzika AC, Helaers R, Schramm G, Milinkovitch MC (2011) Reptilian-transcriptome v1.0, a glimpse in the brain transcriptome of five divergent Sauropsida lineages and the phylogenetic position of turtles. EvoDevo 2: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lyson TR, Sperling EA, Heimberg AM, Gauthier JA, King BL, et al. (2012) MicroRNAs support a turtle+lizard clade. Biol Lett 8: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fong JJ, Brown JM, Fujita MK, Boussau B (2012) A phylogenomic approach to vertebrate phylogeny supports a turtle-archosaur affinity and a possible paraphyletic Lissamphibia. PLoS ONE 7: e48990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiari Y, Cahais V, Galtier N, Delsuc F (2012) Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria). BMC Biol 10: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaffer HB, Minx P, Warren DE, Shedlock AM, Thomson RC, et al. (2013) The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol 14: R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Z, Pascual-Anaya J, Zadissa A, Li W, Niimura Y, et al. (2013) The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet 45: 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaffney ES (1980) Phylogenetic relationships of the major groups of amniotes. In Panchen AL, editor. The Terrestrial Environment and the Origin of Land Vertebrates. London: Academic Press. 593–610. [Google Scholar]
- 8. Gauthier J, Kluge AG, Rowe T (1988) Amniote phylogeny and the importance of fossils. Cladistics 4: 105–209. [DOI] [PubMed] [Google Scholar]
- 9. Laurin M, Reisz RR (1995) A reevaluation of early amniote phylogeny. Zool J Linn Soc 113: 165–223. [Google Scholar]
- 10. Lee MSY (1997) Pareiasaur phylogeny and the origin of turtles. Zool J Linn Soc 120: 197–280. [Google Scholar]
- 11. Lyson TR, Bever GS, Bhullar BS, Joyce WG, Gauthier JA (2010) Transitional fossils and the origin of turtles. Biol Lett 6: 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hill RV (2005) Integration of morphological data sets for phylogenetic analysis of amniota: the importance of integumentary characters and increased taxonomic sampling. Syst Biol 54: 530–547. [DOI] [PubMed] [Google Scholar]
- 13. deBraga M, Rieppel O (1997) Reptile phylogeny and the interrelationships of turtles. Zool J Linn Soc 120: 281–354. [Google Scholar]
- 14. Rieppel O, Reisz RR (1999) The origin and early evolution of turtles. Ann Rev Ecol Syst 30: 1–22. [Google Scholar]
- 15. Becker RE, Valverde RA, Crother BI (2011) Proopiomelanocortin (POMC) and testing the phylogenetic position of turtles (Testudines). J Zool Syst Evol Res 49: 148–159. [Google Scholar]
- 16. Kumazawa Y, Nishida M (1999) Complete mitochondrial DNA sequences of the green turtle and blue-tailed mole skink: Statistical evidence for archosaurian affinity of turtles. Mol Biol Evol 16: 784–792. [DOI] [PubMed] [Google Scholar]
- 17. Meyer A, Zardoya R (2003) Recent advances in the (molecular) phylogeny of vertebrates. Ann Rev Ecol Evol Syst 34: 311–338. [Google Scholar]
- 18. Iwabe N, Hara Y, Kumazawa Y, Shibamoto K, Saito Y, et al. (2005) Sister group relationship of turtles to the bird-crocodilian clade revealed by nuclear DNA–coded proteins. Mol Biol Evol 22: 810–813. [DOI] [PubMed] [Google Scholar]
- 19. Shen XX, Liang D, Wen JZ, Zhang P (2011) Multiple genome alignments facilitate development of NPCL markers: a case study of tetrapod phylogeny focusing on the position of turtles. Mol Biol Evol 28: 3237–3252. [DOI] [PubMed] [Google Scholar]
- 20. Crawford NG, Faircloth BC, McCormack JE, Brumfield RT, Winker K, et al. (2012) More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol Lett 8: 783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mannen H, Li SL (1999) Molecular evidence for a clade of turtles. Mol Phylogenet Evol 13: 144–148. [DOI] [PubMed] [Google Scholar]
- 22. Hedges SB, Poling L (1999) A molecular phylogeny of reptiles. Science 283: 998–1001. [DOI] [PubMed] [Google Scholar]
- 23. Shedlock AM, Botka CW, Zhao S, Shetty J, Zhang T, et al. (2007) Phylogenomics of nonavian reptiles and the structure of the ancestral amniote genome. Proc Natl Acad Sci USA 104: 2767–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cotton JA, Page RDM (2002) Going nuclear: gene family evolution and vertebrate phylogeny reconciled. Proc R Soc Lond B 269: 1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delsuc F, Brinkmann H, Chourrout D, Philippe H (2006) Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439: 965–968. [DOI] [PubMed] [Google Scholar]
- 26. Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, et al. (2008) Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452: 745–749. [DOI] [PubMed] [Google Scholar]
- 27. Philippe H, Derelle R, Lopez P, Pick K, Borchiellini C, et al. (2009) Phylogenomics revives traditional views on deep animal relationships. Current Biol 19: 706–712. [DOI] [PubMed] [Google Scholar]
- 28. Kocot KM, Cannon JT, Todt C, Citarella MR, Kohn AB, et al. (2011) Phylogenomics reveals deep molluscan relationships. Nature 477: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeffroy O, Brinkmann H, Delsuc F, Philippe H (2006) Phylogenomics: the beginning of incongruence? Trends Genet 22: 225–231. [DOI] [PubMed] [Google Scholar]
- 30. Salichos L, Rokas A (2013) Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497: 327–331. [DOI] [PubMed] [Google Scholar]
- 31. Felsenstein J (1978) Cased in which parsimony and compatibility methods will be positively misleading. Syst Zool 27: 401–410. [Google Scholar]
- 32. Rannala B, Yang Z (2008) Phylogenetic inference using whole genomes. Ann Rev Genomic. Hum Genet 9: 217–231. [DOI] [PubMed] [Google Scholar]
- 33. Nishihara H, Okada N, Hasegawa M (2007) Rooting the eutherian tree: the power and pitfalls of phylogenomics. Genome Biol 8: R199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buth DG (1984) The application of electrophoretic data in systematic studies. Ann Rev Ecol Syst 15: 501–522. [Google Scholar]
- 35. Doyle JJ (1992) Gene trees and species trees: molecular systematics as one-character taxonomy. Syst Bot 17: 144–163. [Google Scholar]
- 36. Ebersberger I, Strauss S, von Haeseler A (2009) HaMStR: Profile hidden markov model based search for orthologs in ESTs. BMC Evol Biol 9: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267. [DOI] [PubMed] [Google Scholar]
- 38. Susko E, Leigh J, Doolittle WF, Bapteste E (2006) Visualizing and assessing phylogenetic congruence of core gene sets: a case study of the γ-proteobacteria. Mol Biol Evol 23: 1019–1030. [DOI] [PubMed] [Google Scholar]
- 39. Nielsen R, Yang Z (1998) Likelihood Models for Detecting Positively Selected Amino Acid Sites and Applications to the HIV-1 Envelope Gene. Genetics 148: 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heimberg AM, Cowper-Sallari R, Sémon M, Donoghue PCJ, Peterson KJ (2010) microRNAs reveal the interrelationships of hagfish, lampreys, and gnathostomes and the nature of the ancestral vertebrate. Proc Natl Acad Sci USA 107: 19379–19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Delsuc F, Brinkmann H, Philippe H (2005) Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6: 361–375. [DOI] [PubMed] [Google Scholar]
- 42. Philippe H, Brinkmann H, Lavrov DV, Littlewood DTJ, Manuel M (2011) Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol 9: e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Queiroz A, Donoghue MJ, Kim J (1995) Separate versus combined analysis of phylogenetic evidence. Ann Rev Ecol Syst 26: 657–681. [Google Scholar]
- 44. Phillips MJ, Delsuc F, Penny D (2004) Genome-scale phylogeny and the detection of systematic biases. Mol Biol Evol 21: 1455–1458. [DOI] [PubMed] [Google Scholar]
- 45.Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1993) Phylogenetic inference. In Hillis FM, Moritz C, Mable BK, ed., Molecular Systematics, 2nd Edition. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 46. Collins TM, Fedrigo O, Naylor GJ (2005) Choosing the best genes for the job: the case for stationary genes in genome-scale phylogenetics. Syst Biol 54: 493–500. [DOI] [PubMed] [Google Scholar]
- 47. Brinkmann H, Philippe H (2008) Animal phylogeny and large-scale sequencing: progress and pitfalls. J Syst Evol 46: 274–286. [Google Scholar]
- 48. Moeller AH, Townsend JP (2011) Phylogenetic informativeness profiling of 12 genes for 28 vertebrate taxa without divergence dates. Mol Phylogenet Evol 60: 271–272. [DOI] [PubMed] [Google Scholar]
- 49. Wagner GP (2001) The character concept in evolutionary biology. Elsevier [Google Scholar]
- 50. Miyamoto MM, Fitch WM (1995) Testing species phylogenies and phylogenetic methods with congruence. Syst Biol 44: 64–76. [Google Scholar]
- 51. Liu L, Yu L, Kubatko L, Pearl DK, Edwards SV (2009) Coalescent methods for estimating phylogenetic trees. Mol Phylogenet Evol 53: 320–328. [DOI] [PubMed] [Google Scholar]
- 52. Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24: 332–340. [DOI] [PubMed] [Google Scholar]
- 53. Siddall ME, Kluge AG (1997) Probabilism and phylogenetic inference. Cladistics 13: 313–336. [DOI] [PubMed] [Google Scholar]
- 54. Oliver MJ, Petrov D, Ackerly D, Falkowski P, Schofield OM (2007) The mode and tempo of genome size evolution in eukaryotes. Genome Res 17: 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eisen JA (1998) Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res 8: 163–167. [DOI] [PubMed] [Google Scholar]
- 56. Lee EK, Cibrian-Jaramillo A, Kolokotronis SO, Katari MS, Stamatakis A, et al. (2011) A functional phylogenomic view of the seed plants. PLoS Genet 7: e1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brown JR, Doolittle WF (1997) Archaea and the prokaryote-to-eukaryote transition. Microbiol Mol Biol Rev 61: 456–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nagashima H, Sugahara F, Takechi M, Ericsson R, Kawashima-Ohya Y, et al. (2009) Evolution of the turtle body plan by the folding and creation of new muscle connections. Science 325: 193–196. [DOI] [PubMed] [Google Scholar]
- 59. Serra F, Arbiza L, Dopazo J, Dopazo H (2011) Natural Selection on Functional Modules, a Genome-Wide Analysis. PLoS Comput Biol 7: e1001093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Surget-Groba Y, Montoya-Burgos JI (2010) Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res 20: 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28: 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Carre W (2006) Chicken genomics resource: sequencing and annotation of 35,407 ESTs from single and multiple tissue cDNA libraries and CAP3 assembly of a chicken gene index. Physiol Genomics 25: 514–524. [DOI] [PubMed] [Google Scholar]
- 64. Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- 65. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 66. Castoe TA, Fox SE, de Koning APJ, Poole AW, Daza JM, et al. (2011) A multi-organ transcriptome resource for the Burmese Python (Python molurus bivittatus). BMC Res Notes 4: 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller H, Biggs P, Voelckel C, Nelson NJ (2012) De novo sequence assembly and characterisation of a partial transcriptome for an evolutionarily distinct reptile, the tuatara (Sphenodon punctatus). BMC Genomics 13: 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang W, Li C, Ge C, Lei L, Gao Y, et al. (2013) De-novo characterization of the soft-shelled turtle Pelodiscus sinensis transcriptome using Illumina RNA-Seq technology. J Zhejiang Univ Sci B 14: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- 72.Swofford D (2002) PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0b10. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 73. Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- 74. Hirt RP, Logsdon JM, Healy B, Dorey MW, Doolittle WF, et al. (1999) Microsporidia are related to Fungi: Evidence from the largest subunit of RNA polymerase II and other proteins. Proc Nat Acad Sci USA 96: 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lartillot N, Philippe H (2004) A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21: 1095–1109. [DOI] [PubMed] [Google Scholar]
- 76. Lartillot N, Lepage T, Blanquart S (2009) PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25: 2286–2288. [DOI] [PubMed] [Google Scholar]
- 77. Pogue MG, Mickevich MF (1990) Character definitions and character state delineation: The bete noire of phylogenetic inference. Cladistics 6: 319–361. [DOI] [PubMed] [Google Scholar]
- 78. Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51 (3) 492–508. [DOI] [PubMed] [Google Scholar]
- 79. Shimodaira H, Hasegawa M (2001) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17: 1246–1247. [DOI] [PubMed] [Google Scholar]
- 80. Shen YY, Liang L, Li GS, Murphy RW, Zhang YP (2012) Parallel evolution of auditory genes for echolocation in bats and toothed whales. PLoS Genet 8: e1002788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Castoe TA, de Koning APJ, Pollock DD (2010) Adaptive molecular convergences: Molecular evolution versus molecular phylogenetics. Commun Integr Biol 3 (1) 67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang Z (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- 83. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Length distribution of transcripts of the pond turtle ( Mauremys reevesii ).
(EPS)
Summary of the transcriptome data for the pond turtle (Mauremys reevesii).
(DOCX)