

Abstract
Suppression of GABAergic neurotransmission in the ventromedial hypothalamus (VMH) is crucial for full activation of counterregulatory responses to hypoglycemia, and increased γ-aminobutyric acid (GABA) output contributes to counterregulatory failure in recurrently hypoglycemic (RH) and diabetic rats. The goal of this study was to establish whether lactate contributes to raising VMH GABA levels in these two conditions. We used microdialysis to deliver artificial extracellular fluid or l-lactate into the VMH and sample for GABA. We then microinjected a GABAA receptor antagonist, an inhibitor of lactate transport (4CIN), or an inhibitor of lactate dehydrogenase, oxamate (OX), into the VMH prior to inducing hypoglycemia. To assess whether lactate contributes to raising GABA in RH and diabetes, we injected 4CIN or OX into the VMH of RH and diabetic rats before inducing hypoglycemia. l-lactate raised VMH GABA levels and suppressed counterregulatory responses to hypoglycemia. While blocking GABAA receptors did not prevent the lactate-induced rise in GABA, inhibition of lactate transport or utilization did, despite the presence of lactate. All three treatments restored the counterregulatory responses, suggesting that lactate suppresses these responses by enhancing GABA release. Both RH and diabetic rats had higher baseline GABA levels and were unable to reduce GABA levels sufficiently to fully activate counterregulatory responses during hypoglycemia. 4CIN or OX lowered VMH GABA levels in both RH and diabetic rats and restored the counterregulatory responses. Lactate likely contributes to counterregulatory failure in RH and diabetes by increasing VMH GABA levels.
Hypoglycemia has long been considered the major limiting factor to attaining proper glycemic control in diabetic patients intensively treated with insulin. Both prior exposure to hypoglycemia and diabetes per se can diminish the glucagon and epinephrine responses that normally correct a fall in blood glucose. While the mechanism(s) responsible for these defects is not well understood, our previous work has shown that the inhibitory neurotransmitter γ-aminobutyric acid (GABA) within the ventromedial hypothalamus (VMH) plays an important role in suppressing glucose counterregulatory responses under these conditions (1). During euglycemia and hyperglycemia, GABA tonically suppresses the release of glucagon and epinephrine, whereas during hypoglycemia, a decrease in VMH GABA output is crucial to allow full activation of the counterregulatory defense responses (2). More importantly, both antecedent hypoglycemia and diabetes lead to increases in GABAergic tone within the VMH that likely contribute to counterregulatory failure (3,4). While these studies demonstrate increased GABAergic tone in the VMH as one potential contributor to counterregulatory failure under these two conditions, the mechanism(s) underlying pathogenesis of the rise in GABA has not been identified.
Several theories have been proposed to explain the phenomenon of counterregulatory failure. One of these postulates is that the brain may adapt to using alternative fuel substrates such as ketones or lactate to sustain its metabolic needs during periods of glucose deprivation and that this, in turn, may prevent it from detecting the onset of hypoglycemia (5,6). Astrocytes play a key role in brain energy metabolism and in maintaining the blood-brain barrier, vascular reactivity, regulation of extracellular glutamate levels, and protection from reactive oxygen species—among other functions (7–9). Over a decade ago, Magistretti and colleagues (10,11) proposed the astrocyte-neuron lactate shuttle hypothesis, which posits that astrocytes provide adjacent neurons with lactate during periods of increased activation to help sustain increased metabolic demands. Lactate can be produced by astrocytes through aerobic glycogenolysis when glucose levels are low or it can be generated from the direct catabolism of glucose when glucose levels are high. As such, it may serve as a key substrate under both glucoprivic and insulin-deficient hyperglycemic conditions, making it an ideal candidate for stimulating or sustaining GABAergic neuronal activation in the VMH after recurrent exposure to hypoglycemia or in diabetes. In support of this, 13C-magnetic resonance spectroscopy studies conducted in type 1 diabetic patients showed that these individuals had a greater propensity to take up and use monocarboxylic acids compared with their nondiabetic counterparts (12). In a similar manner, studies in intact rats showed that acute hypoglycemia decreases expression of the neuronal lactate transporter, monocarboxylic acid transporter (MCT)2, in the VMH, but recurrent exposure to hypoglycemia prevents this drop (13). These studies indicate that both diabetes and recurrent hypoglycemia can alter brain metabolism in a manner that favors the use of alternative fuel substrates, such as lactate, when glucose supplies become scarce. Hence, the use of alternative fuel substrates may preclude the brain from detecting a fall in plasma glucose levels, and therefore, it may continue to activate mechanisms that suppress glucose counterregulatory responses to hypoglycemia. That local delivery of lactate into the VMH was able to suppress the counterregulatory responses to hypoglycemia is consistent with this hypothesis, although the underlying mechanism was never established (14).
In the current study, we examined whether the use of alternate fuel substrates such as lactate by VMH GABAergic neurons can increase GABA output and, subsequently, lead to counterregulatory failure. We showed that lactate dramatically raises extracellular GABA levels in the VMH and suppresses the counterregulatory response in normal, nondiabetic animals. More importantly, our studies show that lactate also serves a pivotal role in stimulating GABA release in the VMH and in the subsequent development of counterregulatory failure in both recurrently hypoglycemic and poorly controlled diabetic animals.
RESEARCH DESIGN AND METHODS
Our studies were carried out using male Sprague-Dawley rats (Harlan Laboratories, South Easton, MA) that started with a body weight of ∼300 g and were individually housed in the Yale Animal Resources Center in temperature- (22–23°C) and humidity-controlled rooms. The animals had free access to rat chow (Harlan Teklab, Indianapolis, IN) and water and were acclimatized to handling and a 12-h light cycle (lights on between 0700 h and 1900 h) before experimental manipulation. Principles of laboratory animal care were followed, and experimental protocols were approved by the Institutional Animal Care and Use Committee at Yale University.
Acute hypoglycemia.
For these mechanistic studies, nondiabetic, hypoglycemia-naïve rats were implanted with vascular catheters and microdialysis guide cannulas 8–10 days prior to being studied.
Recurrent hypoglycemia.
The recurrent hypoglycemia regiment commenced 4 days after surgery for the implantation of vascular catheters and microdialysis probes, as described below. At 0900 h on the three consecutive days prior to the day of study, the rats were injected with regular human insulin (Eli Lilly & Co, Indianapolis, IN) at a dose of 10 units/kg i.p. Food was then withheld for 3 h, and tail vein glucose was assessed every 30 min using the AlphaTRAK rodent glucometer (Abbott Animal Health, Chicago, IL) to ensure sustained hypoglycemia (30–50 mg/dL) for at least 2 h. At the end of this period, the rats were given free access to food again. Control rats received a daily injection of 300 μL 0.9% saline under the same sampling conditions.
Streptozotocin diabetes.
Diabetes was induced using a single injection of streptozotocin (STZ) (65 mg/kg i.p. in citrate buffer). Nondiabetic control animals received a single intraperitoneal injection of an equivalent volume of citrate buffer. A 10% glucose solution was provided in the drinking water for the first 24 h to prevent hypoglycemia. One week after STZ administration, the rats underwent surgery to have catheters and microdialysis probes implanted as described below. One week after surgery, the animals were studied. During the 2 weeks of diabetes, blood glucose was monitored twice daily with the AlphaTRAK glucometer to ensure adequate hyperglycemia (>250 mg/dL) in our diabetic animals. In addition, the diabetic animals were not treated with insulin and therefore not likely to have experienced any bouts of hypoglycemia.
Surgery.
Approximately 8–10 days prior to the experiment, the animals underwent aseptic surgery to have vascular catheters implanted into the left carotid artery for blood sampling and right jugular vein for infusion as previously described (15). Subsequently, the animals are placed into a stereotaxic frame (David Kopf Instruments, Tujunga, CA) and bilateral stainless steel guide cannulas (Eicom, Kyoto, Japan) for microdialysis and microinjection (from bregma: VMH 2.6 mm posterior, 3.8 mm lateral, and 8.9 mm ventral at an angle of 16°) were bilaterally inserted into the brain and secured in place with screws and dental acrylic. These coordinates position the 1-mm microdialysis probes into the ventrolateral portion of the VMH. The animals were then allowed to recover for ~1 week in their home cages before being under a glucose clamp study.
Microdialysis.
All animals were fasted overnight in microdialysis cages to allow sufficient time for acclimation. The following day, the rats were connected to infusion pumps and bilateral microdialysis-microinjection probes were inserted through the implanted guide cannulas. The animals were then left to recover for 2.5–3 h, allowing for GABA levels to stabilize prior to the start of microdialysate and baseline blood sample collection. Artificial extracellular fluid (aECF) was initially perfused through the microdialysis probe at a constant rate of 1.5 μL/min and then continued or switched over to 100 mmol/L l- or d-lactate 45 min prior to the start of the baseline collection period and continued for the remainder of the study (Supplementary Fig. 1). Microdialysate samples were collected at 10-min intervals for the duration of the study. The in vitro probe efficiency for lactate recovery was ∼2%. As a result, the perfusion of 100 mmol/L lactate via microdialysis most likely produced an increase in lactate in the VMH that was slightly above 2 mmol/L, thereby allowing us to raise extracellular lactate concentrations within the upper physiological limits (16). This is an estimation however, based on ideal in vitro conditions, and thus we cannot exclude that we may have raised VMH lactate levels somewhat higher. We determined average baseline extracellular lactate concentrations in nondiabetic hypoglycemia-naïve animals to be ∼1.80 mmol/L (see results below), and, hence, we estimate that extracellular lactate concentrations were initially raised to no greater than ∼3.8 mmol/L.
Microinjection.
As shown in Fig. 1, immediately prior to the hypoglycemic clamping phase of the study and after the 45-min period in which aECF or lactate was delivered via microdialysis, the animals were bilaterally microinjected over the course of 1 min with 0.1 μL of one of the following compounds: aECF; the MCT2 antagonist, α-cyano-4-hydroxycinnamate (4CIN) (15 nmol); the GABAA receptor antagonist, bicuculline methiodide (BIC) (12.5 pmol); the ATP-sensitive K+ channel blocker, diazoxide (DZ) (1 nmol); or oxamate (OX) (50 nmol), an inhibitor of lactate dehydrogenase, using a CMA 402 syringe pump (CMA Microdialysis, North Chelmsford, MA). This microinjection protocol minimizes spread of the compound and has been shown to localize the injection to the ventromedial nucleus (17).
FIG. 1.

Microdialysate and plasma hormone concentrations for acute hypoglycemia studies. Top panel: VMH extracellular GABA concentrations collected under baseline conditions and during the hypoglycemic clamp. Control animals were perfused with aECF (control) (n = 5). l-lactate animals received 100 mmol/L l-lactate by microdialysis and were microinjected with aECF (L-Lactate) (n = 10), 4CIN (L-Lactate + 4CIN) (n = 6), OX (L-Lactate + OX) (n = 5), BIC (L-Lactate + BIC) (n = 8), or DZ (L-Lactate + DZ) (n = 5). A separate group of rats received 100 mmol/L d-lactate (D-Lactate) (n = 5) by microdialysis as an osmolarity control. Peak plasma glucagon (middle panel) and epinephrine (bottom panel) concentrations during the hypoglycemic clamp. Plasma glucagon and epinephrine responses were significantly lower in the l-lactate group compared with controls. Data are presented as means ± SEM. *P < 0.001 vs. control baseline. #P < 0.05 vs. baseline. ¶P < 0.01 vs. control hypoglycemia.
Glucose clamp.
In the acute hypoglycemia studies, after microinjection a constant insulin (50 mU/kg/min) and variable dextrose solution were infused intravenously to lower and maintain plasma glucose levels at 45 ± 5 mg/dL for 90 min. Blood samples were collected at 30-min intervals throughout the hypoglycemic clamp portion of the study for measurement of plasma glucagon and catecholamine responses and at the end of the study for plasma insulin concentrations. After each sample collection, the erythrocytes were resuspended in an equivalent volume of artificial plasma (18) and reinfused back into the animal to prevent volume depletion and anemia. At the end of the study, after collection of the final blood and microdialysate samples, the animals were killed with an overdose of sodium pentobarbital and the brains were removed and frozen on dry ice. Accuracy of probe placement was determined histologically by visual inspection of coronal brain sections. Only data obtained from those animals with correctly positioned microdialysis probes were analyzed.
For the recurrent hypoglycemia studies (Supplementary Fig. 2), aECF, 4CIN, or OX were microinjected into the VMH after the baseline microdialysate collection period, just prior to the start of the insulin and dextrose infusions. Plasma glucose was lowered to ∼45 ± 5 mg/dL for 90 min and a blood sampling regimen similar to that in the acute hypoglycemia studies described above.
For the diabetes studies (Supplementary Fig. 3), after the baseline collection period, a constant insulin (50 mU/kg/min) and variable dextrose infusion were used to induce baseline euglycemia (115 ± 10 mg/dL) in the diabetic animals for at least 30 min. Once plasma glucose levels were stable at euglycemia, blood was withdrawn to assess plasma hormone concentrations during hyperinsulinemic-euglycemia. Nondiabetic control animals were maintained at euglycemic levels during this period. Subsequently, the rats were microinjected with aECF, 4CIN, or OX as described above. After microinjection, the glucose infusion rate was decreased and plasma glucose levels were lowered and maintained at ∼45 ± 5 mg/dL for 90 min. A blood sampling procedure similar to that described above was used during the hypoglycemic clamping period.
Hormone and microdialysate analysis.
Plasma catecholamine concentrations were analyzed by high-performance liquid chromatography using electrochemical detection, while plasma hormone concentrations were determined using commercially available radioimmunoassay kits. VMH GABA concentrations from microdialysate samples were determined using liquid chromatography/tandem mass spectrometry after butylation as previously described (19). Microdialysate lactate concentrations were analyzed using a photometric assay on the CMA600 small clinical chemistry analyzer (CMA Microdialysis, Holliston, MA).
Statistical analysis.
Treatment effects were analyzed using one- or two-way ANOVA for independent or repeated measures as appropriate, followed by post hoc analysis using the Statistica suite of analytical software for personal computers by StatSoft. P < 0.05 was set as the criterion for statistical significance.
RESULTS
Acute hypoglycemia.
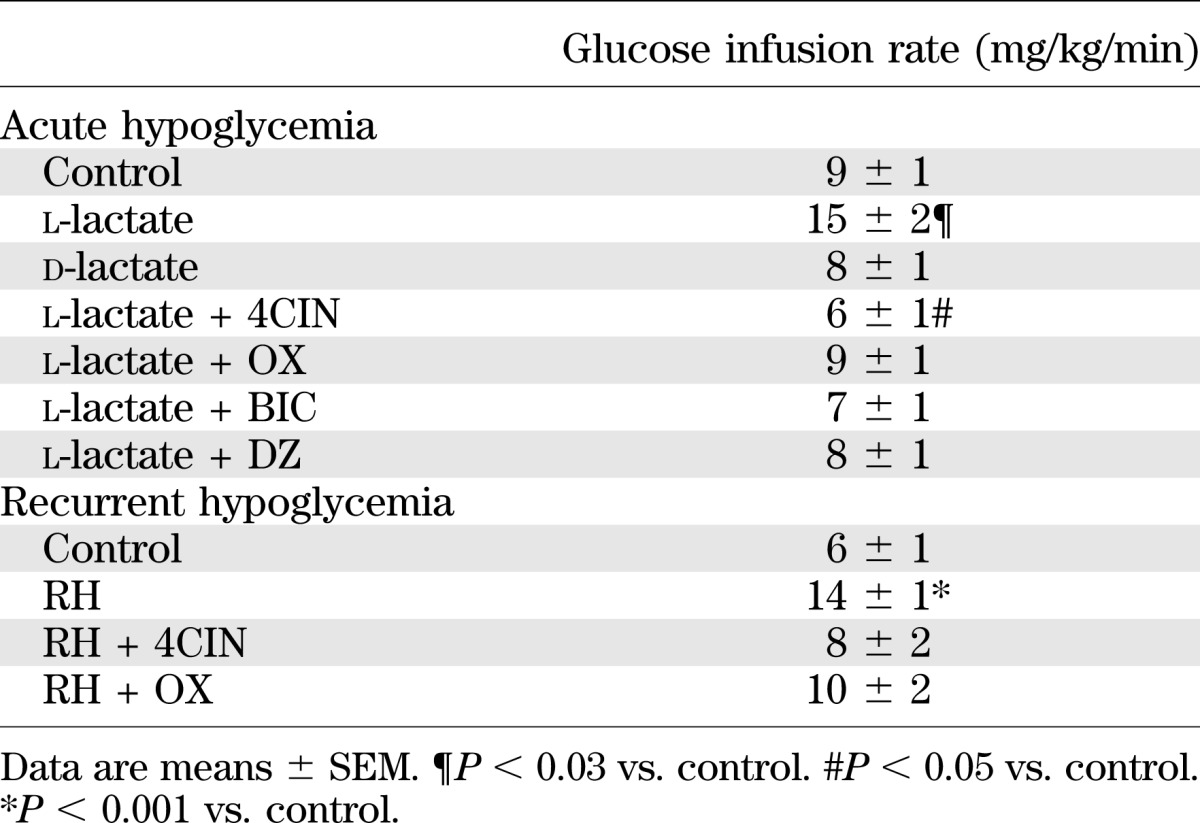
To better understand the mechanism by which lactate raises extracellular GABA levels and in turn impairs glucose counterregulation, we conducted a series of experiments to inhibit uptake or utilization of lactate in nondiabetic hypoglycemia-naïve rats. In the first study, we examined the effects of physiologically raising extracellular lactate levels on VMH GABA levels during hypoglycemia. Plasma glucose (Supplementary Fig. 1) and insulin (Table 1) levels were similar between all of the different treatment groups used. Despite this, exogenous glucose requirements were 67% greater in those animals receiving l-lactate compared with the other treatment groups (Table 2). In response to a hypoglycemic challenge, control animals showed the expected decrease in VMH GABA levels and accompanying activation of the counterregulatory responses (Fig. 1). When l-lactate was delivered into the VMH, local GABA levels dramatically increased by ∼10-fold and these levels remained elevated during the hypoglycemic clamping period (P < 0.001). These higher GABA levels were accompanied by a 48–72% suppression of the glucagon and epinephrine responses (P < 0.01).
TABLE 1.
Plasma insulin concentrations under baseline conditions and during the hypoglycemic clamp
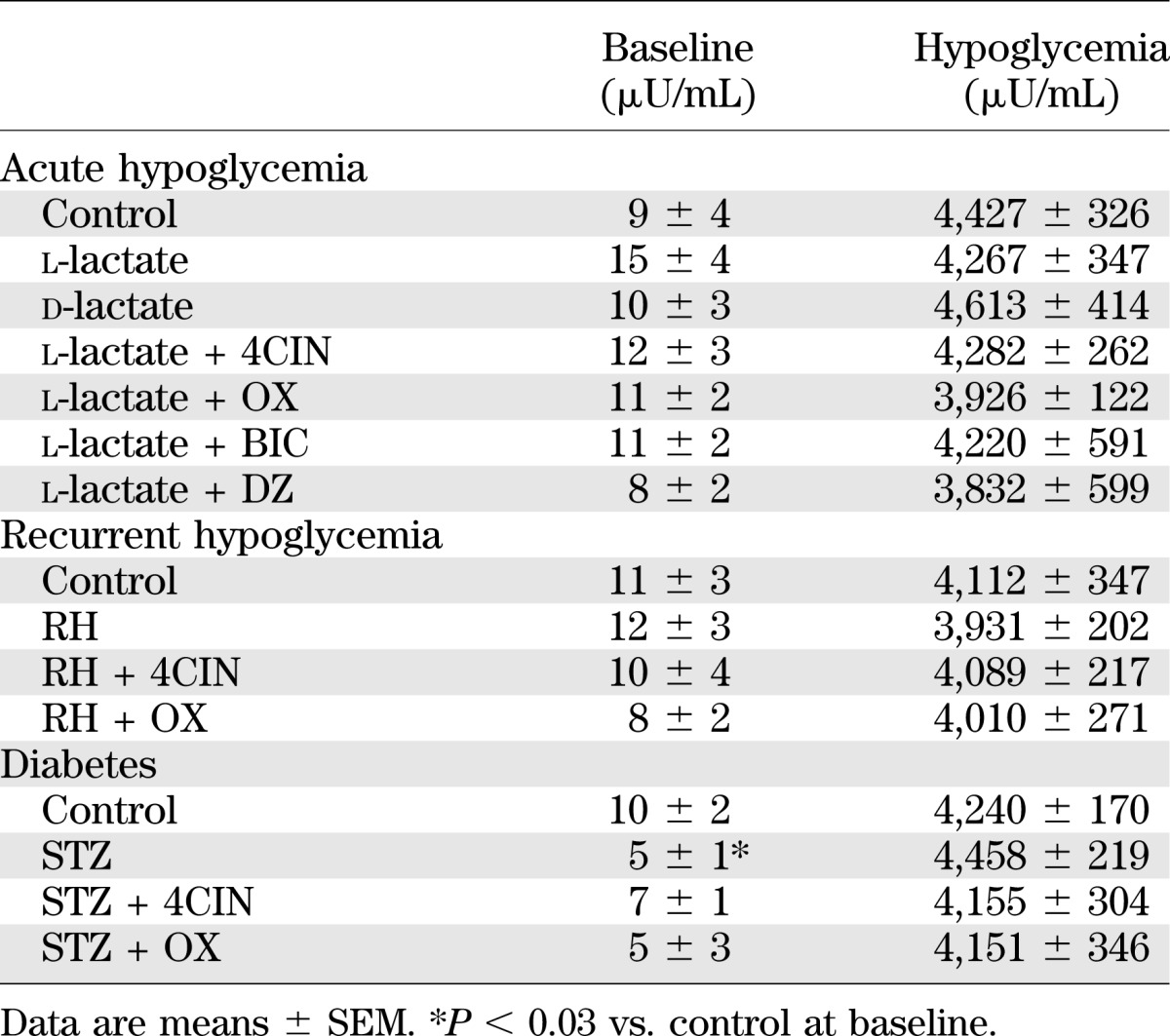
TABLE 2.
Glucose infusion rates during the hypoglycemic clamping period from 30 to 90 min
To control for differences in osmolarity, we microdialyzed d-lactate into the VMH and saw no discernable effect on GABA release or on counterregulatory hormone responses compared with control animals receiving aECF (Fig. 1).
To assess whether the uptake of lactate by neurons contributes to raising VMH GABA levels, we microinjected the MCT2 antagonist 4CIN into the VMH while simultaneously perfusing the region with l-lactate (Fig. 1). When we prevented the entry of lactate into neurons, we dramatically reduced VMH GABA levels during hypoglycemia and restored the counterregulatory responses to normal.
While the above studies show that lactate has a dramatic effect in raising VMH GABA levels and we previously showed that GABA can suppress glucose counterregulatory responses (1), they do not establish a causal link between lactate-mediated GABA release and suppression of the counterregulatory response. To address this question, we microinjected the GABAA receptor antagonist, BIC, into the VMH to block postsynaptic receptors while simultaneously perfusing the region with lactate (Fig. 1). When this was done, we saw a complete restoration of the counterregulatory responses, despite the fact that VMH GABA levels remained significantly elevated (P < 0.001), suggesting that lactate acts to suppress the counterregulatory responses in part through a mechanism involving GABA release.
To examine whether the catabolism of lactate contributes to the release of GABA, we delivered OX into the VMH while physiologically raising extracellular lactate concentrations (Fig. 1). OX inhibits lactate dehydrogenase, effectively preventing the interconversion of lactate to pyruvate in both neurons and astrocytes. OX produced very similar effects to the group given 4CIN. Thus, it is likely that the uptake and subsequent metabolism of lactate mediate the effects on VMH GABA levels. To explore this possibility further, we investigated whether the catabolism of lactate and the subsequent formation of ATP play a role in stimulating GABA release. To address this question, we microinjected diazoxide, an ATP-sensitive potassium (KATP) channel opener, into the VMH, while simultaneously perfusing the region with l-lactate (Fig. 1). When DZ was given, extracellular GABA levels decreased substantially and the counterregulatory defects were restored to normal.
Recurrent hypoglycemia.
Plasma glucose (Supplementary Fig. 2) and insulin (Table 1) concentrations were similar between controls and the three recurrent hypoglycemia (RH) treatment groups at baseline and during the hypoglycemic clamping period. However, exogenous glucose requirements (Table 2) in RH animals were more than twofold greater compared with controls (P < 0.001). As expected, baseline extracellular GABA concentrations in the VMH were nearly threefold higher in RH animals compared with their control counterparts (Fig. 2) (P < 0.03) and extracellular lactate concentrations were 45% higher in RH animals (controls 1.73 ± 0.37 and RH 2.50 ± 0.45 mmol/L, P = 0.065). In response to hypoglycemia, VMH GABA levels decreased by 57% (P < 0.05) in control animals and this was associated with marked increases in glucagon and epinephrine. In contrast, RH animals failed to significantly reduce local GABA levels during the hypoglycemic clamp, and this was accompanied by a 52–57% reduction in both glucagon and epinephrine responses (Fig. 2). To assess whether the utilization of lactate contributed to raising extracellular GABA levels in RH animals, we microinjected either 4CIN or OX into the VMH of RH animals just prior to inducing hypoglycemia on day 4. In both instances, VMH delivery of either 4CIN or OX reduced exogenous glucose requirements closer to control levels, reduced VMH GABA levels, and restored the counterregulatory hormone responses to normal.
FIG. 2.

Microdialysate and plasma hormone concentrations for recurrent hypoglycemia studies. Top panel: VMH extracellular GABA concentrations collected under baseline conditions and during the hypoglycemic clamp. Plasma glucagon (middle panel) and epinephrine (bottom panel) concentrations under baseline conditions during the hypoglycemic clamp. Data collected from normal, hypoglycemia-naïve (control) (n = 4), RH (n = 5), and RH rats microinjected with either 4CIN (RH + 4CIN) (n = 5) or OX (RH + OX) (n = 5) are presented as means ± SEM. *P < 0.03 vs. control baseline. #P < 0.05 vs. control baseline. ¶P < 0.02 vs. control.
Diabetes.
Supplementary Fig. 3 is a schematic depicting the stepped hyperinsulinemic-euglycemic-hypoglycemic clamp protocol used in the diabetic animals. Plasma glucose (Supplementary Fig. 3) and insulin (Table 1) concentrations were matched between nondiabetic controls and diabetic animals during the euglycemic and hypoglycemic clamping phases of the study. As shown in Fig. 3, baseline GABA concentrations in the VMH were approximately twofold higher in the diabetic rats compared with nondiabetic animals (P < 0.04) and lactate levels were 30% higher, though not statistically significant (control 1.87 ± 0.6 and STZ 2.44 ± 0.12 mmol/L, P = 0.33), a picture similar to that seen in rats exposed to RH. In response to hypoglycemia, VMH GABA levels did not fall in the diabetic animals, whereas they did in the control animals (Fig. 3). The high GABA levels in diabetic animals were associated with significant decreases in both glucagon and epinephrine release during hypoglycemia (P < 0.01). Diabetic animals treated with either 4CIN or OX prior to the induction of hypoglycemia exhibited a dramatic reduction in VMH GABA levels during the hypoglycemic clamping phase, and this was accompanied by restoration of counterregulatory hormone responses to values indistinguishable from controls.
FIG. 3.

Microdialysate and plasma hormone concentrations for diabetes studies. Top panel: VMH extracellular GABA concentrations collected under baseline conditions and hypoglycemia. Peak plasma glucagon (middle panel) and epinephrine (bottom panel) concentrations during the hypoglycemic clamp. Data from normal, nondiabetic (control) (n = 5); STZ diabetic (n = 5); and STZ diabetic rats microinjected with either 4CIN (STZ + 4CIN) (n = 4) or OX (STZ + OX) (n = 5) are presented as means ± SEM. *P < 0.04 vs. control baseline. #P < 0.05 vs. control baseline. ¶P < 0.01 vs. control.
DISCUSSION
It has been postulated that under certain conditions such as glucose deprivation, the brain can utilize other fuel substrates to sustain its metabolic demands—one of these substrates being lactate (20). While there is a growing body of evidence suggesting that lactate is a critically sensed fuel within the brain and that it acts centrally to regulate food intake and body weight (21,22), the exact role of lactate and the nature of these adaptations have yet to be elucidated. Lactate itself may serve as an obligatory fuel for energetically demanding neurons under conditions where glucose availability is limited and thereby help to preserve brain function (23–25). This role was underscored by data showing that when lactate was delivered directly into the VMH of normal rats, it dramatically suppressed glucose counterregulatory responses to hypoglycemia (14), suggesting that lactate may be able to replace glucose as a fuel for VMH glucose-responsive neurons and as a result a fuel deficit is not being detected. In the current study, we demonstrate that lactate likely exerts its inhibitory effects, in part, by stimulating GABA release in the VMH, although we cannot exclude its effects on other types of neurons. More importantly, lactate may also be an important contributor to the counterregulatory defects observed in recurrent hypoglycemia and diabetes.
We previously reported that rats exposed to recurrent hypoglycemia and those with insulin deficient diabetes exhibit higher extracellular concentrations of GABA in the VMH and that this increase in tonic inhibition contributes to the counterregulatory defect (3,4). While these studies identified GABA as an important contributor to counterregulatory failure, the mechanisms that led to the increase in GABA were not identified. We noted that in manner similar to GABA, local delivery of lactate into the VMH suppresses the counterregulatory responses to hypoglycemia in nondiabetic, hypoglycemia-naïve rats (14). More importantly, it has been reported that lactate transport and extracellular lactate levels are elevated in recurrent hypoglycemia and diabetes, respectively (16,26). As lactate can serve as both an energy substrate for neurons and a precursor for the synthesis of GABA, we investigated its potential to mediate some of the changes in GABA observed in RH and diabetic animals. We assessed whether lactate contributes to the derangements in hypothalamic GABA release by pharmacologically inhibiting lactate uptake and utilization in the VMH. Indeed, inhibition of lactate utilization with either 4CIN or OX restored the drop in VMH GABA seen normally during hypoglycemia and in doing so also restored the counterregulatory responses to normal in both RH and diabetic animals. These results suggest that brain adaptations leading to the uptake and oxidation of lactate in glucose responsive GABAergic neurons may be important contributors to the deficits observed in these two models of counterregulatory failure. Consistent with this possibility, Herzog et al. (26) recently demonstrated that an enhancement in brain lactate transport with RH is necessary for preserving glucose oxidation and neuronal tricarboxylic acid (TCA) cycling during subsequent bouts of hypoglycemia. Similarly, increased cerebrospinal fluid lactate levels and increased uptake and utilization of monocarboxylic acids have been reported in patients and rodents with type 1 diabetes as well as for preserving neuronal function in recurrently hypoglycemic animals (12,16,26,27). The availability of lactate in the VMH may allow it to replace glucose as a fuel for glucose-responsive GABAergic neurons during periods of glucose deprivation, acting to enhance the firing of VMH GABAergic neurons even as glucose levels begin to fall and suppress the necessary counterregulatory responses.
It is intriguing that both recurrent hypoglycemia and diabetes, two seemingly different conditions, culminate in a similar pathophysiological outcome. The fact that inhibition of lactate uptake or metabolism is able to reverse the defects in both models of counterregulatory failure suggests that lactate may be a common link that leads to counterregulatory failure in the two conditions. We speculate that in the case of recurrent hypoglycemia, lower cerebral glucose concentrations during insulin-induced hypoglycemia is accompanied by a rise in lactate concentrations stemming from greater utilization of glucose and/or the breakdown of glial glycogen stores (26,28). On the other hand, in poorly controlled diabetes, excess glucose in the central nervous system is likely taken up by astrocytes and converted into lactate, which, in turn, is exported into the extracellular space (29). In both scenarios, an increase in extracellular lactate can be taken up and used by nearby neurons, including GABAergic neurons. While our current results demonstrate a profound effect of lactate in raising extracellular VMH GABA concentrations, we cannot eliminate the possibility that lactate may act through indirect pathways or affect other neural inputs that can also influence GABAergic neurons.
To better understand the mechanism by which lactate mediates its effects on GABA, we locally delivered lactate into the VMH of nondiabetic, hypoglycemia-naïve rats using reverse dialysis to raise extracellular lactate levels. The addition of lactate increased baseline GABA levels in the VMH by ~10-fold compared with animals receiving aECF, suggesting that lactate may be one candidate that can potentially increase VMH GABA levels in RH and diabetes. This is in contrast to our previous work, where we showed that acutely raising glucose in the VMH was not sufficient to increase GABA levels above baseline concentrations (2). In a similar manner, Song and Routh (30) demonstrated that raising glucose from 2.5 mmol/L to 5.0 mmol/L did not affect the activity of glucose-excited neurons in brain slice preparations, but the addition of 0.5 mmol/L lactate to the culture medium significantly raised the action potential firing frequency of these neurons. Moreover, studies by Beall et al. (31) using a hypothalamic glucose-sensing cell line showed that lactate has a greater effect on glucose excited (GE) neuron membrane potential compared with glucose. Taken together, this suggests that increased utilization of lactate by VMH glucose-responsive GABA neurons may enhance the firing rate of these neurons, leading to elevated extracellular GABA concentrations.
To test this hypothesis, we inhibited the uptake of lactate with 4CIN or its oxidation with OX while simultaneously delivering lactate into the VMH of normal rats. In both situations, there was a very rapid reduction in extracellular GABA levels that was followed by restoration of the counterregulatory responses. The finding that extracellular lactate concentrations are positively correlated to VMH GABA levels (R = 0.62, P < 0.02) that, in turn, are inversely related to the degree of counterregulatory hormone activation (GABA vs. glucagon, R = −0.41, P < 0.001) is consistent with our previous studies and underscores the likelihood that these two events are tightly coupled (2). The fact that the inhibitory effects of lactate can be reversed with the addition of BIC, a GABAA receptor antagonist, implies that the release of GABA is one means through which lactate exerts its suppressive effects on the counterregulatory responses.
Interestingly, microinjection of DZ into the VMH during lactate perfusion had effects similar to those of 4CIN and OX, suggesting that lactate acts to stimulate TCA cycling in GABA neurons that raises intracellular ATP levels and in turn closes KATP channels. This is consistent with our previous observations suggesting that some VMH GABAergic neurons may be glucose excited in nature (2) as well as those of Song and Routh (30), who showed that lactate can reverse the inhibitory effects of hypoglycemia on VMH glucose-excited neurons by closing the KATP channel. In the latter study, the addition of lactate also increased the action potential frequency of glucose-excited neurons incubated in either 2.5 or 5 mmol/L glucose, which is in keeping with the increase in baseline GABA levels observed after lactate delivery. Furthermore, studies using 13C-labeled lactate to trace the metabolic fate of the fuel in cortical neurons during glucose deprivation show that oxidation of lactate by GABA neurons serves two main purposes: firstly, it provides energy to sustain neuronal activity, and secondly, it provides substrates for the synthesis of GABA (32). If this observation extends into the VMH, then provision of lactate might substantially increase GABA levels by two concomitant mechanisms: de novo synthesis and greater release upon neuronal activation. Hence, when the ability to utilize lactate is removed with 4CIN or OX, the neurons are better able to sense low plasma glucose levels and initiate appropriate responses. It has been suggested that the uptake of lactate and other alternative fuels by the brain may only account for 25% of the brain glucose energy deficit observed during hypoglycemia (33). However, in that study, the contribution from brain lactate was not accounted for—only uptake from the periphery. While the exact contribution from each of these processes cannot be determined based on our pharmacological manipulations, the data are consistent with the hypothesis that increased lactate entry and utilization by glucose-responsive VMH GABAergic neurons may serve not only to support their metabolic demands but also to promote GABA release during periods of glucose deprivation—a mechanism that may be dependent on the KATP channel (34). Furthermore, the data suggest that VMH GABAergic neurons may function more as general metabolic fuel or nutrient sensors as opposed to being specifically glucose sensing in nature.
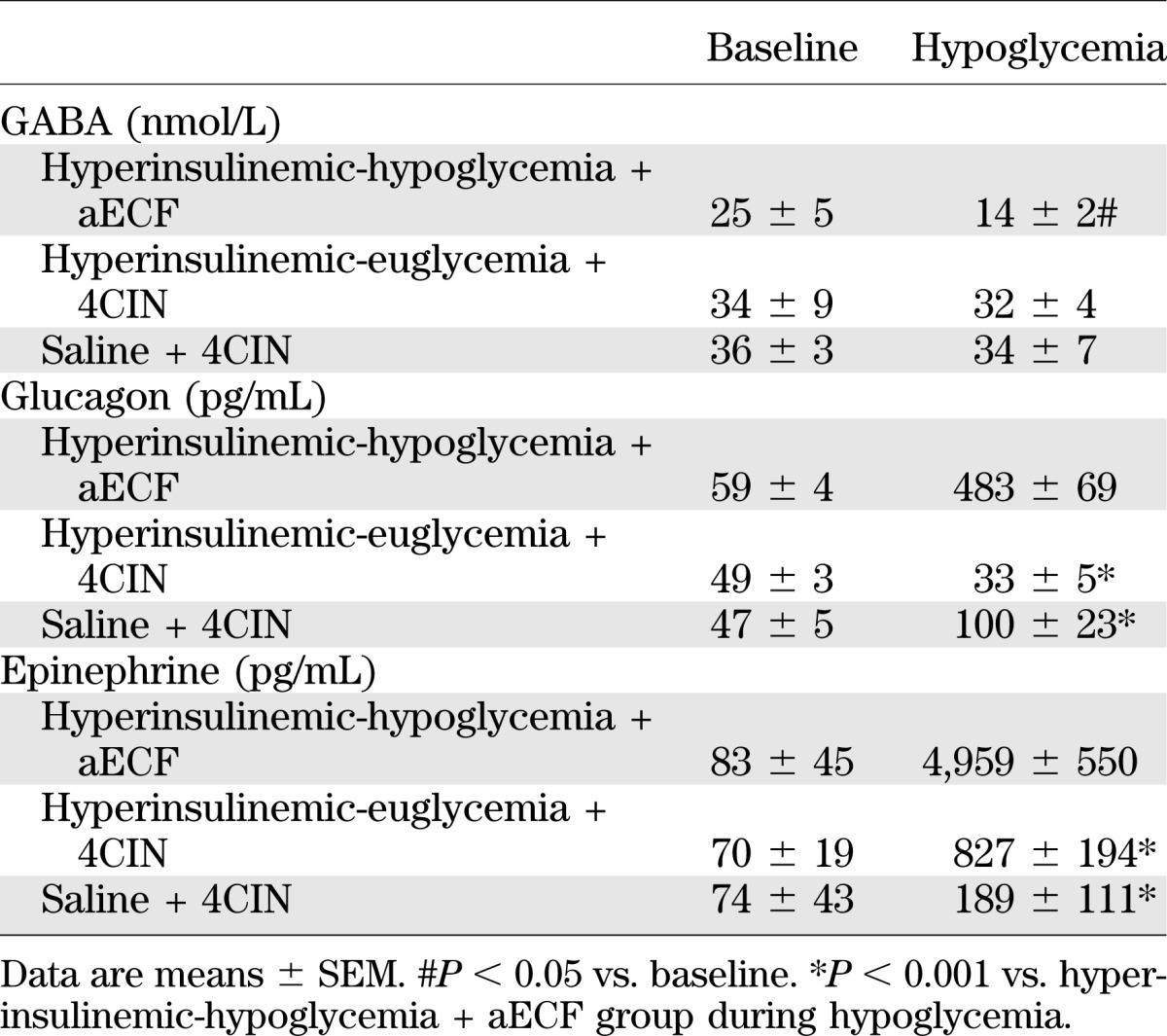
Although 4CIN itself is not a specific inhibitor of MCT2, its specificity for the different MCT isoforms is concentration dependent (35). At lower concentrations, it favors the neuronal isoform, whereas at higher concentrations, it also inhibits astrocytic MCT1. We used a lower concentration of 4CIN, which gives it greater specificity for MCT2, and conducted several control studies to verify the specificity of this dose for the neuronal MCT2 isoform. To assess whether this dose affected other MCTs, we directly injected the compound into the VMH and measured plasma glucagon and epinephrine responses during a peripheral saline infusion or during a hyperinsulinemic-euglycemic glucose clamp. Minimal changes in glucagon and epinephrine levels were observed, suggesting that mitochondrial transport of pyruvate was not appreciably hindered (Table 3). In addition, injection of 4CIN under hypoglycemic conditions without the perfusion of lactate had little effect on counterregulatory responses. To further assess the neuronal specificity of our manipulations, we used another compound, OX, to inhibit mitochondrial conversion of lactate to pyruvate. The effects of 4CIN were replicated in animals given OX, suggesting the conversion of lactate to pyruvate and its subsequent catabolism in the neuron are important for stimulating GABA release. These observations are consistent with a dominant effect on MCT2, but we cannot exclude an effect of 4CIN on MCT1 with complete certainty.
TABLE 3.
Extracellular GABA and plasma hormone concentrations for control studies examining the effects of 4CIN in the absence of VMH lactate delivery
In summary, our data suggest that antecedent hypoglycemia and diabetes may cause similar adaptations in the VMH (though probably via different mechanisms) that lead to the preferential use of lactate as a fuel substrate that diminishes the ability of glucose-sensing neurons to detect immediate falls in blood glucose levels. In GABAergic neurons, lactate likely serves two roles: 1) serving as a substrate in the synthesis of GABA and 2) contributing to TCA cycle activity and thereby stimulating GABA neuronal firing. These studies suggest that the inability of type 1 diabetic patients to detect and respond to hypoglycemia may be mediated by excessive GABA tone in the VMH and that this alteration is driven by accelerated brain lactate metabolism, which is accentuated by exposure to antecedent hypoglycemia. Such data have important implications for the development of new therapeutic targets to reduce hypoglycemia risk in insulin-treated diabetic patients.
Supplementary Material
ACKNOWLEDGMENTS
This work was generously supported by research grants from the American Diabetes Association (7-11-BS-21 to O.C.), the JDRF (2-2008-480 to O.C.), and the National Institutes of Health (P30 DK-45735 and R01 DK-099315 to O.C., and R37 DK-20495 to R.S.S.). O.C. is the recipient of a Career Development Award from the JDRF, and S.A.P. is the recipient of a postdoctoral fellowship from the JDRF.
No potential conflicts of interest relevant to this article were reported.
O.C. conceptualized and designed the studies described in this manuscript, researched and analyzed data, and wrote the manuscript. S.A.P. and W.Z. researched data. A.H. researched and analyzed data. R.S.S. reviewed the manuscript and contributed to discussions. O.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the 70th Scientific Sessions of the American Diabetes Association, Orlando, Florida, 25–29 June 2010.
The authors thank Aida Groszmann, Codruta Todeasa, Maria Batsu, and Ralph Jacob from the Yale Diabetes Research Center for their invaluable assistance with the hormone assays in these studies.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0770/-/DC1.
See accompanying commentary, p. 3999.
REFERENCES
- 1.Chan O, Zhu W, Ding Y, McCrimmon RJ, Sherwin RS. Blockade of GABA(A) receptors in the ventromedial hypothalamus further stimulates glucagon and sympathoadrenal but not the hypothalamo-pituitary-adrenal response to hypoglycemia. Diabetes 2006;55:1080–1087 [DOI] [PubMed] [Google Scholar]
- 2.Zhu W, Czyzyk D, Paranjape SA, et al. Glucose prevents the fall in ventromedial hypothalamic GABA that is required for full activation of glucose counterregulatory responses during hypoglycemia. Am J Physiol Endocrinol Metab 2010;298:E971–E977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan O, Cheng H, Herzog R, et al. Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes 2008;57:1363–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan O, Paranjape S, Czyzyk D, et al. Increased GABAergic output in the ventromedial hypothalamus contributes to impaired hypoglycemic counterregulation in diabetic rats. Diabetes 2011;60:1582–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pellerin L. Lactate as a pivotal element in neuron-glia metabolic cooperation. Neurochem Int 2003;43:331–338 [DOI] [PubMed] [Google Scholar]
- 6.Pellerin L, Bouzier-Sore AK, Aubert A, et al. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 2007;55:1251–1262 [DOI] [PubMed] [Google Scholar]
- 7.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 2000;267:4912–4916 [DOI] [PubMed] [Google Scholar]
- 8.Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci 1996;16:877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zonta M, Angulo MC, Gobbo S, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 2003;6:43–50 [DOI] [PubMed] [Google Scholar]
- 10.Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011;14:724–738 [DOI] [PubMed] [Google Scholar]
- 11.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 1994;91:10625–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mason GF, Petersen KF, Lebon V, Rothman DL, Shulman GI. Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes 2006;55:929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vavaiya KV, Paranjape SA, Briski KP. Testicular regulation of neuronal glucose and monocarboxylate transporter gene expression profiles in CNS metabolic sensing sites during acute and recurrent insulin-induced hypoglycemia. J Mol Neurosci 2007;31:37–46 [DOI] [PubMed] [Google Scholar]
- 14.Borg MA, Tamborlane WV, Shulman GI, Sherwin RS. Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes 2003;52:663–666 [DOI] [PubMed] [Google Scholar]
- 15.Chan O, Paranjape SA, Czyzyk D, et al. Increased GABAergic output in the ventromedial hypothalamus contributes to impaired hypoglycemic counterregulation in diabetic rats. Diabetes 2011;60:1582–1589 [DOI] [PMC free article] [PubMed]
- 16.Jacob RJ, Fan X, Evans ML, Dziura J, Sherwin RS. Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: no evidence for protective adaptation by the blood brain barrier. Metabolism 2002;51:1522–1524 [DOI] [PubMed] [Google Scholar]
- 17.Cheng H, Zhou L, Zhu W, et al. Type 1 corticotropin-releasing factor receptors in the ventromedial hypothalamus promote hypoglycemia-induced hormonal counterregulation. Am J Physiol Endocrinol Metab 2007;293:E705–E712 [DOI] [PubMed] [Google Scholar]
- 18.McDermott JC, Hutber A, Tan MH, Bonen A. The use of a cell-free perfusate in the perfused rat hindquarter: methodological concerns. Can J Physiol Pharmacol 1989;67:1450–1454 [DOI] [PubMed] [Google Scholar]
- 19.Bolteus AJ, Garganta C, Bordey A. Assays for measuring extracellular GABA levels and cell migration rate in acute slices. Brain Res Brain Res Protoc 2005;14:126–134 [DOI] [PubMed] [Google Scholar]
- 20.Pellerin L, Pellegri G, Bittar PG, et al. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci 1998;20:291–299 [DOI] [PubMed] [Google Scholar]
- 21.Lam CK, Chari M, Wang PY, Lam TK. Central lactate metabolism regulates food intake. Am J Physiol Endocrinol Metab 2008;295:E491–E496 [DOI] [PubMed] [Google Scholar]
- 22.Patil GD, Briski KP. Lactate is a critical “sensed” variable in caudal hindbrain monitoring of CNS metabolic stasis. Am J Physiol Regul Integr Comp Physiol 2005;289:R1777–R1786 [DOI] [PubMed] [Google Scholar]
- 23.Izumi Y, Benz AM, Zorumski CF, Olney JW. Effects of lactate and pyruvate on glucose deprivation in rat hippocampal slices. Neuroreport 1994;5:617–620 [DOI] [PubMed] [Google Scholar]
- 24.Maran A, Cranston I, Lomas J, Macdonald I, Amiel SA. Protection by lactate of cerebral function during hypoglycaemia. Lancet 1994;343:16–20 [DOI] [PubMed] [Google Scholar]
- 25.Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 1988;240:1326–1328 [DOI] [PubMed] [Google Scholar]
- 26.Herzog RI, Jiang L, Herman P, et al. Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Invest 2013;123:1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao H, Sadoshima S, Nishimura Y, et al. Cerebrospinal fluid lactate in patients with diabetes mellitus and hypoglycaemic coma. J Neurol Neurosurg Psychiatry 1989;52:372–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang L, Herzog RI, Mason GF, et al. Recurrent antecedent hypoglycemia alters neuronal oxidative metabolism in vivo. Diabetes 2009;58:1266–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grill V, Gutniak M, Björkman O, et al. Cerebral blood flow and substrate utilization in insulin-treated diabetic subjects. Am J Physiol 1990;258:E813–E820 [DOI] [PubMed] [Google Scholar]
- 30.Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2005;54:15–22 [DOI] [PubMed] [Google Scholar]
- 31.Beall C, Hamilton DL, Gallagher J, et al. Mouse hypothalamic GT1-7 cells demonstrate AMPK-dependent intrinsic glucose-sensing behaviour. Diabetologia 2012;55:2432–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waagepetersen HS, Bakken IJ, Larsson OM, Sonnewald U, Schousboe A. Metabolism of lactate in cultured GABAergic neurons studied by 13C nuclear magnetic resonance spectroscopy. J Cereb Blood Flow Metab 1998;18:109–117 [DOI] [PubMed] [Google Scholar]
- 33.Lubow JM, Piñón IG, Avogaro A, et al. Brain oxygen utilization is unchanged by hypoglycemia in normal humans: lactate, alanine, and leucine uptake are not sufficient to offset energy deficit. Am J Physiol Endocrinol Metab 2006;290:E149–E153 [DOI] [PubMed] [Google Scholar]
- 34.Chan O, Lawson M, Zhu W, Beverly JL, Sherwin RS. ATP-sensitive K(+) channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes 2007;56:1120–1126 [DOI] [PubMed] [Google Scholar]
- 35.Erlichman JS, Hewitt A, Damon TL, et al. Inhibition of monocarboxylate transporter 2 in the retrotrapezoid nucleus in rats: a test of the astrocyte-neuron lactate-shuttle hypothesis. J Neurosci 2008;28:4888–4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.