

Abstract
In utero exposure to gestational diabetes mellitus (GDM) is associated with an increased risk of type 2 diabetes and cardiovascular disease in later life, yet the underlying mechanisms remain to be elucidated. We examined the effects of GDM on the proteome, redox status, and nuclear factor erythroid 2–related factor 2 (Nrf2)-mediated antioxidant gene expression in human fetal endothelial cells. Proteomic analysis revealed that proteins involved in redox homeostasis were significantly altered in GDM and associated with increased mitochondrial superoxide generation, protein oxidation, DNA damage, and diminished glutathione (GSH) synthesis. In GDM cells, the lipid peroxidation product 4-hydroxynonenal (HNE) failed to induce nuclear Nrf2 accumulation and mRNA and/or protein expression of Nrf2 and its target genes NAD(P)H:quinone oxidoreductase 1 (NQO1), Bach1, cystine/glutamate transporter, and glutamate cysteine ligase. Although methylation of CpG islands in Nrf2 or NQO1 promoters was unaltered by GDM, decreased DJ-1 and increased phosphorylated glycogen synthase kinase 3β levels may account for impaired Nrf2 signaling. HNE-induced increases in GSH and NQO1 levels were abrogated by Nrf2 small interfering RNA in normal cells, and overexpression of Nrf2 in GDM cells partially restored NQO1 induction. Dysregulation of Nrf2 in fetal endothelium may contribute to the increased risk of type 2 diabetes and cardiovascular disease in offspring.
Endothelial cells are primary targets of hyperglycemia-induced oxidative damage (1), and elevated intracellular glucose levels increase mitochondrial superoxide generation, leading to activation of the polyol and hexosamine pathways and, ultimately, endothelial dysfunction (2). The risk of hypertension is increased in diabetic patients (3), and interestingly, endothelial dysfunction correlates significantly with insulin resistance in young offspring of first-degree relatives of patients with type 2 diabetes (4).
Gestational diabetes mellitus (GDM) is defined as glucose intolerance first diagnosed during pregnancy and affects ∼3–10% of all births (5). Similar to type 2 diabetes, fetal exposure to GDM is strongly associated with a higher risk of insulin resistance in adulthood (6–8), with some but not all studies confirming elevated blood pressure and other metabolic risk factors in GDM offspring (9,10). Streptozotocin-induced diabetes in animal models has been reported to elevate blood pressure and reduce endothelium-dependent vasodilation in offspring (11), potentially a consequence of in utero fetal programing (5–7).
We previously reported that fetal umbilical vein endothelial cells (HUVEC) cultured from GDM pregnancies exhibit reduced rates of cell proliferation and protein and DNA turnover, which are maintained during culture in vitro (12). Our findings of a sustained membrane hyperpolarization, altered Ca2+ signaling, and insulin resistance provided further evidence for an altered phenotype of GDM HUVEC (12,13). Levels of reactive oxygen species (ROS) and lipid peroxidation products are elevated in cord blood (14) and in the embryo (15) of pregnancies affected by diabetes. However, the consequences of increased placental oxidative stress in GDM (5) on the redox environment of fetal endothelial cells, a potential factor influencing the risk of vascular disease in later life, remain to be investigated.
The redox-sensitive nuclear factor erythroid 2–related factor 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1) pathway plays a key role in transcriptional activation of antioxidant defense genes and restoration of vascular redox homeostasis (16,17). Nrf2 is normally targeted for proteasomal degradation via its cytosolic binding protein Keap1 (18), but in response to oxidative or electrophilic stress, accumulates in the nucleus and binds to the antioxidant/electrophile response element (ARE/EpRE) in the promoter region of genes encoding phase II detoxifying enzymes, such as NAD(P)H quinone oxidoreductase 1 (NQO1) (19), and glutamate cysteine ligase (GCLM) and the cystine/glutamate transporter (xCT) involved in glutathione (GSH) synthesis (16,20).
We sought to determine the effects of GDM on the redox status and Nrf2 antioxidant defenses in fetal endothelial cells challenged with the lipid peroxidation product 4-hydroxynonenal (HNE). We report the first proteomic analysis of HUVEC from normal and GDM pregnancies and demonstrate that GDM alters proteins involved in oxidative stress and Nrf2-linked phase II enzyme detoxification and GSH synthesis. We also identified markers of oxidative stress in GDM HUVEC, including increased mitochondrial superoxide generation, protein carbonylation, and DNA damage, consistent with a pro-oxidative redox environment in utero in GDM pregnancies. Although basal gene expression was altered negligibly by GDM, nuclear translocation of Nrf2 and induction of its target genes NQO1, Bach1, GCLM, and xCT by the lipid peroxidation product HNE was abrogated in GDM cells. Our findings of decreased DJ-1 and increased phosphorylated glycogen synthase kinase 3β (p-GSK3β) expression in GDM cells may contribute to the deficits in Nrf2 nuclear accumulation and signaling.
In view of accumulating evidence that pregnancies affected by GDM are associated with an increased risk of cardiovascular disease in later life (6,7), our study provides novel insights into the mechanisms underlying dysregulation of redox homeostasis in fetal endothelium, which may contribute to endothelial dysfunction and type 2 diabetes in the offspring of GDM pregnancies (21).
RESEARCH DESIGN AND METHODS
Additional experimental data are provided in the Supplementary Data online.
Materials.
Chemicals and tissue culture media were purchased from Sigma-Aldrich unless stated otherwise. HNE was from Alexis Biochemicals (San Diego, CA), the Nucleospin 96 RNA Kit was from Macherey-Nagel (Duren, Germany), the QuantiTect reverse transcription and QuantiFast PCR reaction kits were from Qiagen (Hilden, Germany), the DNA fragmentation ELISA kit was from Roche (Mannheim, Germany), and the TransAM Nrf2 binding activity assay kit was from Active Motif (Carlsbad, CA). MitoSox Red (M36008) and Alexa Fluor goat anti-rabbit-488 (A11008) were from Invitrogen Molecular Probes (Carlsbad, CA). Nrf2 (SC-722, 110 kDa), NQO1 (SC-16464), lamin A/C (SC-6254), and donkey anti-goat secondary antibody (SC-2020) were from Santa Cruz Biotechnology (Santa Cruz, CA). DJ-1 (#2134) was from Cell Signaling, and G6PD (ab993) and p-GSK3β(Y216) (ab4797) were from Abcam (Cambridge, U.K.). α-Tubulin (mab1864) and goat anti-rabbit (abn117 HRP), goat anti-rat (abn192 HRP), and goat anti-mouse (abn106 HRP) secondary antibodies were from Millipore (Darmstadt, Germany). GCLM was a gift from Terrance Kavanagh (University of Washington, Seattle, WA), and adenovirus (Ad)Nrf2 was a gift from Jeffry Johnson (University of Wisconsin, Madison, WI).
Patients and umbilical cords.
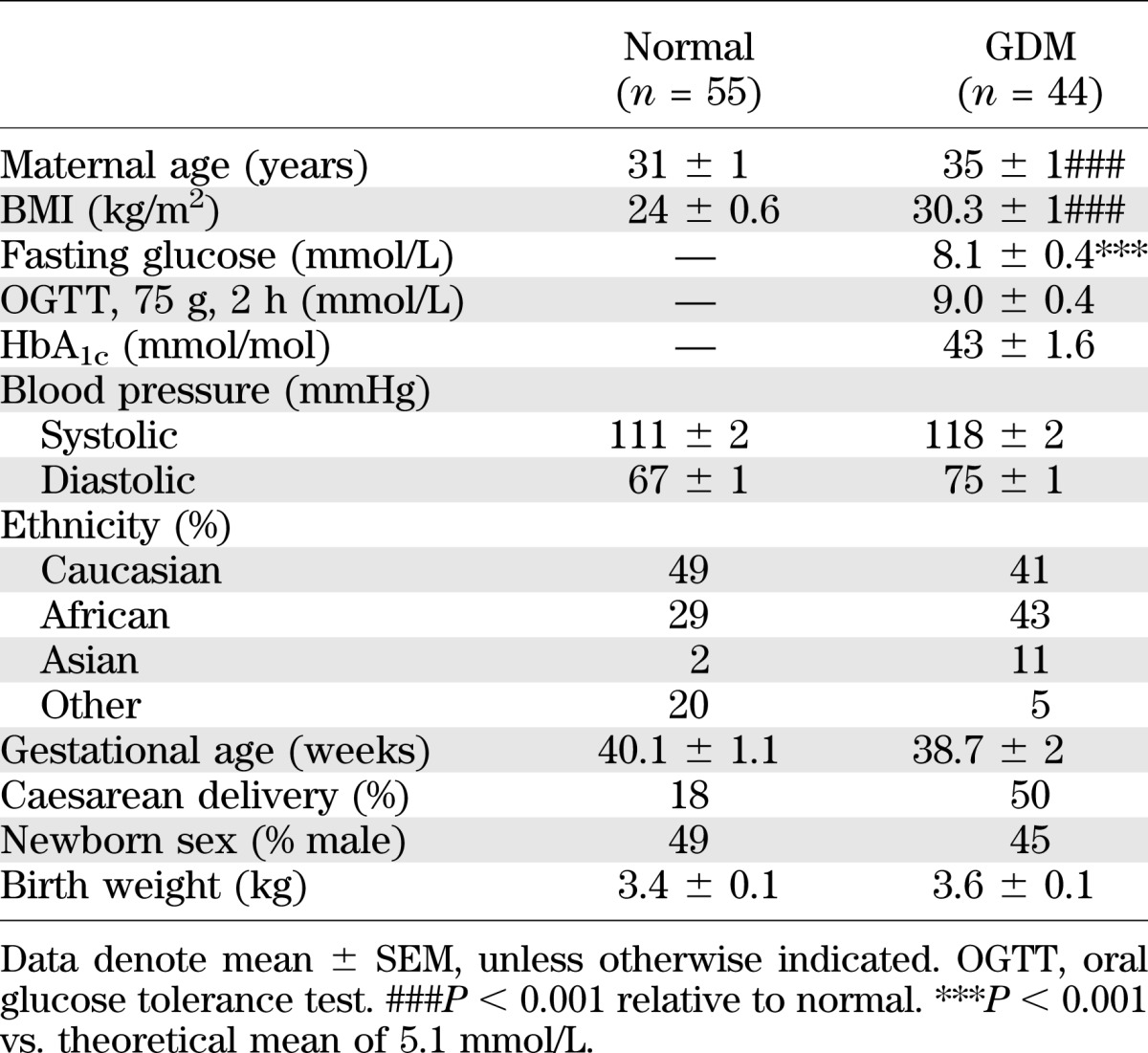
Umbilical cords were obtained from normal (n = 55) and GDM (n = 44) pregnancies from St Thomas’ Hospital (London, U.K.) with informed patient consent and research ethics approval. GDM was defined as glucose intolerance (fasting venous plasma glucose concentration ≥5.1 mmol/L and/or 2-h venous plasma glucose concentration ≥8.5 mmol/L after a 75-g oral glucose tolerance test) first diagnosed during pregnancy. Clinical characteristics of all subjects and pregnancies are summarized in Table 1, and Supplementary Table 2 summarizes clinical data for the proteomic analysis of normal and GDM HUVEC.
TABLE 1.
Clinical characteristics of normal and GDM pregnancies
Endothelial cell culture.
HUVEC were cultured in M199 containing 5 mmol/L d-glucose, as previously described (12). Experiments were conducted with passage 3 HUVEC serum-deprived in 1% FCS M199 for 4 h before treatments. An endothelial phenotype was confirmed by a characteristic cobblestone morphology and von Willebrand factor immunostaining (12).
Proteomic analysis of HUVEC from normal and GDM pregnancies.
Proteomic comparison of normal (n = five donors) and GDM (n = five donors) unstimulated HUVEC was conducted using differential in-gel electrophoresis (DIGE) combined with nanoliquid chromatography tandem mass spectrometry, as previously described (22). Identified proteins were analyzed using the Ingenuity Pathway Knowledge Base (Ingenuity System, Mountain View, CA) to determine the most relevant interaction networks and biological functions. See Supplementary Table 1 for proteome profiling of normal versus GDM HUVEC and Supplementary Table 2 for clinical characteristics of the subjects included in the proteomic analysis.
Samples from different normal and GDM HUVEC cultures were paired for two-dimensional separation (23), and fluorescent gel images were captured using an Ettan DIGE Imager (GE Healthcare) and analyzed using DeCyder software to detect differentially expressed proteins between normal and GDM samples. Spots exhibiting statistical differences (P < 0.05, unpaired Student t test) were excised for trypsin digestion and tandem mass spectrometry. Identification of proteins was performed using TurboSEQUEST software (Bioworks Browser version 3.3.1, Thermo Fisher Scientific) against a UniProt database with the following filter: for charge state 1, XCorr >1.5; for charge state 2, XCorr >2.0; for charge state 3, XCorr >2.5.
Intracellular GSH measurements.
HUVEC were treated with vehicle (0.08% v/v hexane) or HNE (20 μmol/L) for 0–24 h before GSH extraction with 6.5% trichloroacetic acid on ice for 10 min. Total cellular GSH levels were then measured using a fluorometric assay (16).
DNA fragmentation assay.
DNA damage in HUVEC was assessed using a BrdU-based DNA fragmentation ELISA kit (Roche). HUVEC were incubated in 10 μmol/L BrdU-labeling solution for ∼16 h and treated for 6 h with HNE (20 μmol/L) or vehicle (0.08% v/v hexane) in 1% FCS M199. Cytosolic fractions containing BrdU-labeled DNA fragments were extracted for ELISA, and DNA fragmentation was expressed as the average optical density per milligram of protein.
Protein oxidation.
Oxidation of proteins was assessed using an OxyBlot kit (Millipore, Darmstadt, Germany). Cell lysates were denatured and derivatized using 2,4-dinitrophenylhydrazine, and carbonylated proteins were detected by immunoblotting with an anti-dinitrophenyl (DNP) antibody relative to α-tubulin.
Mitochondrial ROS generation.
Basal mitochondrial ROS levels were measured using MitoSox Red, a mitochondrial targeted derivative of hydroethidine, as previously described (24). Cells were loaded with MitoSox Red (5 µmol/L) for 30 min. Fluorescence (excitation/emission 560/625 nm) was detected in 4% paraformaldehyde-fixed cells using a Zeiss Axiovert 200 confocal microscope and quantified using MetaFluor software, and background was fluorescence subtracted.
Chemiluminescence detection of ROS generation.
HUVEC were incubated at 37°C in Krebs buffer containing L-012 and challenged with vehicle or HNE in the absence or presence of the mitochondrial complex I inhibitor rotenone (1 μmol/L) (24). Luminescence was monitored over 40 min after addition of L-012 in a Chameleon V microplate luminometer (Hidex) and expressed per milligram of protein.
Immunoblotting.
Whole-cell and nuclear proteins were extracted using lysis buffer or a nuclear extraction kit (Active Motif), and denatured samples were separated by gel electrophoresis and probed with primary antibodies using α-tubulin or lamin C as reference proteins (17). Protein expression was determined by enhanced chemiluminescence, with images captured in a G-Box gel documentation system (InGenius, Syngene Bioimaging), and densitometry was conducted using ImageJ software (National Institutes of Health, Baltimore, MD).
Nrf2 immunofluorescence.
Cells treated with HNE (20 μmol/L) for 1–4 h were fixed in PBS containing paraformaldehyde (4%), permeabilized with Triton X-100 (0.1%), and examined by immunofluorescence using an anti-Nrf2 primary and Alexa Fluor 488 secondary antibody. Nuclei were colabeled with propidium iodide (1 mg/mL) for 10 min. Cells were visualized in an inverted Nikon Eclipse TE2000-U microscope, and images were captured using a charged-coupled device Nikon DXM1200F digital camera. Densitometry analysis is expressed as ratios of nuclear to cytosolic Nrf2 fluorescence intensity.
Nrf2 binding activity assay.
Cells were treated with HNE (20 μmol/L) for 2 h, nuclear protein was extracted, and binding of nuclear Nrf2 to an ARE sequence (5′-GTCACAGTGACTCAGCAGAATCTG-3′) was determined by ELISA (TransAM Nrf2 kit, Active Motif).
Quantitative RT-PCR.
RNA was extracted and isolated using a NucleoSpin RNA kit (Macherey-Nagel). RNA integrity was assessed by microcapillary electrophoresis using a nano chip (RNA 6000 Nano Assay) and analyzed using an Agilent 2100 Bioanalyser system (Agilent Technologies, Santa Clara, CA). Total RNA was reverse-transcribed using the QuantiTect reverse transcription kit (Qiagen). Nrf2-linked and Nox4 (data not shown) gene expression was assessed using a QuantiFast Probe PCR kit (Qiagen) in a real-time PCR system (Corbett Rotor-Gene). mRNA levels were normalized to the geometric mean of three housekeeper genes: β-actin, ribosomal protein L13a, and succinate dehydrogenase unit complex A. See Supplementary Table 3 for primer sequences.
Nrf2 knockdown by small interfering RNA and adenoviral overexpression of Nrf2.
Normal HUVEC were transfected with 40 pmol/24 well Nrf2 small interfering RNA (siRNA) (Santa Cruz Biotechnology) for 24 h using a siRNA kit (Roche, Mannheim, Germany) before treatment with HNE (20 μmol/L) or vehicle for 20 h, followed by measurements of Nrf2 and NQO1 protein or intracellular GSH levels. GDM HUVEC were transfected for 12 h using adenoviral vectors (50 mol/multiplicity of infection) coordinating expression of Ad-containing green fluorescent protein or active AdNrf2. Cells were then treated with HNE (20 μmol/L) or vehicle and Nrf2, and NQO1 protein expression was determined.
Statistical analysis.
Data denote the mean ± SEM of experiments with HUVEC isolated from three to six independent normal or GDM umbilical cords. Comparison of the proteome of normal (n = five) and GDM (n = five) HUVEC was conducted using an unpaired Student t test. Comparisons of more than two conditions in the same experiment were evaluated using one-way or two-way ANOVA with the Tukey or Bonferroni post hoc test, and P < 0.05 was considered significant.
RESULTS
Proteomic profiling of HUVEC from normal and GDM pregnancies.
The proteome of normal and GDM HUVEC was compared using DIGE (Fig. 1A), and differentially expressed proteins were identified by liquid chromatography tandem mass spectrometry (Fig. 1B). As summarized in Supplementary Table 1, GDM is associated with reduced levels of peroxiredoxin (Prx)-1 and GSH-S-transferase and increased levels of Prx5 and protein disulfide-isomerase A3. Prx1 and GSH-S-transferase are direct downstream target genes of the Nrf2/ARE pathway, and decreased ratios indicate diminished Nrf2 activity. Increased ratios for Prx5 and protein disulfide-isomerase A3, critical redox regulators in mitochondria and endoplasmic reticulum, respectively, are indicative of increased oxidative stress that may affect antioxidant signaling (25,26).
FIG. 1.

Proteomic analysis of normal and GDM HUVEC using DIGE. A: In this representative image of DIGE, proteins from unstimulated normal and GDM HUVEC are stained with Cy3 (red) and Cy5 (green), respectively. B: Fluorescent images of DIGE gels shown in panel A were analyzed using DeCyder 7.0 software to detect differentially expressed proteins in GDM HUVEC (n = 5 pairs of HUVEC from different normal and GDM donors). Differentially expressed proteins were excised and identified by liquid chromatography tandem mass spectroscopy. The numbered spots show significant differences for 39 proteins (ratio of normal-to-GDM >1.1 or <1.1; P < 0.05, unpaired Student t test). Results are summarized in Supplementary Table 1.
When differentially expressed proteins (n = 39, Fig. 1B) were submitted to Ingenuity Pathway Analysis (Supplementary Fig. 1), the computational algorithms generated a primary protein network highlighting oxidative stress, depletion of GSH, phase II defense enzymes, peroxisome proliferator–activated receptor-α/retinoid X receptor-α, and Nrf2-mediated oxidative stress responses as key pathways altered in GDM HUVEC. These links were confirmed by a canonical pathway analysis showing that alterations in oxidative stress signaling were the major phenotypic changes detected in GDM cells (Supplementary Table 1).
Increased mitochondrial ROS generation and protein carbonylation in GDM HUVEC.
Because mitochondrial proteins were altered in GDM cells (Supplementary Table 1), it is noteworthy that basal mitochondrial superoxide generation (Fig. 2A and B) and HNE-stimulated superoxide generation (Fig. 2C and D) were increased in GDM HUVEC. The inhibition of HNE-stimulated superoxide generation by rotenone and negligible changes in Nox4 expression (data not shown) suggests that mitochondria are the most likely source of increased ROS generation. To confirm that GDM was associated with oxidative modification of proteins, normal and GDM HUVEC lysates were immunoblotted for carbonylated proteins, and as shown in Fig. 2E, basal levels of protein oxidation were increased significantly in GDM cells.
FIG. 2.

Increased mitochondrial ROS generation, protein oxidation, and HNE-induced DNA fragmentation in GDM HUVEC. Normal and GDM cells were loaded with MitoSox Red, and mitochondrial ROS generation was measured by fluorescence. Representative basal MitoSox Red fluorescence (grayscale) images in cells from normal (n = 6) or GDM (n = 6) donors (A), with average fluorescence obtained by densitometry from >100 cells on duplicate coverslips for each culture (B). Basal and HNE (20 μmol/L)-stimulated ROS generation after 1 h (C) and after 24 h (D) in normal and GDM HUVEC measured by L-012 chemiluminescence in the absence or presence of the mitochondrial complex I inhibitor rotenone (1 μmol/L). E: Basal protein carbonylation in dinitrophenylhydrazine-derivatized extracts expressed relative to α-tubulin in cells from normal (n = 3) and GDM (n = 3) donors. Representative immunoblot of normal vs. GDM lysates run on a single membrane, with nonrelevant lanes omitted shown in Supplementary Fig. 2A. F: Cells were treated for 6 h with HNE (20 μmol/L) or vehicle (0.08% v/v hexane), and DNA fragmentation was measured in four replicates per culture from different normal (n = 5) and GDM (n = 5) donors. Absorbance expressed per milligram of protein. Data denote mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Increased DNA damage induced by HNE in GDM HUVEC.
GDM and diabetes are characterized by increased lipid peroxidation, and HNE is known to induce DNA damage (27,28). To determine whether GDM increased the sensitivity of fetal endothelial cells to DNA damage, normal and GDM cells were treated with HNE (20 μmol/L) or vehicle for 6 h (Fig. 2D). Although basal DNA fragmentation was not altered, HNE-induced DNA damage was significantly elevated in GDM HUVEC, highlighting potentially compromised antioxidant defenses.
GDM impairs HNE-induced adaptive increases in GSH synthesis and expression of xCT, GCLM, and NQO1.
Because depletion of GSH leads to an increased susceptibility to oxidative stress and apoptosis (21), we compared basal and HNE-stimulated GSH levels in normal and GDM cells. HNE elicited a biphasic adaptive response in GSH levels in normal HUVEC (Fig. 3A), with GSH (nmol/mg protein) initially decreasing to 52 ± 5 at 1.5 h (66 ± 5 in vehicle-treated HUVEC, P < 0.05) and then increasing to 112 ± 9 after 24 h (76 ± 5 in vehicle-treated HUVEC, P < 0.01). However, basal and HNE-induced adaptive increases in GSH were significantly diminished in GDM cells (Fig. 3B and C). Adaptive increases in GSH involve induction of the Nrf2-regulated genes xCT and GCLM (17,20), and notably, upregulation of xCT mRNA and GCLM protein and mRNA levels by HNE in normal cells was absent in GDM cells (Fig. 3D–F and Supplementary Fig. 2B). In contrast, glucose-6-phosphate dehydrogenase (G6PD) expression and enzyme activity were unaffected by GDM (Supplementary Fig. 2C–E).
FIG. 3.
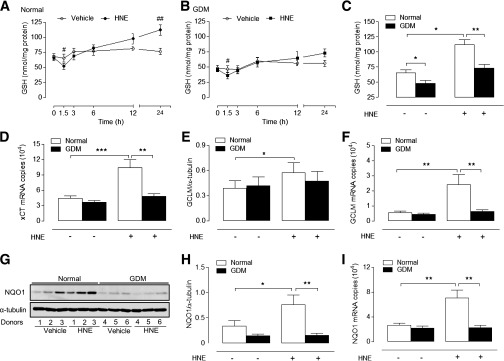
HNE-stimulated adaptive increases in GSH levels and xCT, GCLM, and NQO1 expression are abrogated in GDM HUVEC. Time-dependent changes in intracellular total GSH levels in normal (A) and GDM (B) HUVEC treated with HNE (20 µmol/L) or vehicle. C: Basal and HNE-stimulated GSH levels in normal and GDM HUVEC after 24-h treatment with HNE (20 µmol/L) or vehicle. Basal and HNE (20 µmol/L)-stimulated xCT mRNA (4 h) (D), GCLM protein (12 h) (E), and mRNA (4 h) (F) expression in normal and GDM cells. G–I: Basal and HNE-stimulated NQO1 protein (12 h) and mRNA (4 h) expression in normal and GDM cells. Data denote mean ± SEM of measurements in cultures from normal (n = 4–6) and GDM (n = 4–6) donors. #P < 0.05, ##P < 0.01; *P < 0.05, **P < 0.01, ***P < 0.001 vs. normal HUVEC.
NQO1 is a key phase II defense enzyme regulated by Nrf2 (19), and induction of NQO1 in response to HNE was maximal after 12 h in normal cells but notably abrogated in GDM cells (Fig. 3G and H). Similar to xCT and GCLM, NQO1 mRNA levels were not increased in GDM cells treated with HNE (Fig. 3I).
Impaired Nrf2 activation by HNE in GDM HUVEC.
Given the deficits in GSH synthesis and absence of xCT, GCLM, and NQO1 induction in GDM cells by HNE, we hypothesized that activation of Nrf2 may be compromised. Immunoblot analysis of nuclear-enriched cellular fractions confirmed that HNE increased nuclear Nrf2 levels in normal HUVEC maximally after 2 h (2.2 ± 0.4-fold, P < 0.05), whereas nuclear translocation of Nrf2 was abrogated in GDM HUVEC. Moreover, HNE-induced binding of nuclear Nrf2 to an ARE consensus sequence was abolished in GDM cells (Fig. 4C). Immunofluorescence analysis of nuclear to cytosolic Nrf2 levels under basal and HNE-stimulated conditions further demonstrated that HNE-induced nuclear accumulation of Nrf2 was abrogated in GDM cells (Fig. 4D).
FIG. 4.

Effects of GDM on basal and HNE-stimulated Nrf2 nuclear translocation and ARE binding and expression. Normal and GDM HUVEC were treated with HNE (20 μmol/L) or vehicle (0.08% v/v hexane). A: Representative nuclear immunoblot of nuclear Nrf2 with densitometric analysis relative to lamin C shown in B. C: Nrf2 binding to immobilized ARE on a TransAM ELISA plate, with data expressed as absorbance values at 450 nm. D: Quantification of Nrf2 immunofluorescence in normal and GDM HUVEC. Average nuclear:cytosolic Nrf2 fluorescence values from 100 cells per condition were used for analysis. Basal and HNE-stimulated Nrf2 mRNA (E) and total protein expression (F) in normal and GDM HUVEC. Data denote mean ± SEM in cultures from different normal (n = 3–6) and GDM (n = 3–6) donors. *P < 0.05, **P < 0.01.
To determine whether deficits in HNE-induced Nrf2 activation in GDM cells were due to alterations in Nrf2 expression, we measured basal and HNE-induced Nrf2 mRNA and protein levels in normal and GDM cells. Basal Nrf2 expression was similar in both cell types, but HNE only enhanced total Nrf2 protein (12 h) levels in normal cells (Fig. 4F). When we examined whether impaired Nrf2 activation in GDM was affected by its cytosolic regulator Keap1 or nuclear transcriptional repressor Bach1 (29), basal mRNA levels were unaffected by GDM (Fig. 5A and B), and HNE only upregulated Bach1 mRNA in normal cells (Fig. 5B). Thus, GDM cells may also have lost a key feedback mechanism to regulate Nrf2-linked gene expression via Bach1 (29,30).
FIG. 5.
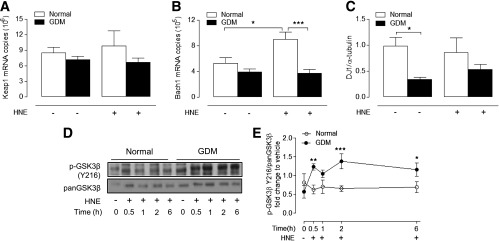
Effects of GDM on DJ-1, Keap1, Bach1, and p-GSK3β expression. Basal and HNE-stimulated protein (12 h) and/or mRNA (4 h) expression of DJ-1 (A), Keap1 (B), and Bach1 (C) in normal and GDM HUVEC. D and E: Basal and HNE (20 μmol/L)-stimulated p-GSK3β protein levels in normal and GDM HUVEC. Data denote mean ± SEM of measurements in cultures from different normal (n = 4–6) and GDM (n = 4–6) donors. *P < 0.05, **P < 0.01, ***P < 0.001 vs. normal HUVEC.
Because disruption of DJ-1, a Parkinson disease–associated protein, leads to decreased Nrf2 protein stability and antioxidant enzyme expression (31), we measured basal DJ-1 expression in normal and GDM cells and found that DJ-1 protein levels were decreased in GDM cells (Fig. 5C). We further investigated whether GDM affects cellular levels of p-GSK3β, which is known to enhance nuclear export of Nrf2 (32). Treatment of GDM HUVEC with HNE led to a significant and sustained increase in p-GSK3β expression, but p-GSK3β levels were unaffected in normal cells (Fig. 5D and E).
Nrf2 siRNA abrogates HNE-induced increases in GSH and NQO1 levels in normal cells, and Nrf2 overexpression upregulates NQO1 levels in GDM cells.
To confirm that adaptive increases in GSH and NQO1 levels induced by HNE were mediated by Nrf2, we used Nrf2 siRNA to knockdown transcriptional activity in normal cells (Fig. 6A and B). Although Nrf2 gene-silencing had negligible effects on basal GSH and NQO1 levels, it significantly attenuated HNE-induced increases in GSH (Fig. 6C) and NQO1 expression (Fig. 6D), mimicking the phenotype of GDM HUVEC. Moreover, transient transfection of GDM HUVEC with an Ad coordinating overexpression of Nrf2 significantly increased basal Nrf2 and NQO1 expression in GDM cells (Fig. 6E and F), suggesting that impaired Nrf2 redox signaling in GDM HUVEC can be partially restored.
FIG. 6.

Effects of Nrf2 knockdown in normal cells and Nrf2 adenoviral overexpression in GDM cells. Normal HUVEC were transfected for 24 h with Nrf2 siRNA or scrambled (Scr) siRNA and then treated with HNE (20 μmol/L) or vehicle (0.08% v/v hexane) for a further 20 h. Representative immunoblot (A) and densitometric analysis (B) of Nrf2 protein levels relative to α-tubulin. C: Basal and HNE-stimulated (24 h) GSH levels in HUVEC lysates. D: Densitometric analysis of HNE-induced NQO1 protein expression in normal HUVEC transfected with Nrf2 siRNA. Representative immunoblot (nonrelevant lanes omitted) (E) and densitometric analysis (F) showing adenoviral (Ad) overexpression of Nrf2 in GDM HUVEC partially rescues NQO1 expression. Data denote mean ± SEM of measurements in independent cultures from four different normal or GDM donors. AdGFP, adenovirus-containing green fluorescent protein; Veh, vehicle. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
To date, there are no reports of the effects of GDM on redox signaling in human fetal endothelium. The current study provides the first whole-cell proteome analysis of the effects of GDM on fetal endothelial cells, characterizing phenotypic alterations in proteins involved in redox signaling. We further demonstrate that Nrf2 nuclear accumulation and Nrf2-mediated adaptive increases in GSH and induction of NQO1, xCT, GLCM and Bach1 expression by the lipid peroxidation product HNE are abrogated in GDM HUVEC. These findings are consistent with increased levels of protein oxidation, enhanced sensitivity to HNE-induced DNA damage, and elevated mitochondrial ROS generation in GDM cells.
Previous studies reported decreased GSH levels in umbilical cord (14) and neonatal (15) blood from GDM pregnancies and correlated inflammatory markers, such as C-reactive protein, intracellular adhesion molecule-1, and interleukin-6, in cord plasma with fetal triglycerides and/or neonatal fat mass (33). Moreover, adiposity in young children is strongly correlated with the extent of methylation of DNA encoding endothelial nitric oxide synthase and retinoid X receptor-α in HUVEC at birth (30), suggesting that our comparison of DNA methylation (Supplementary Fig. 3) and redox regulation in normal and GDM HUVEC may provide valuable insights into the epigenetic effects of GDM in utero.
Markers of oxidative stress involving altered mitochondrial function and endoplasmic reticulum stress, as indicated by our proteomic data (Supplementary Table 1), were validated by our finding that mitochondrial ROS generation was markedly elevated in GDM cells. An elevated glucose load, leading to mitochondrial dysfunction and increased superoxide production, has been widely implicated in the development of glucose intolerance and vascular dysfunction in diabetes (2). Studies using a rodent model of intrauterine growth retardation have shown that elevated intrauterine stress and mitochondrial dysfunction are associated with type 2 diabetes and metabolic disease in offspring (25,34). In our study, fetal endothelial cells from GDM pregnancies exhibited increased basal protein carbonylation and enhanced DNA damage (Fig. 2) in response to physiologically relevant HNE concentrations that rarely exceed 100 µmol/L in cells and tissues (28).
Our proteomic network analysis revealed that GSH and phase II detoxification pathways were altered in GDM cells, suggesting that Nrf2 activation may be compromised in GDM. Because HNE-mediated adaptive increases in GSH and in xCT and GCLM mRNA and/or protein levels were significantly decreased in GDM cells, these findings highlight the importance of Nrf2 and enzymes related to GSH synthesis in promoting cell survival and adaptation in oxidative stress (25). Although lower basal GSH levels in GDM HUVEC may contribute to the increased sensitivity to HNE-mediated damage, adaptive increases in Nrf2-linked gene expression in normal HUVEC appear to be independent of mitochondrial ROS generation (Fig. 2). In normal HUVEC, initial (1.5 h) depletion of intracellular GSH was paralleled by maximal Nrf2 nuclear translocation over 1–2 h, suggesting that HNE-induced Nrf2 activation is a consequence of the altered cellular redox environment rather than a direct effect of acute ROS production per se. Cardiomyocytes exposed to HNE exhibit higher levels of apoptosis in the absence of adaptive increases in GSH (35), findings consistent with our observation of increased HNE-induced DNA damage in GDM HUVEC. Because HNE-mediated induction of xCT, GCLM, NQO1, and Bach1 was abrogated in GDM cells, it seems unlikely that impaired activation of Nrf2 in GDM cells was due to downregulation by Bach1 (29).
Disruption of DJ-1 leads to decreased Nrf2 stability and antioxidant enzyme expression (31), and reduced DJ-1 levels in GDM HUVEC may explain the lack of Nrf2-linked gene induction by HNE. Moreover, increased p-GSK3β levels in GDM HUVEC (Fig. 5) may also contribute to impaired Nrf2-mediated antioxidant gene expression in GDM HUVEC, because activated GSK3β has been reported to phosphorylate Fyn tyrosine kinase, leading to enhanced nuclear export of Nrf2 and proteasomal degradation via the adaptor protein β-TrCP independent of Keap1 (32,36). Because GSK-3β activity is increased in fibroblasts from rats with type 2 diabetes (37), it is possible that HNE-stimulated nuclear accumulation of Nrf2 and induction of NQO1, xCT, GCLM, and Bach1 in GDM endothelial cells are negatively regulated by GSK-3β activation and/or the loss of DJ-1.
Induction of NQO1 by HNE is primarily mediated by Nrf2 (19), and in this context, we demonstrated that knockdown of Nrf2 in normal HUVEC abrogates HNE-induced increases in GSH levels and NQO1 expression, whereas overexpression of Nrf2 in GDM cells partially restores NQO1 levels (Fig. 6). NQO1-null mice exhibit insulin resistance accompanied by higher levels of glucose, triglycerides, lactate, and pyruvate in liver (38), whereas induction of NQO1 suppresses oxidative stress and lipid peroxidation (19), underpinning the importance of NQO1 in cellular defense against oxidative stress.
There are conflicting reports concerning the activation of Nrf2 in endothelial cells exposed to hyperglycemia. Acute exposure of human dermal microvascular endothelial cells to high glucose (30 mmol/L) causes negligible changes in nuclear accumulation of Nrf2 and NQO1 expression (39), whereas elevated glucose enhances Nrf2/ARE-driven luciferase activity and NQO1 mRNA levels in human coronary artery endothelial cells (40). However, Nrf2 knockdown exacerbated glucose-induced ROS generation in both of these studies. We found that elevated glucose (25 mmol/L for 24 h) elicited only marginal increases in ROS generation and Nrf2-mediated antioxidant responses in HUVEC and observed negligible differences between normal and GDM cells (data not shown). Although Nrf2-null mice are not diabetic, they exhibit significantly increased glucose intolerance, urine output, and serum ketones and triglycerides after streptozotocin-induced diabetes (38). Conversely, chemical inducers of Nrf2 markedly attenuate vascular dysfunction and glucose intolerance and prevent obesity and dyslipidemia in mice fed a high-fat diet (41).
Epigenetic modifications have been implicated in altered prenatal programming (30,34,42) and may potentially modulate Nrf2-linked antioxidant responses in GDM endothelial cells. Although methylation of CpG islands in the Nrf2 promoter appears to inhibit transcriptional activity in transgenic adenocarcinoma mouse prostate tumor cells (43), we found no significant differences in the methylation status of CpG islands in the promoters of Nrf2 (Supplementary Fig. 2) or NQO1 (data not shown) between normal and GDM cells. Serum levels of some microRNAs are decreased in mothers with GDM compared with normal pregnancies (44), and microRNAs may be partly involved in developmental priming of type 2 diabetes (45). By targeting “epigenetic machinery,” such as DNA methyltransferases and histone deacetylases, microRNAs may affect Nrf2-mediated antioxidant gene expression and thereby influence the risk of cardiovascular disease.
Vascular Nrf2 levels and adaptive antioxidant defenses have been shown to decline with aging (24). Because we have demonstrated that Nrf2 signaling is impaired in GDM fetal endothelial cells and others have reported that vascular cells in aged rodents (46) exhibit altered Nrf2 signaling, we hypothesize that the GDM endothelial redox phenotype may reflect in utero aging as a consequence of sustained oxidative stress. Premature aging in utero has been implicated in spontaneously hypertensive rats, in which a diabetic intrauterine environment was associated with a significantly reduced life span of offspring (47).
As illustrated in Fig. 7, we speculate that intrauterine exposure to maternal diabetes (GDM) alters the redox proteome of fetal endothelial cells, leading to impaired Nrf2-mediated antioxidant defenses. Nrf2 plays an important role in maintaining mitochondrial function (22,23) and protecting cells against endoplasmic reticulum stress (28). Persistent deficits in redox signaling in fetal endothelial cells in GDM will undermine cellular defenses against oxidative stress in utero, predisposing offspring to an increased risk of type 2 diabetes and cardiovascular disease in later life. The Nrf2 antioxidant defense pathway may, therefore, provide a therapeutic target for ameliorating oxidative stress associated with diabetes, aging, and nephropathy (48).
FIG. 7.

Impaired Nrf2 activation and antioxidant gene expression in fetal endothelial cells in GDM. The Nrf2-Keap1 antioxidant defense pathway is compromised in fetal endothelial cells exposed to maternal diabetes in utero, potentially as a consequence of decreased DJ-1 and increased p-GSK3β expression. Decreased stabilization of Nrf2 by DJ-1 and/or enhanced nuclear export of Nrf2 by p-GSK3β renders cells more vulnerable to a disturbed redox balance. The altered fetal endothelial redox phenotype may predispose offspring of GDM mothers to cardiovascular disease (CVD) and type 2 diabetes in later life.
Supplementary Material
ACKNOWLEDGMENTS
This study received support from the British Heart Foundation (FS/10/59/28533) and the China Scholarship Council PhD Award (X.C.). M.M. is a Senior British Heart Foundation Fellow.
No potential conflicts of interest relevant to this article were reported.
X.C., S.J.C., B.P., and W.P. researched the data. D.S. provided advice for quantitative PCR measurements. X.Y. and M.M. provided advice for the proteomic data analysis and interpretation. R.C.M.S. and G.E.M. designed the research. All authors edited the draft manuscript. G.E.M. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the data analysis.
The authors thank the St Thomas’ Hospital Research Midwives for the collection of umbilical cords.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0169/-/DC1.
R.C.M.S. and G.E.M. are joint senior authors.
REFERENCES
- 1.Ceriello A, dello Russo P, Amstad P, Cerutti P. High glucose induces antioxidant enzymes in human endothelial cells in culture. Evidence linking hyperglycemia and oxidative stress. Diabetes 1996;45:471–477 [DOI] [PubMed] [Google Scholar]
- 2.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010;107:1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011;123:2799–2810 [DOI] [PubMed]
- 4.Balletshofer BM, Rittig K, Enderle MD, et al. Endothelial dysfunction is detectable in young normotensive first-degree relatives of subjects with type 2 diabetes in association with insulin resistance. Circulation 2000;101:1780–1784 [DOI] [PubMed] [Google Scholar]
- 5.Lappas M, Hiden U, Desoye G, Froehlich J, Hauguel-de Mouzon S, Jawerbaum A. The role of oxidative stress in the pathophysiology of gestational diabetes mellitus. Antioxid Redox Signal 2011;15:3061–3100 [DOI] [PubMed] [Google Scholar]
- 6.Krishnaveni GV, Veena SR, Hill JC, Kehoe S, Karat SC, Fall CH. Intrauterine exposure to maternal diabetes is associated with higher adiposity and insulin resistance and clustering of cardiovascular risk markers in Indian children. Diabetes Care 2010;33:402–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobngwi E, Boudou P, Mauvais-Jarvis F, et al. Effect of a diabetic environment in utero on predisposition to type 2 diabetes. Lancet 2003;361:1861–1865 [DOI] [PubMed] [Google Scholar]
- 8.Fraser A, Tilling K, Macdonald-Wallis C, et al. Associations of gestational weight gain with maternal body mass index, waist circumference, and blood pressure measured 16 y after pregnancy: the Avon Longitudinal Study of Parents and Children (ALSPAC). Am J Clin Nutr 2011;93:1285–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tam WH, Ma RC, Yip GW, et al. The association between in utero hyperinsulinemia and adolescent arterial stiffness. Diabetes Res Clin Pract 2012;95:169–175 [DOI] [PubMed] [Google Scholar]
- 10.Patel S, Fraser A, Davey Smith G, et al. Associations of gestational diabetes, existing diabetes, and glycosuria with offspring obesity and cardiometabolic outcomes. Diabetes Care 2012;35:63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holemans K, Gerber RT, Meurrens K, De Clerck F, Poston L, Van Assche FA. Streptozotocin diabetes in the pregnant rat induces cardiovascular dysfunction in adult offspring. Diabetologia 1999;42:81–89 [DOI] [PubMed] [Google Scholar]
- 12.Sobrevia L, Cesare P, Yudilevich DL, Mann GE. Diabetes-induced activation of system y+ and nitric oxide synthase in human endothelial cells: association with membrane hyperpolarization. J Physiol 1995;489:183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sobrevia L, Yudilevich DL, Mann GE. Elevated D-glucose induces insulin insensitivity in human umbilical endothelial cells isolated from gestational diabetic pregnancies. J Physiol 1998;506:219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajdl D, Racek J, Steinerová A, et al. Markers of oxidative stress in diabetic mothers and their infants during delivery. Physiol Res 2005;54:429–436 [PubMed] [Google Scholar]
- 15.Sakamaki H, Akazawa S, Ishibashi M, et al. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes 1999;48:1138–1144 [DOI] [PubMed] [Google Scholar]
- 16.Ishii T, Itoh K, Takahashi S, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 2000;275:16023–16029 [DOI] [PubMed] [Google Scholar]
- 17.Ishii T, Itoh K, Ruiz E, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 2004;94:609–616 [DOI] [PubMed] [Google Scholar]
- 18.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem 2004;279:31556–31567 [DOI] [PubMed] [Google Scholar]
- 19.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys 2010;501:116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasaki H, Sato H, Kuriyama-Matsumura K, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 2002;277:44765–44771 [DOI] [PubMed] [Google Scholar]
- 21.Cheng X, Siow RC, Mann GE. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal 2011;14:469–487 [DOI] [PubMed] [Google Scholar]
- 22.Pula G, Mayr U, Evans C, et al. Proteomics identifies thymidine phosphorylase as a key regulator of the angiogenic potential of colony-forming units and endothelial progenitor cell cultures. Circ Res 2009;104:32–40 [DOI] [PubMed] [Google Scholar]
- 23.Pula G, Perera S, Prokopi M, Sidibe A, Boulanger CM, Mayr M. Proteomic analysis of secretory proteins and vesicles in vascular research. Proteomics Clin Appl 2008;2:882–891 [DOI] [PubMed] [Google Scholar]
- 24.Rowlands DJ, Chapple S, Siow RC, Mann GE. Equol-stimulated mitochondrial reactive oxygen species activate endothelial nitric oxide synthase and redox signaling in endothelial cells: roles for F-actin and GPR30. Hypertension 2011;57:833–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 2004;279:20108–20117 [DOI] [PubMed] [Google Scholar]
- 26.Boden G, Duan X, Homko C, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 2008;57:2438–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev 2008;28:569–631 [DOI] [PubMed] [Google Scholar]
- 28.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81–128 [DOI] [PubMed] [Google Scholar]
- 29.Jyrkkänen HK, Kuosmanen S, Heinäniemi M, et al. Novel insights into the regulation of antioxidant-response-element-mediated gene expression by electrophiles: induction of the transcriptional repressor BACH1 by Nrf2. Biochem J 2011;440:167–174 [DOI] [PubMed] [Google Scholar]
- 30.Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011;60:1528–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malhotra D, Thimmulappa R, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med 2008;178:592–604 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 2006;281:14841–14851 [DOI] [PubMed] [Google Scholar]
- 33.Nelson SM, Sattar N, Freeman DJ, Walker JD, Lindsay RS. Inflammation and endothelial activation is evident at birth in offspring of mothers with type 1 diabetes. Diabetes 2007;56:2697–2704 [DOI] [PubMed] [Google Scholar]
- 34.Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood disease. Biochem J 2010;427:333–347 [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Sano M, Shinmura K, et al. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J Mol Cell Cardiol 2010;49:576–586 [DOI] [PubMed] [Google Scholar]
- 36.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol 2011;31:1121–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bitar MS, Al-Mulla F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am J Physiol Endocrinol Metab 2011;301:E1119–E1129 [DOI] [PubMed] [Google Scholar]
- 38.Gaikwad A, Long DJ, 2nd, Stringer JL, Jaiswal AK. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol Chem 2001;276:22559–22564 [DOI] [PubMed] [Google Scholar]
- 39.Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008;57:2809–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ungvari Z, Bailey-Downs L, Gautam T, et al. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol 2011;300:H1133–H1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin S, Wakabayashi J, Yates MS, et al. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur J Pharmacol 2009;620:138–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinney SE, Simmons RA. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab 2010;21:223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu S, Khor TO, Cheung KL, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS ONE 2010;5:e8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao C, Dong J, Jiang T, et al. Early second-trimester serum miRNA profiling predicts gestational diabetes mellitus. PLoS ONE 2011;6:e23925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferland-McCollough D, Ozanne SE, Siddle K, Willis AE, Bushell M. The involvement of microRNAs in Type 2 diabetes. Biochem Soc Trans 2010;38:1565–1570 [DOI] [PubMed] [Google Scholar]
- 46.Collins AR, Lyon CJ, Xia X, et al. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res 2009;104:e42–e54 [DOI] [PubMed] [Google Scholar]
- 47.Iwase M, Wada M, Shinohara N, Yoshizumi H, Yoshinari M, Fujishima M. Effect of maternal diabetes on longevity in offspring of spontaneously hypertensive rats. Gerontology 1995;41:181–186 [DOI] [PubMed] [Google Scholar]
- 48.de Haan JB. Nrf2 activators as attractive therapeutics for diabetic nephropathy. Diabetes 2011;60:2683–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.