

Abstract
The drug fenofibrate has received major attention as a novel medical treatment for diabetic retinopathy (DR) and other diabetes-induced microvascular complications. This interest stems from two recent large, well-designed clinical trials that demonstrated large reductions in the progression of DR and the need for laser intervention, in addition to a reduction in renal and neurological outcomes, in patients with type 2 diabetes. In both trials, the greatest benefit on DR progression was observed in those patients with DR at baseline. Originally considered a lipid-modifying drug, it now appears that multiple mechanisms may underpin the benefit of fenofibrate on diabetic microvascular end points. Fenofibrate regulates the expression of many different genes, with a range of beneficial effects on lipid control, inflammation, angiogenesis, and cell apoptosis. These factors are believed to be important in the development of DR regardless of the underlying diabetes etiology. Cell experiments have demonstrated improved survival of retinal endothelial and pigment epithelial cells in conjunction with reduced stress signaling under diabetic conditions. Further, fenofibrate improves retinal outcomes in rodent models of diabetes and retinal neovascularization. Given the results of these preclinical studies, further clinical trials are needed to establish the benefits of fenofibrate in other forms of diabetes, including type 1 diabetes. In DR management, fenofibrate could be a useful adjunctive treatment to modifiable risk factor control and regular ophthalmic review. Its incorporation into clinical practice should be continually revised as more information becomes available.
Diabetic retinopathy (DR) and other microvascular complications remain a major source of disability in patients with diabetes. Better control of glucose, blood pressure, and lipids in recent years has reduced the risk of DR, but many patients continue to experience progressive eye damage and require specialist ophthalmic care. Fenofibrate, the structure of which is shown in Fig. 1, has shown promise in the prevention of diabetic microvascular complications. The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) (1) and the Action to Control Cardiovascular Risk in Diabetes (ACCORD) (2) trials demonstrated that daily oral fenofibrate significantly reduced the progression of DR and other microvascular end points in patients with type 2 diabetes. Fenofibrate is best recognized for its ability to produce large reductions in triglyceride levels and small to modest increases in HDL-cholesterol (HDL-C) levels. However, its microvascular benefits may be mediated by other novel mechanisms.
FIG. 1.
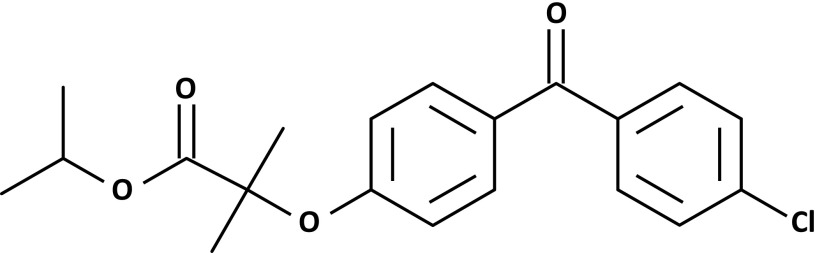
Chemical structure of fenofibrate. Lines indicate carbon bonds. O, oxygen; Cl, chlorine.
FENOFIBRATE PHARMACODYNAMICS
Fenofibrate is an orally administered fibric acid derivative that is conventionally used to treat hypertriglyceridemia, low HDL-C levels, or as an adjunct to statins in dyslipidemia. In addition to its lipid effects, fenofibrate is known to affect many other pathways involved in inflammation, angiogenesis, and cell survival (Fig. 2). During absorption, fenofibrate is rapidly metabolized by tissue and plasma esterases to its active metabolite, fenofibric acid, which is a peroxisome proliferator–activated receptor-α (PPARα) agonist (3). Classically, bound PPARα undergoes a conformational change to form a heterodimer complex with another nuclear receptor, the retinoid X receptor. The PPARα-retinoid X receptor complex then binds with specific DNA peroxisome proliferator response elements to activate (or in some cases repress) target gene transcription (Fig. 3).
FIG. 2.

Identified lipid and nonlipid molecular actions of fenofibrate and its active metabolite, fenofibric acid. IGF1R, IGF-1 receptor; LRP-6, LDL receptor–related protein-6; NF-κB, nuclear factor-κB.
FIG. 3.

Classical fenofibrate signaling pathway. Fenofibrate is rapidly converted to fenofibric acid (FA) in vivo by tissue and plasma esterases before entering the cell. Fenofibric acid binds to PPARα and forms a heterodimer complex with retinoid X receptor (RXR). This complex then binds to specific peroxisome proliferator response elements (PPREs) to activate target gene transcription. RA, 9-cis retinoic acid.
Lipid effects.
PPARα is expressed in several metabolically active tissues with a high turnover of fatty acids (4). Activated PPARα lowers free fatty acids by upregulating synthesis of proteins responsible for fatty acid transport and β-oxidation, which inhibits the formation of triglycerides and VLDL. Triglyceride levels are further reduced due to upregulation of the synthesis of lipoprotein lipase and apolipoprotein (Apo)-V and downregulation of Apo-CIII. A consequence of these changes is a shift in the balance of LDL species from small, dense particles toward larger, more buoyant particles that are more easily cleared by the LDL receptor and less likely to become oxidized (5–7). In addition, increased Apo-AI and Apo-AII expression increases vasoprotective HDL-C levels and facilitates reverse cholesterol transport (4).
Anti-inflammatory effects.
Fenofibrate attenuates inflammation, as demonstrated by its ability to inhibit interleukin (IL)-1–induced IL-6 expression in vascular smooth muscle cells and lipopolysaccharide (LPS)-induced IL-6 expression in mouse aortic explants (8). Further, aortas from PPARα knockout mice showed an exaggerated inflammatory response to LPS that was not inhibited by fenofibrate. In another study using human aortic smooth muscle cells, fenofibrate inhibited IL-1–induced production of IL-6 and prostacyclin and reduced expression of the inflammatory enzyme cyclooxygenase (COX)-2 (9). The anti-inflammatory effects of fenofibrate appear to involve direct agonism of PPARα, with suppression of the nuclear factor-κB and activator protein 1 (AP-1) transcription factors (8,9).
Fenofibrate also inhibits tumor necrosis factor-α (TNF-α)–induced vascular cell adhesion molecule (VCAM)-1 expression and C-reactive protein (CRP)–induced monocyte chemoattractant protein (MCP)-1 expression in endothelial cells (10,11). Further, it can reduce LPS-induced matrix metalloproteinase-9 (MMP-9) production by monocyte-like cells, suggesting that it may reduce extracellular matrix degradation (12).
Antiangiogenic effects.
Retinal angiogenesis is an important pathological event in the development of proliferative DR (PDR). Fenofibrate inhibited basic fibroblast growth factor–induced proliferation of bovine capillary endothelial cells and vascular endothelial growth factor (VEGF)–induced human umbilical vein endothelial cell proliferation and migration (13). Further, fenofibrate reduced human umbilical vein endothelial cell expression of VEGF receptor-2 via PPARα-dependent inhibition of Sp-1 (14). Clofibrate, another PPARα agonist, reduced tumor VEGF levels in a mouse model of ovarian cancer (15). This reduction in VEGF levels may be by inhibition of Wnt signaling, as reported with fenofibrate in the retina (16).
Antiapoptotic effects.
Fenofibrate reduces apoptosis in vitro, as demonstrated by inhibition of glucose-induced apoptosis of cultured human glomerular endothelial cells (17). This appeared to be mediated by activation of AMP-activated protein kinase (AMPK) and endothelial nitric oxide synthase. The antiapoptotic effects of fenofibrate were prevented by inhibition of AMPK and endothelial nitric oxide synthase. Interestingly, these results were not replicated by the PPARα agonists bezafibrate or WY-14643.
Antioxidant effects.
Emerging evidence suggests that fenofibrate may affect antioxidant pathways. Fenofibrate reduced the development of nephropathy and the accumulation of renal reactive oxygen species in streptozotocin-induced diabetic rats (18). In addition, PPARα activation with clofibrate reduced oxidative stress and upregulated the expression of cytoplasmic and mitochondrial superoxide dismutase in a rodent model of myocardial ischemia (19). Upregulation of antioxidant enzymes in the retina could also occur with fenofibrate, but this is yet to be shown directly.
EFFECTIVENESS OF FIBRATES ON DR END POINTS
Hard exudates.
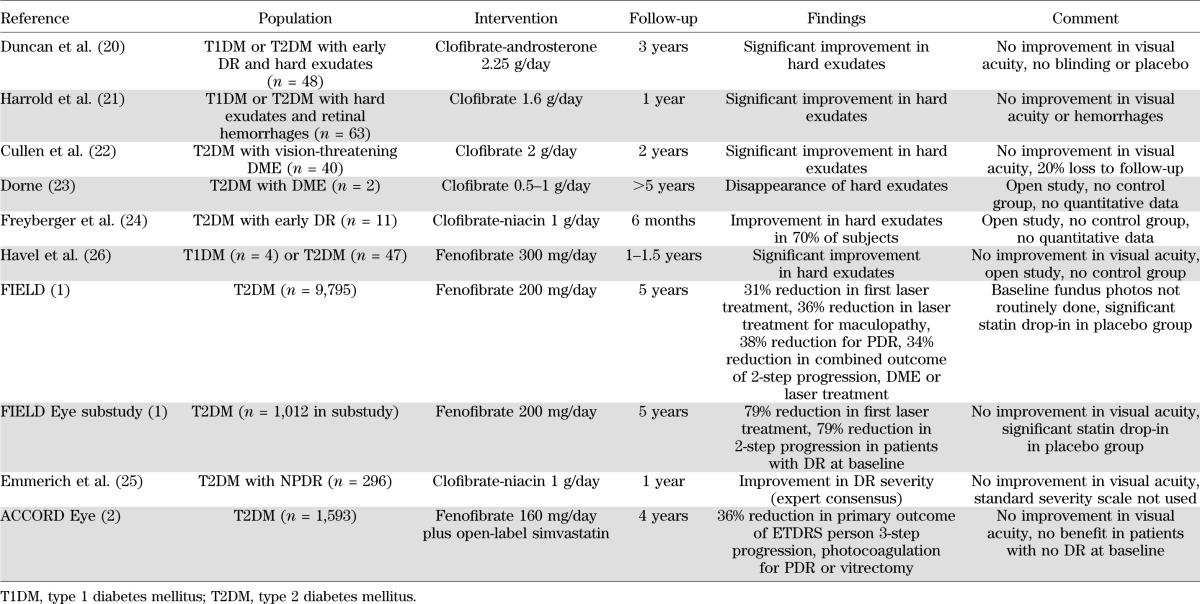
Hard exudates are lipid deposits within the retina and a sign of diabetic macular edema (DME). They form as a result of lipid leakage through permeable retinal vessels damaged during the course of diabetes. Hard exudates have been used as a clinical marker of DR in several fibrate trials (Table 1). Trials of clofibrate in combination with androsterone (Atromid) or niacin (etofibrate) reported regression of hard exudates and other retinal lesions in patients with DR from type 1 or 2 diabetes (20–25). Similar improvements in hard exudates were later reported in patients with type 1 or 2 diabetes treated with fenofibrate for ∼1 year (26). Fenofibrate had no significant effect on hard exudates or DME in the FIELD ophthalmology substudy, but a composite end point of two-step DR progression, DME, or laser treatment was significantly reduced by 34% relative to placebo (1).
TABLE 1.
Fibrates in DR clinical trials
Progression of retinopathy.
In the FIELD ophthalmology substudy, fundus photographs were taken serially over 5 years in 1,012 participants and graded based on the Early Treatment Diabetic Retinopathy Study (ETDRS) severity scale. Fenofibrate reduced two-step progression on the ETDRS scale for individual eyes by 79% in patients with existing DR at baseline over 5 years, but not in those without baseline DR (1).
In the ACCORD-Eye substudy, 2,856 type 2 diabetic patients were monitored for 4 years with fundus photographs that were graded based on the ETDRS severity scale (2). Of these, 1,593 patients with dyslipidemia were evaluated for the effect of fenofibrate on DR outcomes. The DR severity level for each eye was combined to give a single patient score using the “worse eye emphasized” method. The primary outcome was a composite end point of the occurrence of three-step progression of DR on the combined patient (17-point) severity scale, photocoagulation for PDR, or vitrectomy. The ACCORD trial found that treatment with fenofibrate over 4 years reduced this composite outcome by 36% compared with the placebo group. In comparison, intensive glycemic control reduced this outcome by only 30%. Fenofibrate reduced the primary outcome by 25 and 43% in patients assigned to intensive and standard glycemic control, respectively, which suggests that the benefit of fenofibrate may increase with worsening glycemic control. Analysis according to baseline DR status again revealed that fenofibrate was most effective in patients with existing DR. Fenofibrate reduced the odds of the primary outcome by about half in those with baseline DR but had no effect in those without DR. Most primary outcome events included three-step progression of DR, which is not surprising because patients with worse DR are more likely to need laser or vitrectomy surgery. DR progression occurred in 41 patients taking fenofibrate and in 70 patients taking the placebo, translating to an unadjusted relative risk of 0.57 with fenofibrate (P = 0.003).
Laser treatment.
The need for laser treatment signals a failure of conservative medical management to halt the progression of diabetic retinal damage and a need to intervene to prevent visual loss. Laser is generally aimed at preserving central vision, often at the expense of the peripheral retina. After 5 years of fenofibrate treatment, the main FIELD study found a highly significant overall 31% reduction in the need for the first laser treatment for any retinopathy, with a number needed to treat of just 17 to avoid at least 1 individual requiring laser among those with known DR at study entry (1). Almost identical reductions in the first laser treatment were observed for any maculopathy (31%) and PDR (30%). Total laser treatments for any retinopathy were reduced by 37% with fenofibrate, and similar reductions were observed for any maculopathy (36%) and PDR (38%).
A remarkable 79% reduction in the number of patients requiring laser treatment was observed with fenofibrate in the FIELD ophthalmology substudy (1). As expected, patients in the substudy with higher baseline ETDRS grades were more likely to need laser treatment. Twenty-eight patients required a first episode of laser treatment, most of whom had minimal to moderate nonproliferative DR (NPDR) at baseline.
A reduction in laser treatment for PDR was included as part of the primary outcome in the ACCORD-Eye substudy (2). Thirteen patients (1.6%) in the fenofibrate group and 21 (2.7%) in the placebo group required laser therapy for PDR, although the unadjusted relative risk was not significant (P = 0.150) (27). The absence of a significant reduction in laser treatment with fenofibrate in the ACCORD-Eye substudy compared with the FIELD trial may have been due to a shorter follow-up of 4 years, more aggressive glycemic control in the intensive arm, or different treatment patterns at participating trial sites.
Vitrectomy and cataract.
Vitrectomy events were reported in the FIELD ophthalmology substudy and in the ACCORD-Eye substudy (1,27). Neither found any significant differences between fenofibrate and placebo treatment, although numbers were low. Two patients on fenofibrate and one patient on placebo required vitrectomy surgery in the FIELD substudy, whereas five patients on fenofibrate and six on placebo required vitrectomies in the ACCORD-Eye substudy.
The incidence of cataract or cataract surgery was assessed in the FIELD substudy. Unsurprisingly, no significant difference was found between those treated with fenofibrate (37 patients) or placebo (28 patients) (1).
Visual acuity.
The ophthalmic outcome of most interest to patients is how well they can see. However, no fibrate trial has found any statistically significant benefit on visual acuity. In the FIELD ophthalmology substudy, 97 patients (30.7%) on fenofibrate and 90 (29.1%) on placebo experienced two-line worsening of visual acuity (Snellen chart) (1). In the ACCORD-Eye substudy, 227 patients (23.7%) on fenofibrate and 233 (24.5%) on placebo experienced three-line worsening of visual acuity on the ETDRS chart (2).
Fenofibrate was not expected to significantly reduce PDR-related vision loss in the FIELD substudy over 5 years or the ACCORD-Eye substudy over 4 years. Patients were excluded if they had any known baseline indication for laser treatment, putting them at low risk for vision loss from PDR. To illustrate this point, the ETDRS found that progression to high-risk PDR within 5 years occurred in 25% of eyes with a baseline DR severity level of 43, corresponding with moderate NPDR (28). After laser treatment for high-risk PDR, about one in five of these eyes would experience moderate visual loss within another 5 years (29). For this reason, ETDRS two-step progression for individual eyes or three-step progression for combined eyes are considered clinically meaningful trial end points.
DME is another common cause of vision loss in patients with DR. Some cases of visual acuity decline in the FIELD and ACCORD-Eye substudies were likely due to the development of DME, but this cannot be determined from the currently published data. Fenofibrate did appear to reduce the occurrence of any DME in the FIELD substudy (4 with fenofibrate vs. 10 with placebo), but the total events were small and this difference was not significant (1). The most common cause of visual acuity decline in the FIELD substudy was probably cataract, given that 65 patients experienced this outcome, which fenofibrate treatment would not have improved. Specific studies are needed to determine whether fenofibrate can preserve visual acuity related to the development of DME in patients with DR.
It is tempting to compare these findings with recent trials of anti-VEGF agents that showed large benefits on visual acuity with relatively small participant numbers and short follow-up (30). Several important distinctions must be made here. First, anti-VEGF agents were used to treat DME-related vision loss, whereas fenofibrate was used to reduce DR risk. Second, anti-VEGF agents have not been shown to reduce DR progression and may increase cardiovascular events if used long-term in patients with less severe DR. Third, the long-term efficacy and safety of anti-VEGF agents have not been established. Also worth noting is that anti-VEGF agents are generally much more expensive than off-patent fenofibrate.
Other microvascular outcomes.
Additional microvascular benefits have been reported with fenofibrate in people with type 2 diabetes from the FIELD and ACCORD trials. Fenofibrate reduced albuminuria and preserved the estimated glomerular filtration rate, despite causing a small increase in circulating creatinine that reversed on withdrawal of the drug (31,32). Nontraumatic amputations were reduced by 37% and minor amputations without known large-vessel disease were significantly reduced by 47% with fenofibrate in the FIELD trial (33). This reduction in amputations may be related to a protective effect on peripheral neuropathy. The combined findings of these two major clinical trials strongly suggest that fenofibrate is protective against the renal and neurological complications of type 2 diabetes.
Limitations of the FIELD and ACCORD trials.
A major limitation of the FIELD study was that only 10.3% of participants (1,012) were included in the ophthalmology substudy in which fundus photographs were routinely collected. Indications for laser treatment may therefore have been missed in the main study. Another potential confounder was the disproportionate uptake of statin drugs in the placebo group, presumably due to a greater need for dyslipidemia therapy. In the substudy, only a small number of laser events were reported in the two groups (5 with fenofibrate vs. 23 with placebo), and the observed reduction in DR progression was based on a small number of events (3 with fenofibrate vs. 14 with placebo). Fenofibrate had no statistically significant effect on hard exudates or macular edema, although detailed assessment of these features with optical coherence tomography was not done and event numbers were small. Visual acuity declined at similar rates regardless of treatment over 5 years; however, causes of reduced acuity were not attributed and the substudy sample was not sufficiently powered to detect differences in this outcome.
The ACCORD-Eye substudy planned to recruit 4,065 participants from the main ACCORD trial (34). However, the substudy lagged behind the main trial and early termination due to increased cardiovascular mortality in the intensive glycemic control group meant that only 2,856 participants were included (2). Of these, 1,593 were evaluated for the effect of fenofibrate on DR outcomes. The greatest benefit of fenofibrate on DR progression was observed in those with baseline DR, but the interaction of specific DR severity levels was not reported, making it hard to judge when fenofibrate treatment should begin. Events such as the occurrence or progression of hard exudates, macular edema, or cataract were not reported, despite intentions to study these outcomes (34). Visual acuity again declined at similar rates regardless of treatment over 4 years, but causes were not attributed, and the study was not powered to detect significant changes in this outcome.
Fenofibrate safety.
Fenofibrate was generally well tolerated in the FIELD trial (35). Patients on fenofibrate were at greater risk of pancreatitis, but numbers were small (40 with fenofibrate vs. 23 with placebo). Venous thromboembolism events were slightly higher with fenofibrate (n = 120) than with placebo (n = 80). Rhabdomyolysis occurred in one patient on placebo and in three patients on fenofibrate; however, none of these patients were taking statins and all fully recovered. In addition, plasma creatinine and homocysteine were 11 μmol/L and 2.7 μmol/L higher on average, respectively, in patients treated with fenofibrate at the end of the trial. These levels declined to the same as placebo-treated patients within 8 weeks after cessation of fenofibrate. Adverse events were not increased with fenofibrate in patients with moderate renal impairment and end-stage renal events were similar between groups (31,36).
Similar safety data were reported from 5,518 patients in the ACCORD-Lipid substudy (37). Fenofibrate did not significantly increase the risk of serious adverse events, including rhabdomyolysis, hepatitis, or gall bladder-related events. In addition, no venous thromboembolism events were reported in either group. Raised alanine aminotransferase greater than five times the upper limit of normal was slightly more common in fenofibrate-treated patients, although numbers were small (16 with fenofibrate vs. 6 with placebo). Serum creatinine was 11 μmol/L higher on average after 4 years in patients treated with fenofibrate but declined to normal after cessation of the drug (32,37).
PROPOSED MECHANISMS OF THE BENEFITS OF FENOFIBRATE IN DR
Lipid pathways.
Despite being best recognized for its lipid actions, the retinal benefits of fenofibrate in the FIELD and ACCORD trials did not appear to be due to quantitative lipid improvements. After 4 months of fenofibrate treatment in the FIELD trial, levels of total cholesterol decreased by 11.4%, LDL-C levels decreased by 12%, triglyceride levels decreased by 28.6%, and HDL-C levels increased by 5.1% (35). After 5 years, however, levels of total cholesterol were 6.9% lower, LDL-C were 5.8% lower, and triglycerides were 21.9% lower in those receiving fenofibrate, with no difference in HDL-C levels. Neither baseline lipid levels nor changes with treatment appeared to affect the ocular response to fenofibrate. In contrast, the ACCORD trial used open-label simvastatin in all individuals to lower LDL-C levels. After 4 years, average LDL-C levels in the ACCORD participants were ∼2.0 mmol/L in both groups (2). Similar to the FIELD trial, HDL-C levels initially increased with fenofibrate but were not significantly different at the conclusion of the ACCORD trial, whereas triglyceride levels remained ∼16% lower in the fenofibrate group. As in the FIELD trial, baseline lipid levels did not affect the study ocular outcomes (2).
Notwithstanding these findings, the interaction of fenofibrate with systemic lipids should not be ruled out as an important mechanism of its benefit in DR. For instance, improvements in lipid size and composition may reduce the risk of microvascular complications without a material change in overall lipid mass. At the close of the FIELD trial, Apo-B levels were 6.9% lower, whereas Apo-A1 and Apo-AII levels were 1.6 and 27.2% higher, respectively, in patients on fenofibrate compared with placebo (38). In addition, the ratio of Apo-B to Apo-AI was 8.1% lower on average in those on fenofibrate. Apo-AI is overexpressed in the retinas of type 2 diabetic donors and may be a compensatory mechanism of reverse cholesterol transport (39). Fenofibrate might therefore increase intraretinal reverse cholesterol transport by upregulation of Apo-AI and reduce the potential for lipid-mediated oxidative stress. Further, serum Apo-AI, Apo-B, and the ratio of Apo-B to Apo-AI were recently found to be stronger predictors of DR than conventional lipids (40). Apo-B is the structural protein of LDL, thus the reduction in Apo-B by fenofibrate in the FIELD trial may reflect a change from small, dense LDL particles toward larger particles less prone to oxidation and less likely to cause oxidative stress. Additional studies are required to investigate the effect of fenofibrate on Apo species in the eyes, kidneys, and nerves.
Vascular cell survival.
A loss of pericyte and endothelial cells occurs early in DR, which may lead to microaneurysm formation and fluid extravasation. Apoptosis of human retinal endothelial cells by serum deprivation was dose-dependently inhibited by fenofibrate in vitro (41). In these cells, fenofibrate induced activation of AMPK and upregulation of VEGF, whereas inhibition of AMPK by compound C attenuated the survival benefit of fenofibrate. Fenofibrate-induced survival was not prevented by the PPARα antagonist MK-886 or replicated by the selective PPARα agonist WY-14643, indicating that inhibition of vascular cell apoptosis may be mediated by PPARα-independent pathways. VEGF upregulation with fenofibrate was surprising, given reports of reduced retinal VEGF expression with fenofibrate in type 1 diabetic models (42). It is unclear whether this finding is unique to these experimental conditions or whether direct upregulation of VEGF with fenofibrate in retinal endothelial cells provides a negligible contribution to net retinal VEGF expression.
Activation of AMPK appears to be a common mechanism of fenofibrate action in endothelial cells in the retina and in other vascular tissues (17,41). Pericyte apoptosis may also be inhibited by fenofibrate, given the recent finding that specific AMPK activation prevented lipotoxicity in bovine retinal pericytes (43). Fenofibrate may additionally protect against damage to retinal vascular cells by reducing the formation of modified LDL particles. Modified LDL is toxic to retinal capillary endothelial cells and pericytes (44,45). Oxidized LDL has been found in retinal samples from patients with DR and may increase retinal permeability by reducing the expression of tissue inhibitor of MMP-3 in retinal pericytes (45,46).
Retinal pigment epithelium survival and permeability.
Cells of the retinal pigment epithelium (RPE) form the outermost layer of the retina and a protective barrier against fluid extravasation. Cultured ARPE-19 cells, a human RPE cell line, developed hyperpermeability, cell breakdown, and disorganization of tight junction proteins in the presence of high glucose and IL-1β via activation of AMPK (47). Fenofibric acid prevented AMPK activation and hyperpermeability, while silencing AMPK also prevented IL-1β–induced hyperpermeability. AMPK inhibition in RPE cells contrasts with AMPK activation in retinal endothelial cells (41) and highlights that fenofibrate may act differently in different cell types. In a related study, fenofibric acid reduced monolayer permeability and overexpression of the basement membrane components fibronectin and collagen IV in ARPE-19 cells cultured with high glucose and IL-1β (48). Further, production of reactive oxygen species and activation of stress and apoptotic cell markers were increased in ARPE-19 cells cultured in the presence of high glucose and hypoxia (49). Fenofibric acid inhibited these changes and increased IGF-1 receptor survival signaling.
Wnt pathway inhibition.
Wnt pathway inhibition appears to be a novel mechanism of DR benefit with fenofibrate. In retinal endothelial cells, fenofibrate inhibited phosphorylation of the Wnt coreceptor LDL receptor-related protein-6 and inhibited β-catenin accumulation within the cytoplasm (Fig. 4) (16). This may involve upregulated expression of the VLDL receptor, given that this receptor negatively regulates Wnt signaling (50). In streptozotocin-induced diabetic rats and Akita mice, two rodent models of type 1 diabetes, fenofibrate prevented retinal vascular leakage, leukostasis, and inflammation (42). These effects were observed both with oral and intravitreal fenofibrate, suggesting the drug target is present in ocular tissues. Intravitreal fenofibrate was also reported to reduce retinal neovascularization in rats using the classical oxygen-induced retinopathy model. Further, PPARα knockout abolished the beneficial effects of fenofibrate, suggesting that these effects are PPARα-dependent.
FIG. 4.

Canonical Wnt signaling in an endothelial cell. A: Wnt ligand induces recruitment of dishevelled (Dsh) and Axin to the Frizzled receptor and LDL receptor–related protein (LRP) coreceptor, respectively, and inhibition of the β-catenin destruction complex. This allows β-catenin to accumulate in the cytoplasm and translocate to the nucleus, where it binds with T-cell factor (TCF) and activates transcription of proangiogenic genes. B: Fenofibrate (F) appears to inhibit Wnt signaling through inhibition of LRP6 phosphorylation. The mechanism is not completely understood but may involve upregulation of the VLDL receptor (VLDLR). APC, adenomatous polyposis coli; GSK3, glycogen synthase kinase-3; P, phosphate.
CONCLUSIONS
Fenofibrate has multiple potential mechanisms of action that may account for its benefit on diabetic microvascular complications in patients with type 2 diabetes. Effects on lipid characteristics and inhibition of inflammation, angiogenesis, apoptosis, and oxidative stress may all provide protection. These mechanisms may similarly protect against microvascular complications in non–type 2 diabetes, but additional trials are required to prove this hypothesis.
Consistent clinical evidence indicates that fenofibrate is protective against the progression of DR and other diabetic microvascular complications in patients with type 2 diabetes. The greatest benefit on DR appears to be in those patients with at least minimal NPDR at the time treatment is started. Fenofibrate is well tolerated and does not appear to significantly increase the risk of long-term renal complications, despite modest short-term rises in circulating creatinine. In DR management, fenofibrate could be a useful adjunctive treatment to modifiable risk factor control and regular ophthalmic review. Its incorporation into clinical practice should be continually revised as more information becomes available.
ACKNOWLEDGMENTS
The Centre for Eye Research Australia receives Operational Infrastructure Support from the Victorian Government. J.E.N. is supported by a National Health and Medical Research Council (NHMRC) Postgraduate Medical Scholarship (ID1038701). A.C.K. is supported by an NHMRC Fellowship (ID1024105) and Program Grant (ID1037786). E.L.L. is supported by the Australian NHMRC Senior Research Fellowship (ID1045280).
A.J.J. and A.C.K. have performed clinical research in the FIELD study funded by the manufacturers of fenofibrate, Abbott Pharmaceuticals, and have been reimbursed for lectures by Abbott. A.C.K. has also participated in advisory meetings for Abbott Pharmaceuticals. A.J.J., J.-X.M., and A.C.K. are listed individuals on a patent application related to the topical ocular use of fenofibrate. No other potential conflicts of interest relevant to this article were reported.
J.E.N. reviewed literature, wrote the manuscript, and produced the figures. A.J.J., A.C.K., and E.L.L. contributed to discussion and reviewed the manuscript. J.-X.M. and J.J.W. reviewed the manuscript.
REFERENCES
- 1.Keech AC, Mitchell P, Summanen PA, et al. FIELD study investigators Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 2007;370:1687–1697 [DOI] [PubMed] [Google Scholar]
- 2.Chew EY, Ambrosius WT, Davis MD, et al. ACCORD Study Group. ACCORD Eye Study Group Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 2010;363:233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balfour JA, McTavish D, Heel RC. Fenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemia. Drugs 1990;40:260–290 [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre P, Chinetti G, Fruchart J-C, Staels B. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J Clin Invest 2006;116:571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guérin M, Bruckert E, Dolphin PJ, Turpin G, Chapman MJ. Fenofibrate reduces plasma cholesteryl ester transfer from HDL to VLDL and normalizes the atherogenic, dense LDL profile in combined hyperlipidemia. Arterioscler Thromb Vasc Biol 1996;16:763–772 [DOI] [PubMed] [Google Scholar]
- 6.Nigon F, Lesnik P, Rouis M, Chapman MJ. Discrete subspecies of human low density lipoproteins are heterogeneous in their interaction with the cellular LDL receptor. J Lipid Res 1991;32:1741–1753 [PubMed] [Google Scholar]
- 7.Dejager S, Bruckert E, Chapman MJ. Dense low density lipoprotein subspecies with diminished oxidative resistance predominate in combined hyperlipidemia. J Lipid Res 1993;34:295–308 [PubMed] [Google Scholar]
- 8.Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem 1999;274:32048–32054 [DOI] [PubMed] [Google Scholar]
- 9.Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature 1998;393:790–793 [DOI] [PubMed] [Google Scholar]
- 10.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation 1999;99:3125–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasceri V, Cheng JS, Willerson JT, Yeh ETH. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation 2001;103:2531–2534 [DOI] [PubMed] [Google Scholar]
- 12.Shu H, Wong B, Zhou G, et al. Activation of PPARα or γ reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem Biophys Res Commun 2000;267:345–349 [DOI] [PubMed] [Google Scholar]
- 13.Panigrahy D, Kaipainen A, Huang S, et al. PPARα agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A 2008;105:985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meissner M, Stein M, Urbich C, et al. PPARα activators inhibit vascular endothelial growth factor receptor-2 expression by repressing Sp1-dependent DNA binding and transactivation. Circ Res 2004;94:324–332 [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama Y, Xin B, Shigeto T, et al. Clofibric acid, a peroxisome proliferator-activated receptor alpha ligand, inhibits growth of human ovarian cancer. Mol Cancer Ther 2007;6:1379–1386 [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Hu Y, Mott R, et al. Mechanisms for the therapeutic effect of fenofibrate on diabetic retinopathy in type 1 diabetes models. Presented at the 71st Scientific Sessions of the American Diabetes Association, 24–28 June 2011, San Diego, California [Google Scholar]
- 17.Tomizawa A, Hattori Y, Inoue T, Hattori S, Kasai K. Fenofibrate suppresses microvascular inflammation and apoptosis through adenosine monophosphate-activated protein kinase activation. Metabolism 2011;60:513–522 [DOI] [PubMed] [Google Scholar]
- 18.Kadian S, Mahadevan N, Balakumar P. Differential effects of low-dose fenofibrate treatment in diabetic rats with early onset nephropathy and established nephropathy. Eur J Pharmacol 2013;698:388–396 [DOI] [PubMed] [Google Scholar]
- 19.Ibarra-Lara L, Hong E, Soria-Castro E, et al. Clofibrate PPARα activation reduces oxidative stress and improves ultrastructure and ventricular hemodynamics in no-flow myocardial ischemia. J Cardiovasc Pharmacol 2012;60:323–334 [DOI] [PubMed] [Google Scholar]
- 20.Duncan LJ, Cullen JF, Ireland JT, Nolan J, Clarke BF, Oliver MF. A three-year trial of atromid therapy in exudative diabetic retinopathy. Diabetes 1968;17:458–467 [DOI] [PubMed] [Google Scholar]
- 21.Harrold BP, Marmion VJ, Gough KR. A double-blind controlled trial of clofibrate in the treatment of diabetic retinopathy. Diabetes 1969;18:285–291 [DOI] [PubMed] [Google Scholar]
- 22.Cullen JF, Town SM, Campbell CJ. Double-blind trial of Atromid-S in exudative diabetic retinopathy. Trans Ophthalmol Soc U K 1974;94:554–562 [PubMed] [Google Scholar]
- 23.Dorne PA. [Exudative diabetic retinopathy. The use of clofibrate in the treatment of hard exudates using a reduced but prolonged dosage over several years (author’s transl)]. Arch Ophtalmol (Paris) 1977;37:393–400 [PubMed] [Google Scholar]
- 24.Freyberger H, Schifferdecker E, Schatz H. Regression of hard exudates in diabetic background retinopathy in therapy with etofibrate antilipemic agent. Med Klin (Munich) 1994;89:594–597, 633 [PubMed]
- 25.Emmerich KH, Poritis N, Stelmane I, et al. [Efficacy and safety of etofibrate in patients with non-proliferative diabetic retinopathy]. Klin Monatsbl Augenheilkd 2009;226:561–567 [in German] [DOI] [PubMed] [Google Scholar]
- 26.Havel E, Rencova E, Novak J, et al. Serum lipoproteins lowering and diabetic exudative retinopathy. Atherosclerosis 1997;134:309 [Google Scholar]
- 27.Morita H, Nagai R. Retinopathy progression in type 2 diabetes. N Engl J Med 2010;363:2171–; author reply 2173–2174. [DOI] [PubMed] [Google Scholar]
- 28.Early Treatment Diabetic Retinopathy Study Research Group Fundus photographic risk factors for progression of diabetic retinopathy. ETDRS report number 12. Ophthalmology 1991;98(Suppl.):823–833 [PubMed] [Google Scholar]
- 29.Early Treatment Diabetic Retinopathy Study Research Group Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Ophthalmology 1991;98(Suppl.):766–785 [PubMed] [Google Scholar]
- 30.Ho AC, Scott IU, Kim SJ, et al. Anti-vascular endothelial growth factor pharmacotherapy for diabetic macular edema: a report by the American Academy of Ophthalmology. Ophthalmology 2012;119:2179–2188 [DOI] [PubMed] [Google Scholar]
- 31.Davis TME, Ting R, Best JD, et al. Fenofibrate Intervention and Event Lowering in Diabetes Study investigators Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia 2011;54:280–290 [DOI] [PubMed] [Google Scholar]
- 32.Mychaleckyj JC, Craven T, Nayak U, et al. Reversibility of fenofibrate therapy-induced renal function impairment in ACCORD type 2 diabetic participants. Diabetes Care 2012;35:1008–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajamani K, Colman PG, Li LP, et al. FIELD study investigators Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): a prespecified analysis of a randomised controlled trial. Lancet 2009;373:1780–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chew EY, Ambrosius WT, Howard LT, et al. ACCORD Study Group Rationale, design, and methods of the Action to Control Cardiovascular Risk in Diabetes Eye Study (ACCORD-EYE). Am J Cardiol 2007;99:103i–111i [DOI] [PubMed] [Google Scholar]
- 35.Keech A, Simes RJ, Barter P, et al. FIELD study investigators Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005;366:1849–1861 [DOI] [PubMed] [Google Scholar]
- 36.Ting RD, Keech AC, Drury PL, et al. FIELD Study Investigators Benefits and safety of long-term fenofibrate therapy in people with type 2 diabetes and renal impairment: the FIELD Study. Diabetes Care 2012;35:218–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ginsberg HN, Elam MB, Lovato LC, et al. ACCORD Study Group Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010;362:1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taskinen MR, Barter PJ, Ehnholm C, et al. FIELD study investigators Ability of traditional lipid ratios and apolipoprotein ratios to predict cardiovascular risk in people with type 2 diabetes. Diabetologia 2010;53:1846–1855 [DOI] [PubMed] [Google Scholar]
- 39.Simo R, Garcia-Ramirez M, Higuera M, Hernandez C. Apolipoprotein A1 is overexpressed in the retina of diabetic patients. Am J Ophthalmol 2009;147:319–325.e311 [DOI] [PubMed]
- 40.Sasongko MB, Wong TY, Nguyen TT, et al. Serum apolipoprotein AI and B are stronger biomarkers of diabetic retinopathy than traditional lipids. Diabetes Care 2011;34:474–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Ahn J-H, Kim J-H, et al. Fenofibrate regulates retinal endothelial cell survival through the AMPK signal transduction pathway. Exp Eye Res 2007;84:886–893 [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Hu Y, Lin M, et al. Therapeutic effects of PPARα agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013;62:261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cacicedo JM, Benjachareonwong S, Chou E, Yagihashi N, Ruderman NB, Ido Y. Activation of AMP-activated protein kinase prevents lipotoxicity in retinal pericytes. Invest Ophthalmol Vis Sci 2011;52:3630–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyons TJ, Li W, Wells-Knecht MC, Jokl R. Toxicity of mildly modified low-density lipoproteins to cultured retinal capillary endothelial cells and pericytes. Diabetes 1994;43:1090–1095 [DOI] [PubMed] [Google Scholar]
- 45.Wu M, Chen Y, Wilson K, et al. Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Invest Ophthalmol Vis Sci 2008;49:2679–2685 [DOI] [PubMed] [Google Scholar]
- 46.Barth JL, Yu Y, Song W, et al. Oxidised, glycated LDL selectively influences tissue inhibitor of metalloproteinase-3 gene expression and protein production in human retinal capillary pericytes. Diabetologia 2007;50:2200–2208 [DOI] [PubMed] [Google Scholar]
- 47.Villarroel M, Garcia-Ramírez M, Corraliza L, Hernández C, Simó R. Fenofibric acid prevents retinal pigment epithelium disruption induced by interleukin-1β by suppressing AMP-activated protein kinase (AMPK) activation. Diabetologia 2011;54:1543–1553 [DOI] [PubMed] [Google Scholar]
- 48.Trudeau K, Roy S, Guo W, et al. Fenofibric acid reduces fibronectin and collagen type IV overexpression in human retinal pigment epithelial cells grown in conditions mimicking the diabetic milieu: functional implications in retinal permeability. Invest Ophthalmol Vis Sci 2011;52:6348–6354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miranda S, González-Rodríguez Á, García-Ramírez M, et al. Beneficial effects of fenofibrate in retinal pigment epithelium by the modulation of stress and survival signaling under diabetic conditions. J Cell Physiol 2012;227:2352–2362 [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Hu Y, Lu K, Flannery JG, Ma JX. Very low density lipoprotein receptor, a negative regulator of the wnt signaling pathway and choroidal neovascularization. J Biol Chem 2007;282:34420–34428 [DOI] [PubMed] [Google Scholar]