

Abstract
Erythropoietin (EPO) has shown beneficial effects in the regulation of obesity and metabolic syndrome; however, the detailed mechanism is still largely unknown. Here, we created mice with adipocyte-specific deletion of EPO receptor. These mice exhibited obesity and decreased glucose tolerance and insulin sensitivity, especially when fed a high-fat diet. Moreover, EPO increased oxidative metabolism, fatty acid oxidation, and key metabolic genes in adipocytes and in white adipose tissue from diet-induced obese wild-type mice. Increased metabolic activity by EPO is associated with induction of brown fat–like features in white adipocytes, as demonstrated by increases in brown fat gene expression, mitochondrial content, and uncoupled respiration. Peroxisome proliferator–activated receptor (PPAR)α was found to mediate EPO activity because a PPARα antagonist impaired EPO-mediated induction of brown fat–like gene expression and uncoupled respiration. PPARα also cooperates with Sirt1 activated by EPO through modulating the NAD+ level to regulate metabolic activity. PPARα targets, including PPARγ coactivator 1α, uncoupling protein 1, and carnitine palmitoyltransferase 1α, were increased by EPO but impaired by Sirt1 knockdown. Sirt1 knockdown also attenuated adipose response to EPO. Collectively, EPO, as a novel regulator of adipose energy homeostasis via these metabolism coregulators, provides a potential therapeutic strategy to protect against obesity and metabolic disorders.
Obesity and its associated metabolic syndrome, including glucose intolerance and insulin resistance, are well-documented risk factors for cardiovascular disease, type 2 diabetes, and stroke. It is therefore essential to develop new strategies to treat metabolic syndrome and obesity. Recently, erythropoietin (EPO), the cytokine required for the erythrocyte production, has become attractive because of its important protective activity in the nonerythroid system. It is now recognized that EPO has protective effects in animal models of cardiac ischemia/reperfusion injury via stimulating endothelial cells to produce nitric oxide to regulate vascular tone and improve oxygen transport (1–5). EPO activity has also been reported for other nonhematopoietic tissue, including brain protection against ischemia, enhanced neural progenitor production, and anti-inflammatory effects (6–8). We demonstrated that disrupted EPO signaling in all nonerythroid tissues promotes obesity (9). However, the mechanism by which EPO functions as a metabolic regulator to cooperate with other coregulators to regulate energy homeostasis remains largely unknown. Compared with other nonhematopoietic tissue, we found that the EPO receptor (EpoR) is expressed at a high level in white adipose tissue (WAT) (∼60% of hematopoietic tissue), which is secondary to its primary expression site of EpoR, raising the possibility that endogenous EPO action in WAT may contribute importantly to protection against obesity and its associated metabolic disorders.
WAT adipocytes are specialized for the storage of excess energy such as triglycerides. It is now recognized, however, that WAT may also play a central role in energy homeostasis and systemic metabolism (10). Brown adipose tissue (BAT) adipocytes can dissipate calories as heat via uncoupled metabolism due to a pattern of gene expression that results in a high mitochondrial content and elevated cellular respiration (11). Observations that adult humans have functional and metabolically active BAT (12–15) suggest that a higher level of BAT may be protective against obesity and have stimulated interest concerning the therapeutic potential of augmenting brown fat to combat obesity and its associated metabolic disease. Brown adipocytes are found interspersed within the WAT under certain conditions such as cold exposure or after stimulation of the β3-adrenoceptor pathways (16,17). Advances have recently been achieved on identification of several transcriptional factors and coregulators that specifically promote development and acquisition of the BAT-specific gene expression profile, including uncoupling protein 1 (UCP1), PRDM16, peroxisome proliferator–activated receptor γ coactivator 1α (PGC-1α), and peroxisome proliferator–activated receptor α (PPARα) (18–20).
PGC-1α and PRDM16 are transcriptional coactivators involved in the control of energy metabolism, and ectopic expression of PGC-1α and PRDM16 in WAT induces acquisition of BAT features, including expression of mitochondrial and fatty acid oxidation and thermogenic genes to limit weight gain and improve glucose intolerance in response to a high-fat diet (HFD) (20–23). PPARα plays an important role in lipid metabolism, and activation of PPARα in human WAT led to the appearance of brown fat gene expression, including UCP1, PGC-1α, and PRDM16 (19,24). PPARα has been considered a distinctive marker of BAT with respect to the WAT phenotype (25). Another metabolic sensor, Sirt1, an NAD+ dependent type III deacetylase sirtuin, activates PGC-1α and various substrates, including PPARγ and PPARα, in skeletal muscle and adipocytes to contribute to energy expenditure and browning of WAT and resistance to dietary obesity (26–30).
Here, we show that EPO activity in WAT is responsible for energy homeostasis. We confirm that the development of a brown fat–like gene program and an increase in metabolic activity in WAT with EPO stimulation are associated with a rise in whole-body energy expenditure, a suppression of weight gain, and improvement of glucose intolerance and insulin resistance in response to an HFD. In contrast, the loss of EPO activity in WAT leads to opposite effects. Importantly, we demonstrate that PPARα is required for the EPO-stimulated increase in metabolic activity and induction of a thermogenic gene program in white adipocytes. The EPO-promoted increase in the NAD+ level activates Sirt1 activity, which cooperates with PPARα to mediate the adipocyte response to EPO. Our results clearly identify EPO as a critical mediator of energy homeostasis and provide a potential contribution to the protection against obesity and metabolic syndrome.
RESEARCH DESIGN AND METHODS
Animal studies.
For analysis of body weight, body composition, and food intake, C57BL/6 mice (4 weeks; The Jackson Laboratory) were maintained under a 12-h light/dark cycle with free access to food and drinking water, except as indicated for paired-fed mice. Male mice were fed the HFD (60 kcal% fat [high fat]) and separated into different groups. EPO treatment (3,000 units/kg; three times/week for 2.5 to 5 weeks as indicated) was administered by subcutaneous injection. The control and paired-fed mice were injected with PBS as the vehicle control. EpoRaP2KO mice were generated by crossing aP2-cre mice on the C57BL/6 background (The Jackson Laboratory) with EpoRfloxp/floxp mice (7), a C57/129 hybrid mouse strain, but was backcrossed onto a pure C57BL/6 background for at least seven generations in our study. Oxygen consumption (Vo2), respiratory exchange ratios (RER), food intake, and activity levels were monitored by indirect calorimetry using the comprehensive laboratory animal monitoring system as described (9). Body composition was measured using the EchoMRI 3-in-1 (Echo Medical Systems). The National Institute of Diabetes and Digestive and Kidney Diseases Animal Care and Use Committee approved all animal procedures, and studies were conducted in accordance with National Institutes of Health guidelines.
Cell culture.
Adipocyte differentiation of 3T3-L1 fibroblast cells (American Type Culture Collection) was induced as described (9). Primary human preadipocytes (Lonza) were grown to confluence in Preadipocyte Basal Medium-2 (Lonza). Differentiation was induced using commercially available MDI cocktail (Lonza) for 9 days. The stromal-vascular fraction (SVF) from fat depots of mice treated with EPO or saline was prepared and differentiated as described (20). At the beginning of differentiation induction, cells were treated with EPO, at 5 units/mL or at the dosage indicated, or with vehicle (PBS). 3T3-L1 and primary human adipocytes (H-adipocytes) were differentiated with EPO treatment for 9 days. SVF was differentiated to adipocytes with EPO treatment for 6 days.
Glucose tolerance tests and insulin tolerance tests.
Glucose tolerance tests (GTT) and insulin tolerance tests (ITT) were performed as described (9).
Serum parameter analysis.
Insulin and leptin concentrations were determined by ELISA (Crystal Chem, Inc.). Adiponectin concentration was determined by ELISA (BioVision, Inc.). EPO levels were determined by ELISA (R&D Systems).
Quantitative real-time RT-PCR.
Quantitative real-time RT-PCR analyses were carried out using gene-specific primers (available upon request) and fluorescently labeled TaqMan probes or SYBR Green dye (Invitrogen) in a 7900 Sequence Detector (PE Applied Biosystems, Foster City, CA). Plasmid containing the cDNA of interest was used as a template to generate a standard curve. S16 was used as an internal control.
Western blotting.
Cells were lysed in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Lysates were resolved by 4–20% Tris-glycine SDS/PAGE and transferred to nitrocellulose membranes.
Mitochondrial DNA copy measurement.
The amount of mitochondrial DNA (mtDNA) relative to nuclear DNA was determined by quantitative real-time PCR using primers for Nd2 (NADH dehydrogenase subunit 2; mitochondrial genome) and Nme1 (nuclear genome).
Vo2 and fatty acid oxidation.
A Seahorse Bioscience XF24-3 Extracellular Flux Analyzer and Clark electrode were used to measure the oxygen consumption rate (OCR) and fatty acid oxidation.
RNA interference.
For knockdown experiments, small interfering RNA (siRNA) specific for Sirt1 and negative control (Thermo Scientific Dharmacon) were transfected into 3T3-L1 adipocytes and primary mouse adipocytes (M-adipocytes) via the DharmaFECT transfection reagent.
Statistical analyses.
Values are expressed as mean ± SEM. The unpaired two-tailed Student t test was used to determine differences between vehicle- and EPO-treated groups. Results in multiple groups were compared by ANOVA. Significance was at P < 0.05.
RESULTS
Targeted deletion of EpoR in adipocyte tissue leads to obesity, glucose intolerance, and insulin resistance.
To determine the direct adipocyte EPO response, we mated chimeric EpoR floxed mice with mice containing a cre-recombinase gene controlled by the aP2 promoter (31) to generate a fat-specific knockout (KO) of EpoR (EpoRaP2KO mice). EpoR gene expression was analyzed in multiple tissues, including spleen, subcutaneous WAT (S-WAT), and visceral WAT (V-WAT); SVF was analyzed from WAT, BAT, kidney, heart, liver, and muscle (Fig. 1A); and loss of EpoR expression in S-WAT and V-WAT was validated using Western blotting (Fig. 1B). Because aP2 is also expressed in macrophages, we isolated macrophages from WAT and detected EpoR expression in macrophages. The EpoR expression level in macrophages from EpoRaP2KO mice was not significantly different from wild-type (WT) mice (Supplementary Fig. 1A). EpoRaP2KO mice fed normal chow showed a slight but significant increase in body weight and fat mass compared with littermate controls (Fig. 1C and D), and by 30 weeks, EpoRaP2KO mice were 20% more massive, with a 65% increase in fat mass. Total activity, Vo2, and total RER were also significantly lower in EpoRaP2KO mice (Fig. 1E and F and Supplementary Fig. 1B). The difference in EpoRaP2KO body weight was further accentuated when mice were challenged by the HFD (Fig. 1G), although food intake was comparable (Supplementary Fig. 1C). When 10-week-old, EpoRaP2KO mice were fed the HFD for 6 weeks, they exhibited a 1.4-fold increase in fat mass compared with control mice (Fig. 1H), along with glucose intolerance and insulin resistance (Fig. 1I and J). Compared with the HFD control mice, these HFD EpoRaP2KO mice exhibited higher fasting serum leptin and glucose levels, an increased insulin level (Fig. 1K), and a trend toward higher serum adiponectin (Supplementary Fig. 1D). Insulin-stimulated AKT (protein kinase B) activation (phosphorylation) contributes to glucose and energy homeostasis (32). Diet-induced obesity (DIO) reduced the insulin receptor substrate/phosphatidylinositol 3-kinase/AKT intracellular pathway in rats (33). We observed that EPO treatment increased AKT activity, as shown by increased AKT phosphorylation in S-WAT and in V-WAT in control mice. In contrast, EpoRaP2KO mice showed decreased AKT phosphorylation, and AKT activity did not increase with EPO treatment in EpoRaP2KO mice in contrast to control mice (Fig. 1L), suggesting EPO activity modulates AKT activation, which may have an effect on insulin signaling.
FIG. 1.

EpoRaP2KO mice become obese, glucose-intolerant, and insulin-resistant. A: EpoR mRNA levels in tissues from EpoRaP2KO mice and littermate controls were quantified using quantitative PCR. B: EPOR protein level in WAT from EpoRaP2KO mice and littermate controls (Ctrl; EpoRfl/fl). C: Body weight vs. age of EpoRaP2KO mice and littermate controls (Ctrl) are indicated for male mice (n = 10) fed a normal chow (NC) diet. D: Body fat mass content of EpoRaP2KO mice and littermate controls (Ctrl) at indicated age are shown for male mice (n = 6). M, months. Total activity (E) and total Vo2 (F) were determined for male WT and EpoRaP2KO mice (n = 6). Male EpoRaP2KO mice and littermate controls (Ctrl) were treated with an HFD for 6 weeks. Body weight was monitored weekly (G), and body fat mass content (H) was determined at 16 weeks (n = 6). GTT (I) and ITT (J) on male EpoRaP2KO mice and littermate controls (Ctrl) were performed at 16 weeks after HFD treatment for 6 weeks (n = 6). K: Serum insulin, glucose, and leptin levels were measured in EpoRaP2KO mice and littermate controls (Ctrl) fed NC and the HFD (n = 6). L: Total (T)-AKT and phosphorylated (P)-AKT in primary adipocytes of S-WAT and V-WAT isolated from EpoRaP2KO mice (KO) and control (Ctrl) mice were analyzed by Western blotting. WT (M) and EpoRaP2KO (N) mice (8.5 months of age) fed a normal diet were treated with EPO or saline (Ctrl) for 3 weeks (n = 6). Body weight was monitored weekly for up to 4 weeks after treatment. Bar graphs are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
To rule out a role for EpoR on other cell types, we treated EpoRaP2KO and WT mice with EPO for 3 weeks. EpoRaP2KO mice and WT mice showed an increase in hematocrit after EPO treatment (Supplementary Fig. 1E). WT mice also exhibited the expected decrease in body weight of ∼15% at 3 weeks of EPO treatment (P < 0.01), and this decrease continued up to 7 weeks after EPO treatment (Fig. 1M). The EpoRaP2KO mice, however, only exhibited a decreasing trend in body weight of ∼4% at 3 weeks of EPO treatment (Fig. 1N). These data strongly indicate that endogenous and exogenous EPO/EpoR in adipocytes plays an important role in the regulation of obesity and that EPO/EpoR activity in fat tissue is responsible for much of the EPO regulation of body weight, fat mass accumulation, glucose metabolism, and insulin sensitivity.
EPO regulates mitochondrial genes in adipocytes and in WAT.
Increased mitochondrial biogenesis and related gene expression correlate with a reduction of DIO (34). We found that expression of mitochondrial biogenesis genes, including cytochrome c (CytC), isocitrate dehydrogenase 3 α (Idh3α), and cytochrome c oxidase subunit 7A1(Cox7a1) was decreased with the loss of EPO activity in S-WAT from EpoRaP2KO mice compared with littermate control mice (Fig. 2A). The key metabolic regulator, Pgc-1α, and fatty acid utilization gene carnitine palmitoyltransferase 1 (Cpt1) were also downregulated (Fig. 2A). To determine if the effects of EPO are independent of body weight, we performed paired-feeding studies in WT DIO mice without or with EPO treatment (3,000 units/kg, three times/week for 5 weeks). Importantly, although paired-fed mice exhibited a mild reduction in body fat content and fat pad weights, accompanied by modestly improved GTT and ITT compared with the vehicle-treated mice (Supplementary Fig. 2A and B), increased expression of CytC, Idh3α, Cpt1, Pgc-1α, and Cox7a1 in both S-WAT and V-WAT was only observed in EPO-treated DIO mice but not in vehicle-treated and paired-fed DIO mice (Fig. 2B and Supplementary Fig. 2C), suggesting the effects of EPO are independent of differences in body weight.
FIG. 2.
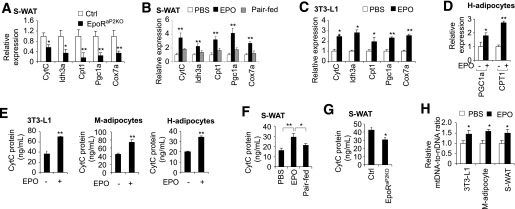
EPO signaling in white fat and adipocytes promotes expression of mitochondrial gene expression and increases mitochondrial DNA level. Expression of mitochondrial activity–related genes was quantified using quantitative PCR in S-WAT from EpoRaP2KO and littermate control mice (A), in S-WAT from DIO mice treated with EPO or PBS (B), and in 3T3-L1 adipocytes with PBS or EPO treatment (5 units/mL) (C). D: Expression of mitochondrial function–related genes, PGC1a and CPT1, without and with EPO treatment, in differentiated H-adipocytes was also determined. CytC protein levels were determined by ELISA in 3T3-L1 adipocytes, M-adipocytes, and H-adipocytes, without and with EPO treatment (E), and in S-WAT from the DIO mice treated for 5 weeks with EPO or PBS or paired-fed (F), and S-WAT from EpoRaP2KO and littermate control mice (n = 6) (G). H: The ratio of mtDNA to nuclear DNA (nDNA) was determined by qPCR in 3T3-L1 adipocytes, M-adipocytes, and S-WAT from DIO mice treated with EPO or PBS (n = 6). One-way ANOVA was used in B and F. All other statistics were performed using the Student t test. Bar graphs are mean ± SEM. In vitro data are means of three independent experiments. *P < 0.05; **P < 0.01.
In vitro, mitochondrial genes and fatty acid oxidation gene were also upregulated by EPO in 3T3-L1 adipocytes and in H-adipocytes (Fig. 2C and D). CytC protein level was also elevated in 3T3-L1 adipocytes, H-adipocytes, and M-adipocytes, differentiated from the SVF of primary cells isolated from S-WAT and induced for adipocyte differentiation for 6 days (Fig. 2E).
In vivo, CytC protein levels in S-WAT and V-WAT in DIO mice were increased with EPO stimulation (Fig. 2F and Supplementary Fig. 2D), but were decreased in S-WAT from EpoRaP2KO mice compared with control mice (Fig. 2G). Lastly, EPO treatment increased the mtDNA level (Fig. 2H) in WAT and in adipocyte cultures, indicating increased mitochondrial number and biogenesis. These data highlight the role for EPO/EpoR activity in the regulation of mitochondrial biogenesis in white adipocytes in vivo and in vitro.
EPO increases metabolic activity, cellular respiration capacity, and fatty acid utilization.
As the key first pacemaking component of the tricarboxylic acid cycle, citrate synthase (CS) activity is an indicator of mitochondrial function. EPO stimulation increased CS activity in 3T3-L1 adipocytes, M-adipocytes, and H-adipocytes (Fig. 3A). In vivo, we also observed that EPO treatment in DIO mice increased CS activity in S-WAT and V-WAT compared with vehicle-treated or paired-fed DIO mice (Fig. 3B). However, CS activity in S-WAT of EpoRaP2KO mice was decreased compared with control mice (Fig. 3C).
FIG. 3.
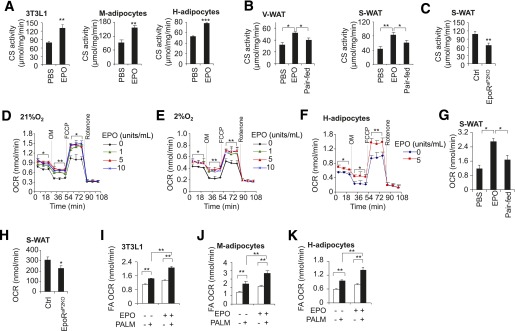
EPO increases adipocyte mitochondrial activity and Vo2 capacity. CS activity in differentiated 3T3-L1 adipocytes, M-adipocytes, and H-adipocytes, without or with EPO treatment (5 units/mL) (A), and in V-WAT and S-WAT of EPO-treated mice (B) and in S-WAT of EpoRaP2KO and littermate control mice (n = 6) (C). OCR at basal conditions and with addition of oligomycin (OM), FCCP, and rotenone in 3T3-L1 adipocytes treated without and with EPO at the indicated EPO dose and cultured at normoxia (21% O2) (D), and hypoxia (2% O2) (E) for 48 h. F: OCRs in H-adipocytes without and with EPO treatment (5 units/mL) at 21% O2 are shown. OCRs in primary adipocytes isolated from S-WAT of the DIO mice treated without (PBS) and with EPO for 5 weeks (G) and in EpoRaP2KO and littermate control mice (H) were determined and compared with samples prepared from paired-fed mice (n = 6). Data are representative of three independent experiments and normalized to total cellular protein. The OCR was assessed for palmitate (PALM)-driven (white bar, without palmitate; black bar, with palmitate), fatty acid (FA) oxidation in cultures of 3T3-L1 adipocytes (I), and in primary M-adipocytes (J) and H-adipocytes (K), without or with EPO treatment (5 units/mL). One-way ANOVA was used in B, D, E, G, and I–K. All other statistics were performed using the Student t test. Bar graphs are mean ± SEM. In vitro data are means of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
To assess mitochondrial oxidative metabolism, we determined the effect of EPO on the OCR as a measure of mitochondrial respiration. EPO increased basal OCR in 3T3-L1 adipocytes (Fig. 3D). The EPO-stimulated increase in OCR was still evident in the presence of oligomycin, which uncouples phosphorylation from mitochondrial respiration by blocking mitochondrial complex V, and with treatment of carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP), which uncouples oxidative phosphorylation from ATP synthesis and is used to assess maximal oxidative phosphorylation capacity (Fig. 3D). Hypoxia associated with WAT of obese mice has been linked to metabolic disorders (28,35,36). Notably, although OCR decreased under hypoxic condition, EPO treatment continued to increase the OCR of 3T3-L1 adipocytes under the basal condition and in the presence of oligomycin and FCCP under the hypoxic condition (Fig. 3E). EPO treatment also increased OCR in H-adipocytes (Fig. 3F). In vivo, adipocytes isolated from S-WAT after 5 weeks of EPO treatment in DIO mice also showed an increase in OCR (Fig. 3G). In comparison, adipocytes isolated from S-WAT of EpoRaP2KO mice exhibited reduced OCR compared with control mice (Fig. 3H). Taken together, these findings highlight a previously unrecognized role for EPO/EpoR activity in increasing cellular mitochondrial respiration and oxidative metabolism capacity beyond its effect of increased erythropoiesis and oxygen transport capacity, leading to increased oxygen utilization capacity and energy oxidative metabolism efficiency, and offset the adverse effect of obesity.
Mitochondrial dysfunction in adipose tissue is linked to obesity and type 2 diabetes in humans, as indicated by reduced oxidative phosphorylation capacity and reduced fatty acid β-oxidation in several tissues, including adipocytes (37,38). We treated adipocytes with palmitate, a substrate for fatty acid oxidation, and observed that EPO stimulation further enhanced the palmitate-stimulated increase in OCR (Fig. 3I–K), suggesting that EPO increases fatty acid oxidation in adipocytes. These data support the view that EPO increases energy expenditure in WAT and also demonstrate that EPO enhances the ability of adipocytes to metabolize fatty acid, which may limit the storage of excess lipid to protect against obesity.
EPO promotes brown fat–like characteristics and uncoupled respiration in white adipocytes.
BAT exhibits a high mitochondrial content, increased oxidative capacity, and elevated cellular respiration compared with WAT, and functionally active BAT is inversely correlated with BMI (12,13). However, EpoR expression in BAT is an order of magnitude lower (Fig. 1A), and expression of BAT-associated factors is unchanged in BAT isolated from EPO-treated mice (Supplementary Fig. 2E). We therefore hypothesized that the EPO-mediated antiobesity activity and associated metabolic improvement observed above may be related to the promotion of brown fat–associated markers in white adipocytes by EPO. We isolated S-WAT after 2.5 weeks of EPO treatment in young DIO mice before the readily detected change in body weight to rule out the effect of body weight difference. As we expected, these mice exhibited similar body weight but improved GTT and ITfcT compared with vehicle-treated mice (Supplementary Fig. 3A and B). With EPO treatment, we observed an increase in the expression of Ucp3 and Pparα in V-WAT (Fig. 4A), whereas S-WAT exhibited a larger scale and more dramatic increase of BAT-associated genes, including Cidea, Prdm16, Ucp1, Ucp3, Pparα, and Pgc-1α in EPO-treated DIO mice (Fig. 4B). We also isolated primary adipocytes from brown interscapular, white inguinal subcutaneous, and white visceral epididymal adipocytes from WT and EpoRaP2KO mice without and with EPO treatment. The expression of BAT-associated factors at the mRNA and protein level was unchanged in brown primary adipocytes (Supplementary Fig. 2E). In inguinal subcutaneous white primary adipocytes, cells harvested from EPO-treated WT mice showed increased expression of Cidea, Prdm16, Ucp1, Ucp3, Pparα, and Pgc-1α compared with untreated WT mice, whereas the expression of these genes was decreased in EpoRaP2KO mice adipocytes and was not different from EPO treated EpoRaP2KO mice (Fig. 4C). In visceral epididymal white adipocytes, cells from EPO-treated WT mice only showed increased expression of Ucp3, Pparα, and Pgc-1α, and the expression of these genes was reduced in adipocytes from EpoRaP2KO mice and remained unchanged in EpoRaP2KO mice treated with EPO (Fig. 4D). Western blotting confirmed that protein expression levels were consistent with the gene expression pattern (Fig. 4E and F). In contrast, expression of WAT-associated genes, Resistin, which is secreted by adipose tissue in rodents and promotes insulin resistance, Wdnm1-like, and Angiotensinogen was decreased by EPO in inguinal and visceral white primary adipocytes isolated from WT mice but was increased in EpoRaP2KO mice compared with WT control mice, and EPO treatment did not show any effect (Fig. 4G). The Resistin protein level was also decreased by EPO treatment in WT mice but not in EpoRaP2KO mice (Fig. 4H). These data suggest suppression of a WAT-associated program of gene expression by EPO.
FIG. 4.
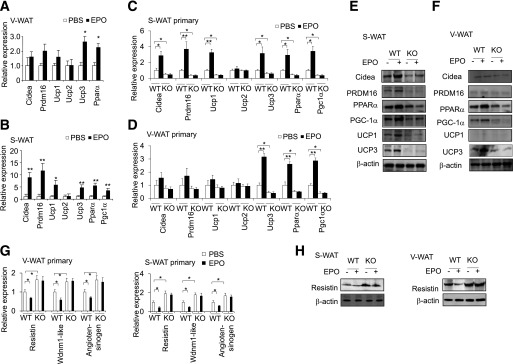
EPO promotes brown fat–associated gene and protein expression in WAT. Expression of BAT-associated genes in V-WAT (A) and S-WAT (B) in DIO mice treated without (PBS) and with EPO for 2.5 weeks was determined (n = 6). Expression of BAT-associated genes in primary adipocytes isolated from S-WAT (C) and V-WAT (D) in EpoRaP2KO (KO) and WT control mice without (PBS) and with EPO treatment were determined and compared (n = 4). The protein level of BAT-associated factors in S-WAT (E) and V-WAT (F) in EpoRaP2KO (KO) and littermate control mice without (PBS) and with EPO treatment (n = 4). Expression of mRNA (G) and protein (H) of white fat–associated genes (G) and Resistin in primary adipocytes isolated from S-WAT and V-WAT in EpoRaP2KO (KO) and WT control mice without (PBS) and with EPO treatment were determined (n = 4). One-way ANOVA was used in C, D, and G. All other statistics were performed using the Student t test. Bar graphs are mean ± SEM. In vitro data are means of three independent experiments. *P < 0.05; **P < 0.01.
We then analyzed CS activity as an indicator of mitochondrial function and bioenergetics marker. We did not observe any change of CS activity in brown primary adipocytes from WT mice treated with EPO or from EpoRaP2KO mice without or with EPO treatment compared with WT mice (Fig. 5A). This is consistent with the gene expression and protein expression analysis in brown primary adipocytes. However, in subcutaneous and visceral white primary adipocytes isolated from WT mice treated with EPO and from EpoRaP2KO mice, CS activity was affected by EPO/EpoR signaling, increasing in WT mice treated with EPO and decreasing in EpoRaP2KO mice with or without EPO treatment compared with WT control mice (Fig. 5A). These data provide further evidence that BAT is not EPO responsive in contrast to S-WAT and V-WAT, with S-WAT exhibiting greater EPO induction of brown fat–associated gene expression. We also performed hematoxylin and eosin staining. The size of inguinal adipocytes was slightly reduced in the EPO-treated WT mice but not in the EpoRaP2KO mice (Fig. 5B). UCP1 immunohistochemistry staining further supports the shift in S-WAT toward brown fat–like characteristics with EPO treatment (Fig. 5C). UCP1 displayed stronger staining in S-WAT from EPO-treated WT control mice but decreased staining in EpoRaP2KO mice without or with EPO treatment compared with untreated WT control mice (Fig. 5C). These results indicated that the EPO promoted WAT to a BAT-like appearance, especially in S-WAT.
FIG. 5.

EPO promotes brown features in WAT. A: CS activity in various primary adipocytes isolated from WT control and EpoRaP2KO mice (KO) without (□, PBS) or with (■) EPO treatment was determined (n = 4). B: Hematoxylin and eosin staining of S-WAT and V-WAT from WT and EpoRaP2KO (KO) mice is shown (scale bar: 20 μm). C: Immunohistochemistry staining for UCP-1 in S-WAT and V-WAT from WT and EpoRaP2KO (KO) mice is shown (scale bar: 20 μm). SVF from the inguinal fat pad was differentiated into adipocytes for 6 days (M-adipocytes) without (PBS) and with EPO treatment (5 units/mL), and expression of BAT associated genes (D) and mitochondrial biogenesis–related genes (E) was determined using quantitative PCR. F: Mitochondria and the nucleus were visualized using MitoTracker (red) and DAPI (blue), respectively, and confocal microscopy. The white arrows indicate lipid droplets. Cells were counterstained with DAPI to detect nuclei (blue). Views are at original magnification ×60. Scale bars: 10 μm. G: Total and uncoupled OCRs in M-adipocytes treated without (PBS) or with EPO (5 units/mL) were monitored. Data are representative of three independent experiments and normalized to total cellular protein. Statistics were performed using the Student t test. Bar graphs are mean ± SEM. Data are averages of three independent experiments. Ctrl, control; OM, oligomycin. *P < 0.05; **P < 0.01.
In culture, EPO stimulation in primary M-adipocytes also increased expression of BAT-associated genes (Fig. 5D), elevated mitochondrial gene expression (Fig. 5E), and mitochondrial density reflected by increased mitochondrial staining and mtDNA (Fig. 5F and 2G). Interestingly, in addition to increased total OCR, a higher uncoupled OCR was observed in EPO-treated M-adipocytes after injection of oligomycin, an index of uncoupled respiration and a characteristic of brown adipocytes (Fig. 5G), providing functional evidence for EPO promotion of BAT-like features in WAT. These findings collectively imply that EPO stimulation in WAT promotes expression of BAT-associated genes and represses WAT-associated genes that may be responsible for the increased mitochondrial biogenesis and oxidative metabolism. Collectively, these effects may partly explain the manner in which EPO counteracts obesity and its associated metabolic disorders during DIO. However, we cannot exclude the possibility of systemic effects of EPO in promoting BAT-like characteristics in WAT in vivo, even if mice do not show any difference in body mass compared with control mice (Supplementary Fig. 3A).
PPARα cooperates with Sirt1 to mediate the adipocyte response to EPO.
Next, we examined the mechanism by which EPO promotes brown features in WAT and increases metabolic activity. Our data showed that EPO increased PPARα expression and its downstream target PGC-1α (Fig. 4). PPARα was reported to promote the expression of the PRDM16 and UCP1, the two regulators determining brown adipocyte lineage. Activation of PPARα in human white adipocytes led to the appearance of a brown adipocyte pattern of gene expression, including induction of PGC-1α and PRDM16 (19). We treated M-adipocytes with the PPARα antagonist GW6471 and found GW6471 abrogated EPO-mediated increases in BAT-enriched genes such as Prdm16, Ucp1, Ucp3, and PGC-1α (Fig. 6A). GW6471 also attenuated EPO-induced basal and uncoupled OCR in M-adipocytes (Fig. 6B), suggesting PPARα is required for EPO activity in increasing mitochondrial activity and promotion of brown features in WAT.
FIG. 6.
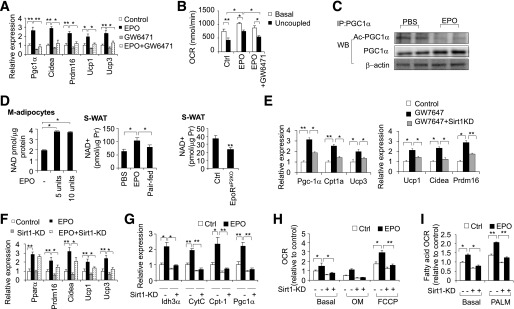
PPARα cooperates with Sirt1 to mediate adipose response to EPO. BAT-associated genes expression (A) and uncoupled OCR (B) were determined in M-adipocytes with EPO and/or GW6471 (10 µm) treatment for 6 days. C: The change in acetylated PGC-1α (Ac-PGC-1α) with EPO treatment (5 units/mL) was determined by immunoprecipitation (IP) of PGC-1α, followed by Western blotting (WB) for acetylated protein. β-Actin was used as the loading control. D: NAD+ level was determined in M-adipocytes treated with EPO at the indicated dosage and in S-WAT from DIO mice with EPO treatment for 5 weeks and compared with PBS control and paired-fed mice and from EpoRaP2KO and littermate control mice (n = 6). E: Mitochondrial and fatty acid oxidation genes and BAT-associated genes were determined in M-adipocytes treated without (Control) or with GW7647 (1 µm) for 6 days without and with knockdown (KD) of Sirt1. Expression of BAT-associated genes (F) and mitochondrial genes (G), OCR (H), and fatty acid oxidation (I) were determined in M-adipocytes with knockdown (KD) of Sirt1, without and with EPO treatment (5 units/mL). PALM, palmitate. One-way ANOVA was used in A, B, and D–I. All other statistics were performed using the Student t test. Bar graphs are mean ± SEM. In vitro data are means of three independent experiments. *P < 0.05; **P < 0.01.
As a downstream target of PPARα, PGC-1α is also activated by its deacetylation via Sirt1, an NAD+ dependent deacetylase and a member of sirtuins that is responsible for PGC-1α deacetylation (39). In EPO-stimulated 3T3-L1 adipocytes overexpressing PGC-1α (Supplementary Fig. 3C), acetylation of PGC-1α was decreased (Fig. 6C), indicating that EPO may regulate Sirt1 deacetylation activity to regulate PGC-1α activity. The increased intracellular NAD+ level in EPO stimulated M-adipocytes and S-WAT from EPO-treated mice and the decreased NAD+ level with the loss of EPO activity in EpoRaP2KO mice (Fig. 6D) further imply EPO modulates the intracellular NAD+ level to activate Sirt1 activity. To elucidate further the role of Sirt1 in PPARα signaling in adipocytes, we treated primary adipocytes with a PPARα agonist GW7647, without or with Sirt1 knockdown. As shown in Fig. 5E, GW7647 treatment induced the expression of mitochondrial genes and BAT fat–enriched genes in M-adipocytes (Fig. 6E). However the induction of these genes, including PPARα targets Pgc1α, Ucp1, Ucp3, and Cpt1α, was significantly lower when Sirt1 was knocked down using siRNA (Supplementary Fig. 3D) in adipocytes (Fig. 6E). Furthermore, Sirt1 knockdown attenuated the EPO-stimulated increase in BAT-enriched factors and mitochondrial genes (Fig. 6F and G) and in basal OCR and uncoupled OCR and fatty acid oxidation (Fig. 6H and I), indicating Sirt1 mediates EPO activity in adipocyte metabolism. Together, these data suggest Sirt1 cooperates with PPARα to respond to EPO to increase metabolic activity and promote brown features in WAT, leading to protection against DIO and its associated metabolic syndrome.
DISCUSSION
Beyond the erythroid-specific effect in the erythrocyte production, EPO has gained interest because of its various nonhematopoietic activities (40,41). These EPO activities were attributed to EPO response in multiple tissues, including pancreatic β-cells, skeletal muscle, and brain. Here, we engineered mice with fat tissue–specific deletion of EpoR to exclude the multiple tissue influence of EPO on energy metabolism and demonstrated that endogenous EPO activity in WAT contributes importantly to protection from obesity and associated metabolic syndromes. Metabolic disturbance due to the imbalance between fat storage and energy expenditure causes the body to store more fat and burn less energy. We demonstrated that targeted deletion of EpoR in adipocyte tissue shows a disproportionate increase in body fat and decreases in whole-body Vo2 and physical activity, with no change in food intake. This phenotype illustrates the importance of WAT in the endogenous metabolic response to EPO compared with other nonhematopoietic tissues. Furthermore, the increase in body weight gain in male adipocyte-specific EpoR KO is comparable to that observed in mice with EpoR deleted in nonhematopoietic tissue (9), suggesting that the loss of EPO signaling in WAT contributes to much of the increase in fat mass observed with loss of EpoR in nonhematopoietic tissue. In contrast, WT mice treated with EPO become leaner, with a decrease in accumulated body fat and an increase in activity. Although EPO treatment can also reduce food intake in WT mice, our paired-feeding experiments demonstrated that reduction in food intake is not sufficient to account for the increase in metabolic activity and the oxidative metabolism observed in WAT with EPO treatment. Collectively, these findings reveal insights into EPO effects on the molecular mechanisms of obesity and related metabolic diseases and point to the therapeutic potential of EPO beyond its known protective effect from tissue ischemia.
Energy metabolism imbalance in multiple tissues, including fat, has been recognized as an inducer of obesity and its associated metabolic disorders (42,43). Mitochondrial dysfunction and the resultant decreased lipid metabolism in WAT and excessive influx of fatty acids and triglycerides in other tissues lead to the excessive fat accumulation and development of obesity-induced insulin resistance and glucose intolerance preceding type 2 diabetes (34,37,38). However, reduced mitochondrial number and activity in WAT in the obese state can be restored to reduce obesity in animal models (44,45). Increased expression of brown adipocyte–enriched factors, such as PRDM6 and PGC-1α in WAT, can increase mitochondrial biogenesis and metabolic activity to prevent obesity and metabolic diseases. Therefore, increasing these BAT-enriched factors may be a plausible strategy for the treatment of obesity. In our study, EPO increased mitochondrial metabolic activity and promoted expression of brown fat–associated genes, such as PRDM16, PGC-1α, and UCPs, possibly through activation of PPARα, especially in inguinal WAT. Our observations suggest a mechanism by which EPO activates metabolic coregulators, PPARα, PGC-1α, and PRDM16, and other factors to contribute to the appearance of brown features, including increased mitochondrial content, oxidative respiration, and fatty acid oxidation in WAT. The transcriptional network by which EPO regulates PPARα, PRDM16, and PGC-1α remains to be determined. We should note that a differential EPO response was found among the various fat depots to EPO treatment in WT mice, along with corresponding opposing changes in adipocyte-specific EpoR KO mice, that reflects the response to endogenous EPO. In contrast to inguinal WAT, epididymal WAT showed an EPO-induced increase only in the expression of UCP3, PPARα, and PGC-1α, whereas BAT exhibited no significant change in BAT-associated gene expression. Among the other fat depots, perianal WAT showed an expression pattern analogous to inguinal WAT, whereas mesenteric WAT showed changes that more closely resembled epididymal WAT, possibly indicating various origins of these fat depots and different physiological function.
Activation of Sirt1 can improve metabolic dysregulation, insulin resistance, and glucose intolerance by enhanced β-oxidation of free fatty acids and promotes mitochondrial biogenesis in obese mice in an NAD+ dependent way (28,29,46,47). NAD+ and NADH metabolism is important in oxidative metabolism and energy homeostasis, has been linked to protection against dietary obesity, and is a therapeutic target for associated metabolic diseases (48–50). Here, we reveal that EPO elevated the intracellular NAD+ level in white adipocytes. In contrast, the loss of EpoR in WAT leads to the opposite effect. This increase in NAD+ leads to changes in PGC-1α deacetylation, likely due to an increase in Sirt1 activity. Furthermore, we show that EPO increases oxidative respiration and fatty acid oxidation in a Sirt1-dependent way in adipocytes, which may improve energy homeostasis and reduce lipid content in adipocytes in a model of obesity induced by an HFD. On the other hand, fatty acid synthesis or export may be altered because Sirt1 has been shown to regulate both processes (51,52). Our results also suggest that Sirt1 may be required for PPARα activity in the adipocyte response to EPO. The mechanism remains to be elucidated. It is likely that EPO may activate other sirtuins via the increased NAD+ level. However, we clearly show that EPO promoted brown remodeling of WAT and stimulated metabolic activity dependent on Sirt1.
Note that involvement of indirect EPO effects during DIO cannot be entirely excluded and that further investigation is warranted. Our study demonstrates for the first time that the loss of EPO/EpoR signaling in adipose tissue is sufficient to develop metabolic syndrome phenotype, including obesity, glucose intolerance, and insulin resistance, and supports the idea that EPO may contribute to protection against obesity and its associated metabolic syndromes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institutes of Health.
No potential conflicts of interest relevant to this article were reported.
L.W. designed and conducted the experiments and wrote the manuscript. R.T., L.D., and H.R. conducted experiments and data analyses. H.W. analyzed data and provided EpoRflox/flox mice. J.B.K. assisted with the Vo2 and fatty acid oxidation experiments. C.T.N. conceived the project, designed the experiments, analyzed data, and wrote the manuscript. C.T.N. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Dr. Gavrilova Oksana, from the National Institute of Diabetes and Digestive and Kidney Diseases Mouse Metabolism Core, for helping test the indirect calorimetry data, and Patricia M. Zerfas, from Division of Veterinary Resources, Office of Research Services, National Institutes of Health, for providing imaging expertise.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0518/-/DC1.
REFERENCES
- 1.Burger D, Lei M, Geoghegan-Morphet N, Lu X, Xenocostas A, Feng Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovasc Res 2006;72:51–59 [DOI] [PubMed] [Google Scholar]
- 2.Ruschitzka FT, Wenger RH, Stallmach T, et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc Natl Acad Sci USA 2000;97:11609–11613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ Res 2010;106:1722–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004;104:2073–2080 [DOI] [PubMed] [Google Scholar]
- 5.Anagnostou A, Liu Z, Steiner M, et al. Erythropoietin receptor mRNA expression in human endothelial cells. Proc Natl Acad Sci U S A 1994;91:3974–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakanaka M, Wen TC, Matsuda S, et al. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A 1998;95:4635–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai PT, Ohab JJ, Kertesz N, et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci 2006;26:1269–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shingo T, Sorokan ST, Shimazaki T, Weiss S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci 2001;21:9733–9743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teng R, Gavrilova O, Suzuki N, et al. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nat Commun 2011;2:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006;444:847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev 2004;84:277–359 [DOI] [PubMed] [Google Scholar]
- 12.Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 2007;293:E444–E452 [DOI] [PubMed] [Google Scholar]
- 13.Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 2009;360:1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med 2009;360:1500–1508 [DOI] [PubMed] [Google Scholar]
- 15.Virtanen KA, Lidell ME, Orava J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med 2009;360:1518–1525 [DOI] [PubMed] [Google Scholar]
- 16.Cousin B, Cinti S, Morroni M, et al. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci 1992;103:931–942 [DOI] [PubMed] [Google Scholar]
- 17.Barbatelli G, Murano I, Madsen L, et al. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab 2010;298:E1244–E1253 [DOI] [PubMed] [Google Scholar]
- 18.Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell Metab 2010;11:257–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hondares E, Rosell M, Díaz-Delfín J, et al. Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem 2011;286:43112–43122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boström P, Wu J, Jedrychowski MP, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012;481:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998;92:829–839 [DOI] [PubMed] [Google Scholar]
- 22.Seale P, Conroe HM, Estall J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 2011;121:96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajimura S, Seale P, Tomaru T, et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 2008;22:1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor α target genes. Cell Mol Life Sci 2004;61:393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villarroya F, Iglesias R, Giralt M. PPARs in the Control of Uncoupling Proteins Gene Expression. PPAR Res 2007;2007:74364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009;9:327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006;444:337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishnan J, Danzer C, Simka T, et al. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev 2012;26:259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006;127:1109–1122 [DOI] [PubMed] [Google Scholar]
- 30.Qiang L, Wang L, Kon N, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012;150:620–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He W, Barak Y, Hevener A, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 2003;100:15712–15717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jordan SD, Krüger M, Willmes DM, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 2011;13:434–446 [DOI] [PubMed] [Google Scholar]
- 33.Akamine EH, Marçal AC, Camporez JP, et al. Obesity induced by high-fat diet promotes insulin resistance in the ovary. J Endocrinol 2010;206:65–74 [DOI] [PubMed] [Google Scholar]
- 34.Bournat JC, Brown CW. Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabetes Obes 2010;17:446–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007;56:901–911 [DOI] [PubMed] [Google Scholar]
- 36.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 2007;293:E1118–E1128 [DOI] [PubMed] [Google Scholar]
- 37.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 38.Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 2003;300:1140–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005;434:113–118 [DOI] [PubMed] [Google Scholar]
- 40.Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y. Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert Rev Mol Med 2008;10:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi D, Schroer SA, Lu SY, et al. Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med 2010;207:2831–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sethi JK, Vidal-Puig AJ. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res 2007;48:1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem 2006;75:367–401 [DOI] [PubMed] [Google Scholar]
- 44.Wilson-Fritch L, Nicoloro S, Chouinard M, et al. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 2004;114:1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rong JX, Qiu Y, Hansen MK, et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 2007;56:1751–1760 [DOI] [PubMed] [Google Scholar]
- 46.Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007;450:712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moynihan KA, Grimm AA, Plueger MM, et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab 2005;2:105–117 [DOI] [PubMed] [Google Scholar]
- 48.Houtkooper RH, Auwerx J. Exploring the therapeutic space around NAD+. J Cell Biol 2012;199:205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 2010;31:194–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cantó C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 2012;15:838–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamazaki Y, Usui I, Kanatani Y, et al. Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am J Physiol Endocrinol Metab 2009;297:E1179–E1186 [DOI] [PubMed] [Google Scholar]
- 52.Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004;429:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.