

Abstract
Signal transducer and activator of transcription (STAT) 4 is one of the seven members of the STAT family. STAT4 has a prominent role in mediating interleukin-12–induced T-helper cell type 1 lineage differentiation. T cells are key players in the maintenance of adipose tissue (AT) inflammation. The role of STAT4 in obesity and AT inflammation is unknown. We sought to determine the role of STAT4 in AT inflammation in obesity-induced insulin resistance. We studied STAT4-null mice on the C57Bl6/J background. We have found that STAT4−/−C57Bl6/J mice develop high-fat diet–induced obesity (DIO) similar to wild-type controls, but that they have significantly improved insulin sensitivity and better glucose tolerance. Using flow cytometry and real-time PCR, we show that STAT4−/− mice with DIO produce significantly reduced numbers of inflammatory cytokines and chemokines in adipocytes, have reduced numbers of CD8+ cells, and display increased alternative (M2) macrophage polarization. CD8+ cells, but not CD4+ cells, from STAT4−/− mice displayed reduced in vitro migration. Also, we found that adipocyte inflammation is reduced and insulin signaling is improved in STAT4−/− mice with DIO. We have identified STAT4 as a key contributor to insulin resistance and AT inflammation in DIO. Targeting STAT4 activation could be a novel approach to reducing AT inflammation and insulin resistance in obesity.
Inflammation and activation of the immune system are emerging as key mechanisms associated with visceral adiposity, type 2 diabetes, and cardiovascular disease. Adipose tissue (AT) inflammation was recently identified as an early indicator of insulin resistance and type 2 diabetes, and as a contributor to disease susceptibility and progression (1). AT contributes to inflammation in obesity by means of increased mass, modified adipocyte phenotype, and increased infiltration of immune cells (2). Strong evidence from mouse models of obesity suggest that AT infiltration with proinflammatory macrophages, T cells and natural killer (NK) cells leads to cytokine and chemokine production and free fatty acid release, which can induce pancreatic β-cell dysfunction, insulin resistance, and atherosclerosis (3–5).
Signal transducers and activators of transcription (STATs) are downstream of the Janus kinase (Jak)/tyrosine kinase, and, upon phosphorylation in response to cytokine and growth factor activation, dimerize and translocate to the nucleus, where they act as transcription factors inducing the expression of genes involved in proliferation and differentiation of various hematopoietic and nonhematopoietic cells (6,7). STAT4 is expressed primarily in T cells and NK cells (8,9). Importantly, recent findings indicate that STAT4 has a determinant role for optimal human T-helper type 1 (Th1) lineage development (10). STAT4-null mice have impaired Th1 lineage development in response to interleukin (IL)-12 stimulation of T cells, have reduced interferon-γ (IFN-γ) production, and display propensity toward the development of Th2 cells (11,12). Also, STAT4 limits the development of regulatory CD4+Foxp3+ cells, suggesting a role in peripheral immune tolerance (13). Importantly, STAT4-null mice are viable and fertile, have normal hematopoiesis, and are resistant to infections by most common pathogens. Several studies have shown that STAT4−/− mice are protected from the development of T-cell–mediated autoimmune diseases and have reduced inflammation in systemic sclerosis (9). However, there is a lack of data indicating in vivo pathogenic roles for any of the STAT family members in insulin resistance and type 2 diabetes.
In this article, we report that STAT4−/−C57Bl6/J mice with diet-induced obesity (DIO) have significantly improved insulin sensitivity and better glucose tolerance compared with wild-type controls. We have shown that DIO STAT4−/− mice produced fewer inflammatory cytokines and chemokines in AT, had reduced CD3+ cells infiltrating AT, had reduced CD8+ T-cell migration in vivo, and displayed increased alternative (M2) macrophage polarization. Also, STAT4-deficient adipocytes and AT have improved insulin signaling compared with wild-type controls after in vivo and in vitro insulin stimulation. In addition, Rag1-null mice lacking T and NK cells after adoptive transfer with STAT4-deficient splenocytes showed improved insulin sensitivity in high-fat diet (HFD)–fed mice compared with C57Bl6 reconstituted counterparts.
Unlike other members of the STAT family, such as STAT1, 2, 5, and 6, shown to have roles in adipogenesis in rodents (14), the expression and roles of STAT4 in adipocytes were not previously shown. A recent article from our laboratory reported that STAT4 activation is increased in AT of obese compared with lean Zucker rats, suggesting a functional role of STAT4 in AT in obesity (15). In this article, we show that STAT4 deficiency is associated with decreased cytokine and chemokine production in adipocytes, improved insulin signaling, and a reduced adipocyte average size. This is the first report showing that STAT4 deficiency in hematopoietic and nonhematopoietic compartments is key to improving insulin sensitivity and reducing AT inflammation in obesity. Therefore, STAT4 modulation may be a novel target to ameliorate AT inflammation in obesity and to improve the associated metabolic and cardiovascular complications.
RESEARCH DESIGN AND METHODS
Animals and diets.
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Eastern Virginia Medical School. Female C57Bl/6J mice, 10 weeks of age, were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained on regular rodent chow or fed an HFD (60 kcal% fat; Research Diets, New Brunswick, NJ) for 16 weeks. Age-matched STAT4−/−C57Bl6/J mice were maintained in similar conditions after the transfer of breeders from Indiana University. These mice were cross-bred periodically on the C57Bl6/J background to avoid genetic drifting. All of the mice were between 26 and 28 weeks of age at euthanasia. Mice were housed in a pathogen-free facility and individually caged, and food and water were provided ad libitum throughout the experiment. Body weight and food intake were measured weekly. Three separate trials were performed under identical experimental conditions for the HFD group with each trial containing 6–9 mice per experimental group. Also, two groups representing STAT4−/− and wild-type mice were maintained on chow diet for 16 weeks and age-matched with the HFD groups (n = 6–7). The obesity index was calculated by dividing the cubic root of the body weight (in grams) by the naso-anal length (in millimeters) × 104 according to Novelli et al. (16) and Simson and Gold (17). Feed efficiency was reported for the last week on the diets, and represents the ratio between the weekly food intake and weight gain. In a separate experiment, age-matched male mice (n = 6/group) were kept either on chow or an HFD for 16 weeks and were used to confirm the metabolic phenotype determined in females. Rag1-null male mice, 8 weeks of age, were purchased from The Jackson Laboratory. Mice were injected via the tail vein with 5 × 106 T splenocytes isolated from either 10-week-old STAT4-deficient (n = 10) or wild-type females (n = 10). One week after the adoptive transfer, all of the mice were placed on an HFD for 15 weeks.
Metabolic measurements.
The intraperitoneal glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs) were performed at the end of the long-term dietary challenge, as previously described (18,19). Plasma insulin level was measured using a commercially available ELISA kit from Mercodia (Uppsala, Sweden), according to the manufacturer’s instructions. Plasma glucose level was measured using a kit from Cayman Chemicals. Free fatty acids, triacylglycerols, and cholesterol were measured with a kit from Wako (Richmond, VA). Adiponectin and leptin were measured by ELISA kits from Millipore (Billerica, MA) and Abnova (Walnut, CA).
Western blotting for in vivo and in vitro insulin signaling and STAT4 activation.
STAT4-deficient and control mice after 16 weeks on HFD were injected intraperitoneally with a bolus of 10 units/kg body wt insulin or with saline, and after 10 min the AT was harvested and homogenized on ice in radioimmunoprecipitation assay buffer with protease/phosphatase inhibitors. To determine the activation of the insulin signaling pathway, insulin receptor (IR), IR substrate-1 (IRS-1), and protein kinase B (Akt) phosphorylation; and total protein expression were measured by Western blotting using the following antibodies: phosphorylated (p) IR (1:500 dilution; Y972; Abcam); IR-β, pAKT (Ser473), Akt (pan) (1:1,000 dilution; Cell Signaling Technology); pIRS-1 (1:500 dilution; Y896; Invitrogen); IRS-1 (1:500 dilution; Cell Signaling Technology). β-actin (1:2,000 dilution; Santa Cruz Biotechnology) was used, in some cases, to normalize for protein loading. Also, total STAT4 (1:500 dilution; Cell Signaling Technology) and activated STAT4 (Y693) (1:500 dilution; BD Transduction Laboratories) were measured in the basal or stimulated state. Activation of the same proteins was also measured by Western blotting in isolated adipocytes from STAT4-deficient or wild-type controls in the basal state and after stimulation with 5 or 50 nmol/L insulin for 30 min.
Pancreatic islet isolation and in vitro glucose-stimulated insulin secretion.
Pancreatic islets were isolated by pancreatic perfusion with collagenase P (Roche Applied Science) through the common bile duct, as previously described (19). For glucose-stimulated insulin secretion, islets were incubated in Krebs-Ringer bicarbonate HEPES buffer supplemented with 3 mmol/L glucose for 1 h followed by an additional 1-h incubation in 28 mmol/L glucose, as previously described (19). The supernatant was collected after each of the treatments, and insulin was measured by an ELISA method (Mercodia).
AT morphometry.
Epididymal tissue was formalin-fixed and paraffin-embedded, and the sections were stained with hematoxylin-eosin. Three representative images were obtained per section, for a total of six tissue sections per mouse. Four mice per experimental group were randomly selected and analyzed. Images were taken using a Zeiss Plan Apochromat ×20 objective, and adipocyte area and number were obtained using the ImageJ software (NIH, Bethesda, MD).
Adipocyte and stromal vascular fraction preparation.
Samples of epididymal AT (0.1–0.3 g) were digested with collagenase, as described previously with minor modifications (20,21). The floating adipocytes were collected and washed, and the infranatant was removed and centrifuged at 500g for 5 min to pellet the stromal vascular fraction (SVF).
Flow cytometry.
Counted SVF cells were incubated for 30 min at room temperature with one of the following combinations of fluorophore-conjugated primary antibodies: Cocktail 1 (for macrophage phenotyping): CD11b-Pacific Blue, CD45-PerCP, Cd11c-PE F4/80-Alexa Fluor 647, and CD206-fluorescein isothiocyanate; Cocktail 2 (for T-cell phenotyping): CD3-Pacific Orange, CD4-APC, CD8-fluorescein isothiocyanate, CD45-PerCP, IFN-γ-PB, and CD25-Cy5. All of the antibodies were from BD Pharmingen (San Jose, CA) or from BioLegend (San Diego, CA). Cells were analyzed on a BD Pharmingen-upgraded FACS Caliber Flow Cytometer (8-colors) using FlowJo software (Tree Star Inc., Ashland, OR). Also, spleens and lymph nodes (inguinal and lumbar, two each) were collected, and single-cell suspensions were prepared by gentle mechanical disruption and stained in a similar manner to the protocols described for SVF.
Real-time PCR.
RNA from total AT, pancreatic islets, adipocytes, or SVF was extracted and reverse-transcribed, as previously described (22). Real-time PCR was performed using Taqman probes from Applied Biosystems (Carlsbad, CA). β-Actin was used to normalize the data. Results were expressed either as 1/ΔCt or as fold change by the 2−ΔΔCT method using the wild-type mice as a control group. The multiplex gene array PCR for adipocytes, and CD4+ and CD8+ T cells was performed using the Insulin Resistance and the Chemokine and Chemokine receptors RT2 Profiler PCR Array from SA Bioscience (Carlsbad, CA).
AT explants and in vitro chemotaxis of T cells.
Epididymal AT was finely minced in ∼1 mm3 pieces and cultured for 24 h in a 5% CO2 incubator. Some of the explants were treated for 6 h with 50 ng/mL mouse IL-12. Media was collected and either analyzed for chemokine production or used for the in vitro migration experiments. For migration experiments, spleens from matched mice were mechanically disrupted, and T cells were isolated using a T-cell enrichment kit from StemCell Technologies (Vancouver, BC, Canada). The purified T cells (105/well) were placed in RPMI medium with 1% FBS in 24-well transwell plates with a 5-μm filter, and the migration was assessed after 4 h of incubation at 37°C in 5% CO2 against either RPMI medium + 1% FBS (negative control), RPMI medium + 10% FBS (positive control), or conditioned media from AT explants cultured for 24 h. Migrated cells from the lower chamber were counted and analyzed by flow cytometry to determine the percentage of CD4+ and CD8+ cells. In a separate experiment, individual CD4+ and CD8+ T-cell subsets were prepared from either STAT4-deficient or wild-type splenocytes using the positive immunoseparation kits from StemCell Technologies. After separation, the PE labeled migrated cells were counted in the lower chamber by flow cytometry.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism Software (GraphPad Software, La Jolla, CA). A Student t test unpaired analysis was used for all data comparisons between the STAT4−/− mice and wild-type controls on either an HFD or chow diet. Data were expressed as the mean ± SEM, and the null hypothesis was rejected for a P value <0.05.
RESULTS
Effect of STAT4 deficiency on the metabolic phenotype.
STAT4-deficient mice on chow diet have similar body weight with wild-type controls, and exposure to high dietary fat (60 kcal% fat) leads to a comparable increase in body weight and perigonadal adiposity; the obesity index (the equivalent of BMI in humans) mirrors the changes in adiposity and body weight (Fig. 1A). Exposure to high-fat feeding resulted in a similar increase in feed efficiency in the STAT4−/−C57Bl6 mice and controls (Fig. 1A). These data indicate that STAT4 deficiency does not alter weight gain after HF exposure, nor does it affect food intake, feed efficiency, or adiposity. This phenotype was present in both female and male mice (data not shown).
FIG. 1.

Anthropometric measurements and metabolic phenotype of the C57Bl6/J/STAT4−/− mice and wild-type controls. A: Body weight, perigonadal adiposity, obesity index, and feed efficiency were significantly increased in both the wild-type and knockout female mice after 16 weeks on an HFD; mice were fed either a 60 kcal% fat HFD for 16 weeks, starting at 10 weeks of age (n = 12–16) or were kept on chow (n = 7–8); the results for the HFD mice are combined from two different but similar experiments (experiments 1 and 2). The obesity index was calculated by dividing the cubic root of the body weight (in grams) by the naso-anal length (in millimeters) × 104. Feed efficiency was calculated by dividing the weight gain by food intake in the last week on high-fat or chow diets (week 16); the results are from six to eight mice per group and represent data from experiment 1. *Significant compared with the chow-fed group. B: Intraperitoneal GTTs and ITTs were performed after 16 weeks of HFD or chow feeding. The AUC was measured in each case. Plotted values in the histograms are the average ± SEM from 7–16 mice per group. *Significant compared with STAT4−/− mice. C: In vitro glucose-stimulated insulin secretion was measured in pancreatic islets of C57Bl6/J/STAT4−/− mice and wild-type controls isolated after 16 weeks on an HFD. Islets were cultured overnight and subsequently treated for 1 h with 3 or 18 mmol/L glucose. Insulin was measured by ELISA in the media of the islets stimulated with a low- or high-glucose concentration. Results represent average ± SEM from sixmice per group; the null hypothesis was rejected for P < 0.05. HF, high fat.
Despite the obesity, STAT4−/− mice showed significant improvements in the metabolic phenotype compared with wild-type controls (Fig. 1B and Table 1). Intraperitoneal GTTs and ITTs were performed in STAT4−/− and control mice on both the HFD and chow diet. No significant differences were detected between the STAT4−/− mice and controls on chow diets. Insulin sensitivity was significantly improved in the HFD STAT4−/− mice compared with controls, and the area under the curve (AUC) showed comparable values with the age-matched STAT4−/− chow fed mice (Fig. 1B). Glucose tolerance was also improved with borderline significance in STAT4-deficient mice on an HFD compared with wild-type controls (Fig. 1B). Levels of fasted plasma glucose and randomly fed plasma insulin were both significantly lower in STAT4−/− mice, whereas the fasted plasma insulin level was not different between groups (Table 1). Also, levels of plasma free fatty acids, triglycerides, and total cholesterol were similar between groups (Table 1). Importantly, the homeostasis model assessment (HOMA) index was significantly lower in STAT4−/− mice compared with controls, reflecting improved insulin sensitivity in the HFD STAT4−/− mice (Table 1).
TABLE 1.
Plasma biochemical profile of STAT4−/− and wild-type mice on HFD
Because female C57Bl6/J mice are reportedly more insulin-sensitive compared with males in response to high-fat feeding, we also performed ITTs and GTTs in STAT4−/− and control males on both a chow and an HFD (Supplementary Fig. 1). The AUCs for both the ITTs and GTTs were similar for STAT4−/− and wild-type males on a chow diet. Similarly to females, the STAT4−/− males also showed improved insulin sensitivity on an HFD (Supplementary Fig. 1). These data show that STAT4−/− deficiency protects against HFD–induced insulin resistance in a sex-independent manner.
Because the intraperitoneal GTT result was improved in the HFD STAT4−/− mice, we also analyzed insulin-stimulated glucose secretion in vitro in islets isolated from STAT4−/− and control mice (Fig. 1C). Both basal (3 mmol/L glucose) and glucose-stimulated (18 mmol/L glucose) insulin secretion were similar between the STAT4−/− and control groups. This result indicates that STAT4 deficiency alone does not have an insulinotropic effect on the pancreatic islets.
Collectively, the phenotype data indicate that STAT4 deficiency improves insulin sensitivity and ameliorates glucose intolerance in response to excess fat intake without changes in body weight, visceral adiposity, circulating lipid levels, or direct effects on pancreatic insulin secretion.
STAT4 is expressed in AT, and STAT4 deficiency improves insulin signaling.
Because STAT4 deficiency showed improved systemic insulin sensitivity, we next investigated whether a deficiency in STAT4 expression improves in vivo insulin signaling in AT after 16 weeks of high-fat feeding (Fig. 2). First, we confirmed that STAT4 is robustly expressed in AT of wild-type mice while absent in the knockout mice, and is activated after in vivo insulin stimulation (Fig. 2A). We next measured phosporylation of IR, IRS-1, and Akt in STAT4-deficient and wild-type controls in the basal state and 10 min after in vivo insulin stimulation (Fig. 2B). In wild-type mice on an HFD, insulin failed to significantly increase activation of IR, IRS-1, and Akt. In contrast, STAT4 deficiency resulted in robust activation of key insulin signals by approximately threefold to eightfold (n = 4, P < 0.05). Also, in the basal unstimulated state the level of activation was similar in STAT4−/− and wild-type mice. Similar data were obtained for trapezius muscle samples obtained from the two groups upon in vivo insulin stimulation (not shown). It is suggested that both reduced adiponectin and increased leptin levels can contribute to impaired insulin signaling and reduced glucose uptake. Although plasma adiponectin levels were similar between the two groups (Table 1), leptin levels were significantly decreased in the STAT4-deficient mice both before and after correction for body weight (Fig. 2C).
FIG. 2.

Insulin signaling and leptin production in AT of C57Bl6/J/STAT4−/− mice and wild-type controls. A: STAT4 is expressed in AT of wild-type but not of knockout mice. In response to a bolus insulin injection of 10 units/kg body wt, STAT4 activation (pSTAT4) was increased in AT of control mice after 30 min (n = 3, P < 0.05). B: STAT4-deficient and control mice were injected intraperitoneally with a 10 units/mL body wt insulin bolus, and tissue was harvested after 10 min. Western blot analysis showed increased activation of pIR, pIRS-1, and pAkt in STAT4-deficient mice compared with controls (n = 4, P < 0.05). Data represent the mean ± SEM. *Significant compared with wild-type mice; #significant compared with nonstimulated (basal) state. C: Fasted plasma leptin levels were measured by ELISA in chow (n = 6) and HFD (n = 9) STAT4−/−C57Bl6 and control mice. In the bottom panel, leptin concentration was normalized to body weight for each mouse. Data represent the mean ± SEM. *Significant compared with chow; #significant compared with control group. P < 0.05 was considered significant. Ins, insulin; KO, knockout.
Roles of adipocyte STAT4 deficiency on adipocyte gene expression, insulin signaling, and adipocyte morphometry.
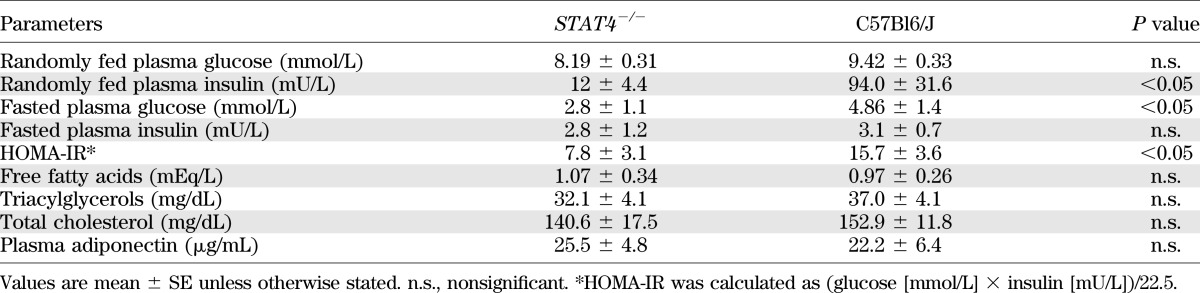
We next evaluated any direct effects on adipocytes in the two mouse groups. The expression of STAT4 in adipocytes has not been previously reported. As shown in Fig. 3A, STAT4 product was detected at the expected size (70 bp) in C57Bl6 mice in both the adipocytes and the SVF. Relative band densitometry showed an average of 5.5-fold higher expression of STAT4 in SVF and a 9.4-fold higher expression in spleen compared with that in adipocytes. Therefore, STAT4 RNA expression is low compared with the lymphoid tissues and hematopoietic cells but clearly detectable. STAT4 protein expression was also detected in adipocytes by Western blot. Similarly to gene expression, STAT4 protein expression was low in adipocytes compared with total AT and positive control (spleen) and was undetectable in tissue from STAT4−/− mice (Fig. 3A). To determine whether there was any direct effect of STAT4 in adipocytes on insulin signaling, isolated adipocytes were treated with insulin in vitro (Supplementary Fig. 2). Both the STAT4-deficient and wild-type adipocytes had significantly increased pAkt levels in response to insulin, but no significant differences were noted between groups (Supplementary Fig. 2). To interrogate further possible changes in adipocytes with STAT4 deficiency, we measured the expression of 84 genes in adipocytes isolated from STAT4-deficient and wild-type mice in the fasted state using the “insulin resistance” array from SABiosciences (Fig. 3B). Ten of the genes were significantly (P < 0.05) downregulated in STAT4-deficient adipocytes versus controls by 2.8-fold to 1.8-fold. Among the downregulated genes, expressions of acetyl CoA carboxylase (Acaca), phosphoenolpyruvate carboxykinase, and pyruvate dehydrogenase kinase were all reduced, suggesting reduced lipogenesis, triglyceride formation, and glucose utilization, overall indicative of improved metabolic response to fasting. Interestingly, retinol binding protein-4 expression was reduced by 2.2-fold in STAT4-deficient adipocytes along with IGF-I receptor, suppressor of cytokine signaling 3 (SOCS3), and Jak2, all of which were positively associated with insulin resistance, thereby suggesting potential mechanisms for enhanced insulin action in vivo (Fig. 2B). Future studies will be needed to investigate whether these changes in gene expression are transcriptionally regulated in a STAT4-dependent manner. STAT4 deficiency also affects the adipocyte morphometry (Fig. 3C). Although adipocyte size increased in both groups on an HFD, adipocytes from STAT4−/− mice showed significantly lower average area versus adipocytes from control mice (Fig. 3C). This suggests a less in vivo hypertrophic response of the AT from HFD STAT4−/− mice, which also reflects a more insulin-sensitive in vivo phenotype.
FIG. 3.
Effect of STAT4 deficiency on adipocyte insulin signaling and morphometry. A: Representative gel showing mRNA (left) and protein (right) expression of STAT4 in adipocytes, SVF, and spleen of STAT4-deficient mice and wild-type controls on chow diet. Relative quantification by real-time PCR shows a 5.6-fold higher STAT4 expression in SVF and an 8.5-fold higher expression in spleen compared with adipocytes. Western blot confirms the absence of STAT4 in adipocytes and in spleens of STAT4−/−C57Bl6 mice. B: Differences in adipocyte gene expression between STAT4-deficient and control mice. Adipocytes were isolated by collagenase digestion from overnight fasted mice, and gene expression was measured using the insulin resistance pathway focused array from SABiosciences (PAMM 22Z). Results are expressed as the fold change in STAT4-deficient adipocytes vs. controls (n = 3). Only the significant (P < 0.05) differences are illustrated. C: Adipocyte size measured by morphometry in paraffin-embedded AT from STAT4−/−C57Bl6 mice and wild-type controls on either chow or an HFD. Results are from four mice per group. Representative micrographs show hemotoxylin-eosin–stained AT sections from knockout and wild-type mice on HFD. Scale bar, 100 μm. KO, knockout. *Significant vs. chow groups; #significant vs. STAT4−/− group.
STAT4 deficiency reduces adipocyte and islet inflammation, and alters T cell and macrophage polarization.
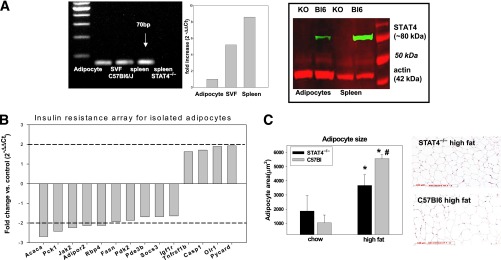
Production of inflammatory cytokines and chemokines by the insulin-resistant adipocytes is a key mechanism leading to immune infiltration and exacerbation of inflammation in AT. Therefore, we next investigated whether STAT4 deficiency changes cytokine and chemokine expression in adipocytes in HFD mice (Fig. 4A). IFN-γ expression was not changed; however, IL-12 subunits and tumor necrosis factor-α (TNF-α) were significantly higher in control mice compared with STAT4−/− mice. In addition, the expression of the potent T-cell chemokines CXC chemokine ligand 10 (CXCL10) and C-C motif ligand (CCL) 5 (regulated on activation normal T-cell expressed and secreted [RANTES]) were significantly higher in wild-type control mice compared with STAT4−/− mice (Fig. 4A). Also, CCL5 was significantly increased in pancreatic islets, indicating a key role of the latter in local inflammation (Fig. 4C). We next measured RANTES production in total AT explants in culture. Indeed, RANTES protein concentration, measured by ELISA, was significantly lower in the conditioned media of 24-h cultured AT explants from STAT4−/− mice compared with those of wild-type controls (Fig. 4B). In addition, 6-h treatment of the explants with 50 ng/mL recombinant IL-12 induced a significant increase in RANTES in the culture media in the C57Bl6 mice but not in the STAT4−/− mice (Fig. 4B). This result suggests that, under proinflammatory conditions, AT from STAT4−/− mice responds by lower production of chemokines compared with C57Bl6 controls. However, the total plasma concentration of RANTES as well as CXCL10 and CCL2 were not different between groups (Fig. 4B), suggesting that differences may be tissue-specific (fat and pancreatic islets) but are not reflected at the systemic level. Additionally, measurements of IL-12p70, IFN-γ, and TNF-α were performed using antibody-conjugated fluorescent beads by flow cytometry; however, the circulating levels of these cytokines were below the detection limit of the assay.
FIG. 4.
Effect of STAT4 deficiency on inflammation and immune cell composition in AT, adipocytes, SVF, secondary lymphoid organs, and pancreatic islets of STAT4−/− and control HFD mice. A: Proinflammatory cytokines and chemokines were measured by real-time PCR in adipocytes isolated from perigonadal fat of STAT4−/−C57Bl6 and control mice on an HFD (n = 7–9). Results are expressed as 1/ΔCt after normalization to actin. Data represent the mean ± SEM. *Significance established for P < 0.05. B: AT explants (∼1–2 mm3 pieces), prepared from STAT4−/−C57Bl6 or control mice (n = 4) on an HFD for 16 weeks, were kept in culture for 24 h and subsequently treated with 50 ng/mL IL-12 for 6 h. Concentration of RANTES (CCL5) was measured in the culture media by ELISA. RANTES was also measured in fasted plasma from the same groups of mice (n = 7–9); CCL2 and CXCL10 were measured in plasma using antibody-conjugated beads by flow cytometry (Flow Cytomix; eBioscience). C: Pancreatic islets were isolated from both groups after 16 weeks of an HFD, and gene expression was measured by real-time PCR. Data are expressed as 1/ΔCt after normalization to β-actin. Results represent average ± SEM from n = 6 mice per group. D: Immune cell composition of the SVF prepared from perigonadal AT of STAT4−/−C57Bl6 mice and wild-type controls on an HFD. SVF was stained with fluorescently labeled anti-mouse antibodies and analyzed by flow cytometry. In the three left panels, T-cell composition was analyzed. Cell numbers were calculated based on the corresponding antibody positivity and normalized to AT weight. To determine the percentage of different T-cell subsets, cells in the CD45+ gate were analyzed. Bottom plots are representative for the CD45+ gating and for the double positivity for CD3+ and CD45+ or CD25+ in each group. In the fourth panel on the left, SVF was additionally stained with an anti–IFN-γ antibody. The percentage of IFN-γ–positive cells in each T-cell subset was calculated after gating for CD45. In the last panel, SVF from the same mice were separately stained with fluorescently labeled anti-mouse CD45, CD11c, CD11b, F4/80, and CD206. The total macrophage numbers were expressed as CD11b+/F4/80+CD11c− cells and were normalized per tissue weight. Relative percentages of CD11b+/F4/80+CD206+ cells were measured within the CD45+ gate; a representative FACS plot for the latter is shown at the bottom. Results are average ± SEM from n = 6–8 mice/group; the null hypothesis was rejected for a P value <0.05. *Significant vs. C57Bl6 group. E: Gene expression for the M2 activation markers arginase and PPAR-γ were measured by real-time PCR in the SVF isolated from n = 4 mice per group. Results are expressed as 1/ΔCt after normalization to 18S ribosomal RNA and represent the mean ± SEM. Immune cell composition of the spleens (F) and lymph nodes (G) was measured by flow cytometry. Spleens as well as inguinal and lumbar (two each) lymph nodes were collected and stained with two different antibody cocktails, as described in research design and methods. Spleens (F) and lymph nodes (G) were analyzed for the relative percentage of CD3+ cells in the CD45+ gate and for relative percentages of CD4+ and CD8+ in the CD45+CD3+ gate; the percentage of CD11b+F4/80+ cells in the CD45+ gate as well as the percentage of CD11b+/F4/80+/CD206+ cells in the CD45+ gate were measured. Data represent the average ± SEM from six to eight mice per group. *Significant for P < 0.05. SSC, side-scattered plot.
Changes in immune cell composition in adipose and lymphoid tissues associated with STAT4 deficiency.
SVF composition of T-cell subsets as well as macrophage number and composition were analyzed by flow cytometry (Fig. 4D). The total number of CD45+ cells as well as the numbers of CD3+ and CD8+ cells were significantly lower in the HFD STAT4−/− mice compared with controls (Fig. 4D). Also, the mean percentage of CD3+ cells of the total CD45+ leukocytes was 43 ± 3.2% in STAT4-deficient mice and was significantly higher (54.8 ± 2.1%) in the wild-type controls. The cellularity data agreed with CD3+ composition, showing a decreased percentage of the CD8+ cells (13.8 ± 1.4% in STAT4−/− vs. 18 ± 1.6% in C57Bl mice) but similar percentages of CD4+ cells (57.1 ± 2.7 in STAT4−/− vs. 53.1 ± 3.6 in C57Bl mice). Also, the percentage of CD3+CD25+ regulatory T cells was significantly increased in STAT4−/− mice (15.7 ± 5.04) compared with controls (1.7 ± 0.38), suggesting improved peripheral immune tolerance with positive effects on local inflammation (Fig. 4D). Next, we examined whether there are differences in IFN-γ protein expression in different CD3+ T-cell subsets. The percentage of IFN-γ–positive events in both the CD4+ and CD8+ cells was significantly decreased in STAT4−/− mice compared with controls (Fig. 4D).
Another important player in AT inflammation in DIO is the macrophage. We measured the number of CD11b+/F4/80+ cells in the perigonadal fat and found a significantly lower number of cells in the STAT4-deficient mice compared with controls (Fig. 4D). The percentage of CD11b+/F4/80+ cells, representing macrophages in the CD45+ leukocyte gate, was similar in the two groups (40.4 ± 3.1% in STAT4−/− vs. 37.6 ± 3.9% in C57Bl mice). However, of the total macrophage population, the percentage of CD206+ cells (likely representing M2 macrophages) was significantly higher in STAT4−/− mice (Fig. 4D), suggesting a bias toward the M2 phenotype in the AT of STAT4−/− mice. Indeed, gene expression of arginase and peroxisome proliferator–activated receptor-γ (PPAR-γ) were also significantly increased in SVF from STAT4-deficient mice versus controls (Fig. 4E).
To establish whether this immune cell profile is recapitulated in the lymphoid organs, we analyzed the spleen (Fig. 4F) and the inguinal/mesenteric lymph nodes (Fig. 4G). Although, the percentage of CD3+ cells in the spleen was significantly higher in STAT4−/− mice, the overall cellularity was comparable between the two groups. Also, the percentages of CD3+CD4+ and CD3+CD8+ cells in the CD45+ leukocyte gate were comparable between the groups (Fig. 4F). No significant differences between groups were found in the numbers of CD3+, CD4+, CD8+, or IFN-γ–producing T-cell subsets in the inguinal/mesenteric lymph nodes (Fig. 4G). Interestingly, the percentage of CD206+ M2 macrophages was consistently reduced in AT, spleen, and lymph nodes of wild-type mice compared with STAT4−/− mice (Fig. 4D, F, G). These data suggest that the T-cell subset composition in AT is unique and may be explained by differences in migration, proliferation, or apoptosis among the T-cell subsets in AT.
Role of STAT4 deficiency in AT cytokine expression and immune composition in lean mice.
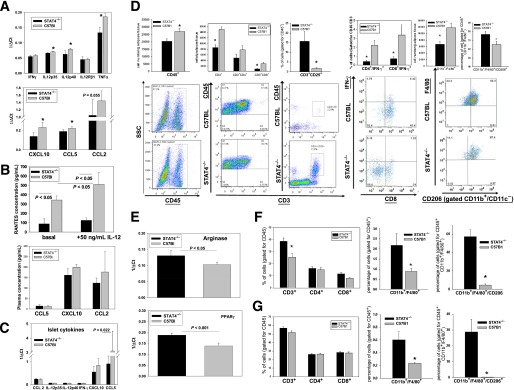
To determine whether STAT4 deficiency results in different inflammatory and immune profiles in the absence of dietary challenge, we also measured cytokine and chemokine expression in adipocytes from STAT4−/− and control mice on a chow diet (Fig. 5). Expression of IL-12 subunits, IFN-γ, CXCL10, and CCL5 were all significantly lower in STAT4−/− mice compared with wild-type mice (Fig. 5A). This result indicates that, even in the absence of a dietary challenge or obesity-associated lipotoxicity, adipocyte cytokine and chemokine production is lower in STAT4−/− mice compared with control mice.
FIG. 5.
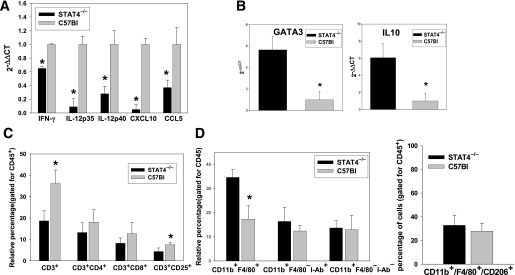
Cytokine expression in adipocytes and SVF, and immune composition of SVF from STAT4−/−C57Bl6 and control mice on chow diet. Gene expression of proinflammatory cytokines in adipocytes (A) and different Th2 markers in SVF (B) of knockout and control chow-fed mice 26–28 weeks of age. Data are expressed as the fold change in STAT4−/−C57Bl6 expression compared with control after normalization to β-actin. Data are the average ± SEM from n = 6–8 mice per group. Relative composition of different T-cell subsets (C) and granulocytes (D) in SVF of STAT4−/−C57Bl6 and C57Bl6 controls on chow diet. In both cases, cells were analyzed in the positive CD45 gate. Data are expressed as the relative percentage of cells and represent the mean ± SEM from n = 6–8 mice per group. *Significant difference for P < 0.05.
In chow-fed STAT4−/− mice, the percentage of CD3+ cells in the CD45+ leukocyte gate was lower compared with wild-type controls, whereas no difference was found in the CD3+CD4+ T-cell subset. Also, the percentage of CD4+CD25+ T-regulatory cells was slightly decreased in the STAT4−/− group on chow diet compared with controls (Fig. 5C). In addition, real-time PCR expression of GATA-binding protein 3 (GATA3) and IL-10 showed increased expression in the STAT4−/− group, suggesting a bias toward a Th2 phenotype in the AT T cells of the latter (Fig. 5B). Macrophage analysis indicated an increased percentage of CD11b+F4/80+ cells in the CD45+ lymphocyte gate in STAT4−/− mice, whereas the number of other granulocytes and the level of macrophage activation assessed by I-Ab positivity or polarization (F4/80+CD206+) were similar among groups (Fig. 5D).
STAT4 deficiency reduces CD8+ but not CD4+ migration.
We next investigated whether there are any differences in the chemotactic properties of different CD3+ cell subsets and tested the potential role played by the AT in T-cell chemoattraction (Fig. 6). Migration of CD3+ splenocytes from STAT4−/− mice and controls was measured against conditioned media from STAT4−/− or C57Bl AT explants cultured for 24 h (Fig. 6A). When migration was measured toward conditioned media from STAT4−/− AT explants, no difference in the CD4+ migration was found among groups. However, the percentage of CD8+ cells that migrated from the STAT4−/− CD3+ cell pool was significantly lower by approximately twofold compared with the C57Bl controls (Fig. 6A). Similar results were obtained when migration was measured against the conditioned media from C57Bl6 AT explants (Fig. 6A). Also, for both the STAT4−/− mice and the controls, the percentage of cells that migrated against the C57Bl6 conditioned media was significantly higher by 1.5-fold to 2.5-fold compared with STAT4−/− conditioned media. This result indicates that CD8+ cells from STAT4−/− mice have a lower migratory capacity and that AT from C57Bl controls provides more potent chemoattractants. Also, the number of STAT4−/− CD8+ cells that migrated against a positive control was lower compared with CD4+ cells and also significantly lower compared with the wild-type CD8+ cells (Fig. 6A). These results indicate a possible intrinsic migratory defect in the CD8+ but not in the CD4+ cells. In a separate experiment, the migration of individual CD8+ and CD4+ cells was assessed against conditioned media from control mice (Fig. 6B). The number of PE+ cells in the lower chamber, determined by flow cytometry (Fig. 6B, bottom panels) showed a strong reduction in migration of CD8+ cells from STAT4−/− mice versus controls. To determine possible mechanisms explaining the lower migratory capacity of the CD8+ versus CD4+ cells in STAT4−/− mice, we measured the expression of 84 chemokines and receptors by real-time PCR gene arrays (Fig. 6C). In CD4+ cells, only nine genes were significantly changed and only two genes were downregulated/upregulated by more than twofold, indicating minor differences between the two groups. However, in the CD8+ cells none of the genes were upregulated in the STAT4-deficient mice, and 17 genes were significantly (P < 0.05) downregulated between 5.8-fold and 1.7-fold. Importantly, two of the most downregulated genes, Ccr3 (5.8-fold) and Ccr5 (2.8-fold), are both RANTES receptors (Fig. 6C). Collectively, gene expression data suggest a robust downregulation of a number of chemokine receptors, along with other relevant genes indirectly involved in chemotaxis, such as Tlr4 and Hif1a, in CD8+ cells from STAT4-deficient mice versus controls. Only minor changes in gene expression were determined for the CD4+ cells.
FIG. 6.
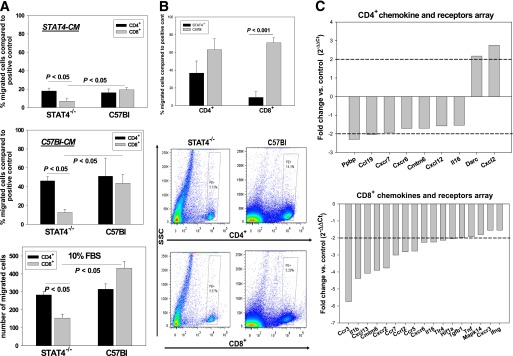
Migration and profiling of the nuclear factor-κB target genes in T cells from STAT4−/−C57Bl6 mice and wild-type controls on an HFD. A: In vitro migration of the splenic CD3+ T cells isolated from knockout and wild-type controls was assessed against conditioned media from AT explants. The AT explants from either STAT4−/−C57Bl6 mice or C57Bl6 controls after 16 weeks of HFD were cultured for 24 h, and conditioned media (STAT4-CM or C57Bl-CM) was used to determine the migratory capacity of splenic CD3+ T cells purified using immunoaffinity-based magnetic separation. Migration of each of the STAT4−/− and C57Bl6 CD3+ cells was determined against each of the homologous AT explant–conditioned media or against the conditioned media of the alternative group. After migration, cells in the lower chamber were counted and phenotyped for CD4+ and CD8+ markers using flow cytometry. Migration was also determined against a positive control of 10% FBS (bottom panel). Results are expressed as the percentage of migrated cells compared with positive control after subtraction of respective negative controls against 1% FBS. Each experiment was performed in duplicate, and results are the mean ± SEM from four to six independent experiments. B: In a separate experiment, CD4+ and CD8+ cells were prepared from splenocytes of STAT4-deficient and control mice on an HFD. Cells were immunoseparated using positive selection with magnetic beads and were labeled with phycoerythrin. The individual migration of CD4+ or CD8+ cells was determined by flow cytometry. Migration was expressed as in A, and conditioned media from AT explants of control mice were used. Each experiment was performed in duplicate, and results are the mean ± SEM from three independent experiments. C: CD4+ and CD8+ cells from STAT4-deficient and control mice on an HFD were immunoseparated using magnetic beads. Cells were analyzed for the expression of 84 chemokines and receptors by real-time PCR using the pathway-focused gene arrays from SABiosciences. Results are expressed as the fold change in STAT4-deficient cells versus controls. Only the significant changes (P < 0.05) are shown. Results are from n = 3 samples per group. SSC, side-scattered plot.
Role of STAT4 deficiency in the hematopoietic compartment on insulin sensitivity and AT immune composition.
Because STAT4 is expressed in both hematopoietic cells and in adipocytes, it will be helpful to understand the individual contribution of STAT4 in each of these compartments to the observed insulin sensitive phenotype. To address this, we used Rag1-null mice, which lack T cells, NKT cells, and B cells. Because STAT4 is expressed in T cells, adoptive transfer of either STAT4-deficient or STAT4-sufficient T cells would address the role of STAT4 in these cell types. To this purpose, we examined the phenotype of Rag1−/− mice reconstituted with splenocytes from either STAT4-deficient (Rag1/STAT4−/−) or wild-type control mice (Rag1/C57Bl), after 15 weeks on an HFD (Fig. 7). The Rag1/STAT4−/− mice had lower fasting insulin levels and HOMA of insulin resistance (HOMA-IR) compared with the Rag1/C57Bl mice, but similar levels of fasted plasma glucose (Fig. 7A). Also, the Rag1/STAT4−/− mice had a slightly lower body weight (by ∼6%) compared with controls, and no difference in average food intake. The ITT showed improved glucose disposal in the Rag1/STAT4−/− mice, but the GTT showed no differences (Fig. 7A). In addition, differences were found in AT immune composition. The largest change was in the percentage of CD3+CD8+ cells, which was significantly decreased in Rag1/STAT4−/− mice, and in the percentage of F4/80+CD206+ macrophages, which was significantly increased (Fig. 7B). These data suggest that the STAT4 deficiency in this hematopoietic compartment is only in part responsible for the improved insulin sensitivity but explains some of the immune changes observed in the global knockout.
FIG. 7.
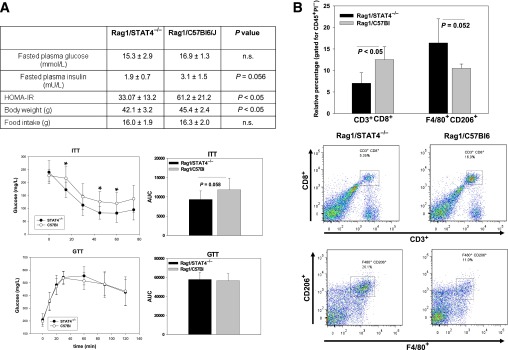
Metabolic phenotype and immune cell composition in AT of Rag1-null mice reconstituted with splenocytes from STAT4-deficient mice and C57Bl6 controls. Eight-week-old Rag1-null mice were injected with splenocytes from either STAT4-deficient mice or wild-type controls and placed on HFD for 15 weeks. A: Fasted plasma glucose and insulin levels, HOMA-IR, body weight, and food intake were measured along with intraperitoneal ITTs and GTTs. AUCs were calculated, and the results represent the mean ± SEM from n = 9–10 mice per group. B: SVF was isolated from AT, and the cells were fluorescently labeled with CD45, CD3, CD8, CD206, and F40/80 anti-mouse antibodies. Cells were gated for CD45 positivity and selected for viability using propidium iodide (PI) exclusion. Representative plots from each group for CD3+CD8+ cells and for CD206+F4/80+ cells are shown in the bottom plots. Results represent the mean ± SEM from 4–5 mice per group. n.s., nonsignificant.
DISCUSSION
Evolving studies suggest that the IL-12 pathway plays a role in AT and islet inflammation in DIO (15,23). STAT4 is the major transcription factor that mediates IL-12–dependent effects on a large number of target genes (8). Many of these gene targets represent cytokines and chemokines that have been identified as important mediators of inflammation in AT, including IFN-γ, IL-1r1, IL-18r1, and TNF-α (24). However, whether STAT4 has a critical role in the initiation and/or maintenance of a proinflammatory response in AT in DIO and insulin resistance was not previously investigated.
Our results indicate that global STAT4 deficiency in C57Bl6/J mice reduced AT inflammation, prevented insulin resistance, and improved glucose homeostasis in DIO. We also showed that selective STAT4 deficiency in certain cells of the hematopoietic compartment is in part responsible for the phenotype. The most notable effect of STAT4 deficiency was on the peripheral glucose clearance in response to an in vivo insulin challenge. The improved peripheral glucose clearance, albeit in the absence of an insulinotropic effect, may explain the reduced fasting glucose and the lower HOMA index in STAT4−/− mice compared with controls. This protective effect in STAT4−/− mice occurs independently of fat accumulation. Both the obesity index and the visceral adiposity are comparable between the STAT4−/− and wild-type mice. Interestingly, this phenotype, characterized by improved insulin sensitivity in the presence of obesity, was also reported for mice in which CD8+ cells, total CD3+ cells or NK cells were selectively and chronically depleted, suggesting that the cellular composition and phenotype rather than the total AT mass is a critical factor involved in insulin resistance during obesity (5,25). Also, we showed that in Rag1 mice with selective T-cell and B-cell STAT4 deficiency the peripheral glucose disposal was improved in response to insulin but the fasted glucose was not improved, suggesting that adipocyte STAT4 deficiency likely also plays an important role.
There were clear differences in both the adipocyte phenotype and the immune cell infiltration in AT of STAT4−/− global mice. STAT4−/− mice had a significantly lower average size of adipocytes compared with controls. Lower adipocyte size is reportedly correlated with better insulin sensitivity and reduced production of proinflammatory adipokines (26–28). We showed that insulin signaling is significantly improved in the AT of STAT4−/− mice upon in vivo insulin stimulation. However, deletion of STAT4 did not modulate in vitro insulin stimulation. This result suggests that the local inflammatory environment, such as exposure to proinflammatory cytokines resulting from immune cell infiltration of AT, may have an important role. Therefore, STAT4-deficient adipocytes may be protected from exposure to proinflammatory cytokines, such as IL-12 or IFN-γ. This may also explain the improved insulin-signaling response in STAT4-deficient mice in the total AT upon in vivo insulin stimulation. We also identified significant expressional differences in a number of genes with key roles in insulin signaling between the STAT4−/− adipocytes and controls. In particular, we found reduced expression of SOCS3 and Jak2 in STAT4−/− adipocytes versus controls. SOCS3 was reportedly causal for insulin resistance in obesity through the inhibition of the IR tyrosine phosphorylation mediated by Jak2/STAT3 and via other mechanisms (29). Another effect of STAT4 deficiency in adipocytes was a seemingly improved overall metabolic response to fasting, based on expressional changes in phosphoenolpyruvate carboxykinase, pyruvate dehydrogenase kinase, and Acaca, which suggest reduced re-esterification of fatty acids, glucose utilization, and lipogenesis. Collectively, the improved insulin signaling, metabolic adaptations, and adipocyte size in STAT4-deficient AT and adipocytes likely result in reduced local inflammation.
Recent data showed that inflammatory signals from adipocytes are key players in inflammation-induced insulin resistance in obesity (30,31). Indeed, our data also showed reduced expression of proinflammatory cytokines and chemokines in adipocytes of lean and obese STAT4−/− mice compared with controls. Notably, CCL5 and CXCL10 expression were significantly reduced in adipocytes. Whether this is a direct effect that involves STAT4 signaling in adipocytes or is due to the reduced local inflammatory milieu remains to be established. Interestingly, STAT4 deficiency reportedly reduced chemokine production, including CCL5, in the lungs of mice with allergic inflammation (32), suggesting that STAT4 signaling is key to triggering and maintaining local inflammatory responses via chemokine production.
We report decreased numbers of CD3+ cells in both lean and obese STAT4−/− AT, which may be explained in part by lower recruitment due to reduced chemokine production. Previous studies indicated that expansion of CD3+ subsets in response to IL-12 stimulation is dependent on STAT4 (33). This opens the possibility that reduced CD3+ cells in AT may also be the result of reduced local proliferation as a consequence of STAT4 deficiency. We found a reduced percentage of CD8+ cells but not CD4+ cells in the AT of STAT4−/− mice. Interestingly, analysis of T-cell number and subsets in the spleen showed increased numbers of CD3+ cells in STAT4−/− mice and no difference in the relative percentage of CD4+ and CD8+ cells among groups. Also, no differences were detected in the mesenteric/inguinal lymph nodes. These results suggest that the proliferation of CD3+ cells is not impaired in the STAT4-deficient mice, at least in the secondary lymphoid tissues, but the migratory capacity of these cells seems to be rather affected. In support of this conclusion, in an in vitro migration experiment against AT-conditioned media, the chemotaxis of STAT4−/− CD8+ cells, but not of the CD4+ cells, was significantly reduced compared with cells from controls. Also, gene expression analysis for 84 chemokines and receptors in CD4+ and CD8+ cells from STAT4-deficient mice and controls revealed minor differences in the CD4+ cells but significant changes in the CD8+ cells. In particular, the expression of two of the Ccl5 receptors (Ccr3 and Ccr5) were significantly downregulated in STAT4−/−CD8+ cells. STAT4 is involved in the regulation of several chemokine receptors that are preferentially expressed by the Th1 cells—for example, CXCR3 and CCR5 (34)—and our gene array data indicating reduced expression for both of these receptors suggest a STAT4-dependent transcriptional effect. Several studies showed that Ccl5 is a critical player in insulin resistance in obesity both in mice and humans (35–37), and our results show that STAT4 deficiency results in the reduced expression of Ccl5 in both adipocytes and islets, suggesting that Ccl5/Ccr3/5 is one of the key pathways underlying reduced inflammation and improved insulin sensitivity in STAT4 deficiency. It is therefore possible that a combined effect of reduced chemokine production in AT and the reduced migratory ability of CD8+ cells, due to a reduction in the chemokine receptors, results in the reduced numbers of CD8+ cells recruited in the AT of STAT4−/− mice. It was shown previously that CD8+ cells in AT play a critical role in local inflammation and depletion of this cell population, resulted in improved insulin sensitivity in DIO mice (5). In addition to the migratory changes, the increased expression of GATA3 and IL-10 in AT of STAT4−/− mice suggests a Th2-biased polarization of the lymphocytes in AT. Indeed, it was previously shown that STAT4-deficient lymphocytes have a propensity toward the formation of Th2 cells (11); however, the phenotype of the lymphocytes residing in AT in STAT4−/− mice was not previously described. In addition, we have found a robust increase of the CD25+ regulatory T cells in AT of STAT4−/− mice in response to an HFD. T-regulatory cells were shown to improve insulin signaling and glucose disposal in DIO mice (38), whereas obesity in mice and humans leads to a depletion of T-regulatory cells in the visceral AT (38,39). Interestingly, STAT4 was shown to limit the development of T-regulatory cells (13), which may have an impact on the reduced numbers in AT in obesity. Therefore, STAT4 deficiency may prevent the reduction in T-regulatory cells in response to increased obesity-induced inflammation, therefore contributing to the maintenance of an insulin-responsive milieu in AT and adipocytes.
We also found reduced levels of circulating leptin in both lean and obese STAT4−/− mice compared with controls. Interestingly, leptin is known to directly affect T cells via signaling through the long form of the leptin receptor and to induce proinflammatory Th1 responses (40,41). Therefore, it is plausible that lower leptin levels in STAT4−/− mice may contribute to the lower T-cell number and Th1 differentiation in AT.
Another important player in AT inflammation in obesity is the infiltration and the predominant M1 proinflammatory phenotype of macrophages (42,43). STAT4−/− mice have reduced numbers of macrophages in the perigonadal depot and a larger percentage of the CD206+ cells, suggesting a bias toward an M2 polarization. M2 macrophage polarization occurs in AT under various conditions, including upon PPAR-γ activation, and we indeed found increased PPAR-γ expression in SVF from STAT4−/− mice (44,45). Previous reports showed that the M2 macrophages secrete less IL-12 compared with the M1 counterparts (46), which, in conjunction with STAT4 deficiency, would result in reduced levels of inflammatory cytokines produced in AT. Interestingly, we have found reduced expression of the retinol-binding protein-4 in STAT4-deficient adipocytes. The latter correlates highly with insulin resistance and induces expression of proinflammatory cytokines in macrophages via Toll-like receptor 4 (47). Also, reduced IFN-γ production in response to IL-12 may account for the reduced AT inflammation and insulin resistance. A recent study shows that, in human adipocytes, IFN-γ signaling via STAT1 impairs insulin signaling, lipid storage, and differentiation (31). Also, Ifng−/− obese mice are protected from AT inflammation and insulin resistance (48). However, in the STAT4−/− mice, the CD4+ and CD8+ cells are still capable of synthesizing IFN-γ, indicating that the beneficial effects on glucose homeostasis and inflammation may be only in part attributed to a reduction in IFN-γ production.
In conclusion, STAT4 deficiency has direct effects on adipocyte inflammation, insulin signaling, macrophage polarization, and CD8+ migration, which all collectively lead to reduced inflammation in AT and improved insulin sensitivity in obesity. Although we did identify a few candidate genes that may explain the beneficial effects of STAT4 deficiency on adipocyte function and inflammation, future studies will be needed to elucidate the STAT4 transcriptome in AT and to understand the comprehensive functional implications of STAT4 modulation. Our data suggest that the reduction of STAT4 activation may be a novel approach to ameliorate AT inflammation and improve the metabolic complications of obesity.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by National Institutes of Health grants R15-HL-114062 to A.D.D., R01-AI-45515 to M.H.K., and RO1-HL-112605 to J.L.N.
A.D.D. designed the experiments, conducted the in vitro migration experiments, participated in the metabolic phenotyping, performed most of the data analysis, and wrote the manuscript. E.V.G. participated in the design and analysis of flow cytometry data and critically read the manuscript. Q.M. and M.H. performed the metabolic phenotyping, glucose-stimulated insulin secretion in pancreatic islets, real-time PCR, ELISA, and flow cytometry. S.M.A. and B.A.H. performed some of the real-time PCR and the gene array. M.J.B. performed some of the flow cytometry experiments. K.M. did the Western blotting experiments. M.H.K. provided the STAT4-null mice and critically read the manuscript. J.L.N. offered critical logistical advice on experimental design and data interpretation and critically read and provided input for the manuscript. A.D.D. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
No potential conflicts of interest relevant to this article were reported.
Parts of this work were published in abstract form in Arterioscler Thromb Vasc Biol 2010;30:e195.
The authors thank Dr. Maggie Morris, Department of Internal Medicine, Eastern Virginia Medical School, for help with tail vein injections in Rag1-null mice; and Kendall Leone, Department of Internal Medicine, Eastern Virginia Medical School, for the assistance with the isolation of pancreatic islets.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1275/-/DC1.
See accompanying commentary, p. 4002.
REFERENCES
- 1.Mori MA, Liu M, Bezy O, et al. A systems biology approach identifies inflammatory abnormalities between mouse strains prior to development of metabolic disease. Diabetes 2010;59:2960–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol 2008;3:545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duffaut C, Galitzky J, Lafontan M, Bouloumié A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun 2009;384:482–485 [DOI] [PubMed] [Google Scholar]
- 4.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 2008;28:1304–1310 [DOI] [PubMed] [Google Scholar]
- 5.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914–920 [DOI] [PubMed] [Google Scholar]
- 6.Darnell JE., Jr STATs and gene regulation. Science 1997;277:1630–1635 [DOI] [PubMed] [Google Scholar]
- 7.Imada K, Leonard WJ. The Jak-STAT pathway. Mol Immunol 2000;37:1–11 [DOI] [PubMed] [Google Scholar]
- 8.Good SR, Thieu VT, Mathur AN, et al. Temporal induction pattern of STAT4 target genes defines potential for Th1 lineage-specific programming. J Immunol 2009;183:3839–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunol Res 2005;31:231–242 [DOI] [PubMed] [Google Scholar]
- 10.Chang HC, Han L, Goswami R, et al. Impaired development of human Th1 cells in patients with deficient expression of STAT4. Blood 2009;113:5887–5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 1996;382:174–177 [DOI] [PubMed] [Google Scholar]
- 12.Thierfelder WE, van Deursen JM, Yamamoto K, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 1996;382:171–174 [DOI] [PubMed] [Google Scholar]
- 13.O’Malley JT, Sehra S, Thieu VT, et al. Signal transducer and activator of transcription 4 limits the development of adaptive regulatory T cells. Immunology 2009;127:587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harp JB, Franklin D, Vanderpuije AA, Gimble JM. Differential expression of signal transducers and activators of transcription during human adipogenesis. Biochem Biophys Res Commun 2001;281:907–912 [DOI] [PubMed] [Google Scholar]
- 15.Chakrabarti SK, Wen Y, Dobrian AD, et al. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am J Physiol Endocrinol Metab 2011;300:E175–E187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novelli EL, Diniz YS, Galhardi CM, et al. Anthropometrical parameters and markers of obesity in rats. Lab Anim 2007;41:111–119 [DOI] [PubMed] [Google Scholar]
- 17.Simson EL, Gold RM. The Lee Obesity Index vindicated? Physiol Behav 1982;29:371–376 [DOI] [PubMed] [Google Scholar]
- 18.Brüning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 1997;88:561–572 [DOI] [PubMed] [Google Scholar]
- 19.Nunemaker CS, Chen M, Pei H, et al. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab 2008;295:E1065–E1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole BK, Keller SR, Wu R, Carter JD, Nadler JL, Nunemaker CS. Valsartan protects pancreatic islets and adipose tissue from the inflammatory and metabolic consequences of a high-fat diet in mice. Hypertension 2010;55:715–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobrian AD, Ma Q, Lindsay JW, et al. Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. Am J Physiol Endocrinol Metab 2011;300:E410–E421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase Products Reduce Insulin Secretion and beta-Cell Viability in Human Islets. J Clin Endocrinol Metab 2010;95:887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafson B. Adipose tissue, inflammation and atherosclerosis. J Atheroscler Thromb 2010;17:332–341 [DOI] [PubMed] [Google Scholar]
- 25.Ohmura K, Ishimori N, Ohmura Y, et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol 2010;30:193–199 [DOI] [PubMed] [Google Scholar]
- 26.Liu A, Sonmez A, Yee G, et al. Differential adipogenic and inflammatory properties of small adipocytes in Zucker Obese and Lean rats. Diab Vasc Dis Res 2010;7:311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundgren M, Svensson M, Lindmark S, Renström F, Ruge T, Eriksson JW. Fat cell enlargement is an independent marker of insulin resistance and “hyperleptinaemia”. Diabetologia 2007;50:625–633 [DOI] [PubMed] [Google Scholar]
- 28.Lönn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J 2010;24:326–331 [DOI] [PubMed] [Google Scholar]
- 29.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 2004;24:5434–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakrabarti SK, Cole BK, Wen Y, Keller SR, Nadler JL. 12/15-Lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity (Silver Spring) 2009;17:1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SJ, Kim JY, Nogueiras R, et al. PKCzeta-regulated inflammation in the nonhematopoietic compartment is critical for obesity-induced glucose intolerance. Cell Metab 2010;12:65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raman K, Kaplan MH, Hogaboam CM, Berlin A, Lukacs NW. STAT4 signal pathways regulate inflammation and airway physiology changes in allergic airway inflammation locally via alteration of chemokines. J Immunol 2003;170:3859–3865 [DOI] [PubMed] [Google Scholar]
- 33.Yoo JK, Cho JH, Lee SW, Sung YC. IL-12 provides proliferation and survival signals to murine CD4+ T cells through phosphatidylinositol 3-kinase/Akt signaling pathway. J Immunol 2002;169:3637–3643 [DOI] [PubMed] [Google Scholar]
- 34.Kim SH, Gunst KV, Sarvetnick N. STAT4/6-dependent differential regulation of chemokine receptors. Clin Immunol 2006;118:250–257 [DOI] [PubMed] [Google Scholar]
- 35.Herder C, Haastert B, Müller-Scholze S, et al. Association of systemic chemokine concentrations with impaired glucose tolerance and type 2 diabetes: results from the Cooperative Health Research in the Region of Augsburg Survey S4 (KORA S4). Diabetes 2005;54(Suppl. 2):S11–S17 [DOI] [PubMed] [Google Scholar]
- 36.Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007;115:1029–1038 [DOI] [PubMed] [Google Scholar]
- 37.Huber J, Kiefer FW, Zeyda M, et al. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab 2008;93:3215–3221 [DOI] [PubMed] [Google Scholar]
- 38.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009;15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS One 2011;6:e16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998;394:897–901 [DOI] [PubMed] [Google Scholar]
- 41.Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C. Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J Immunol 2006;176:7745–7752 [DOI] [PubMed] [Google Scholar]
- 42.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011;121:2111–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 2007;6:137–143 [DOI] [PubMed] [Google Scholar]
- 45.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007;447:1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MJ, Wu Y, Fried SK. Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care 2010;13:371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Norseen J, Hosooka T, Hammarstedt A, et al. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol Cell Biol 2012;32:2010–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res 2008;103:467–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.