

Abstract
Objective
Mutations of the receptor tyrosine kinase Kit occur in several human and canine cancers. While Kit inhibitors have activity in the clinical setting, they possess variable efficacy against particular forms of mutant Kit and drug resistance often develops over time. Inhibitors of heat shock protein 90 (HSP90), a chaperone for which Kit is a client protein, have demonstrated activity against human cancers and evidence suggests they downregulate several mutated and imatinib-resistant forms of Kit. The purpose of this study was to evaluate a novel HSP90 inhibitor, STA-9090, against wild-type (WT) and mutant Kit in canine bone marrow–derived cultured mast cells (BMCMCs), malignant mast cell lines, and fresh malignant mast cells.
Materials and Methods
BMCMCs, cell lines, and fresh malignant mast cells were treated with STA-9090, 17-AAG, and SU11654 and evaluated for loss in cell viability, cell death, alterations in HSP90 and Kit expression/signaling, and Kit mutation. STA-9090 activity was tested in a canine mastocytoma xenograft model.
Results
Treatment of BMCMCs, cell lines, and fresh malignant cells with STA-9090 induced growth inhibition, apoptosis that was caspase-3/7–dependent, and downregulation of phospho/total Kit and Akt, but not extracellular signal-regulated kinase (ERK) or phosphoinositide-3 kinase (PI-3K). Loss of Kit cell-surface expression was also observed. Furthermore, STA-9090 exhibited superior activity to 17-AAG and SU11654, and was effective against malignant mast cells expressing either WT or mutant Kit. Lastly, STA-9090 inhibited tumor growth in a canine mastocytoma mouse xenograft model.
Conclusions
STA-9090 exhibits broad activity against mast cells expressing WT or mutant Kit, suggesting it may be an effective agent in the clinical setting against mast cell malignancies.
Kit is a receptor tyrosine kinase that plays an important role in hematopoiesis, melanogenesis, and mast cell development [1,2]. Dysregulation of Kit is found in a variety of human cancers, primarily through mutation in either the catalytic or juxtamembrane (JM) domains [1,3–5]. Such mutations result in ligand-independent activation of Kit, inducing constitutive signaling that promotes uncontrolled proliferation and survival [6,7]. Cancers in which Kit mutations have been identified include systemic mastocytosis (Asp816Tyr) [7,8], gastrointestinal stromal tumors (GISTs) (primarily JM domain mutations) [3,4], and acute myeloid leukemia (Asp816Tyr and exon 8 mutations) [5,9].
Kit mutations also occur in spontaneous canine tumors [10,11]. Mast cell tumors (MCTs) are the most common skin tumor in dogs and 9% to 30% of malignant MCTs carry internal tandem duplications (ITDs) in the Kit JM domain that induce constitutive phosphorylation [11–14]. A clinical trial of a small-molecule Kit inhibitor, SU11654 (Pfizer, Inc., LaJolla, CA, USA) in dogs with MCTs demonstrated Kit to be a relevant target for therapeutic intervention in this disease [15,16]. Response to therapy was related to the presence of Kit mutation and Kit target modulation could be demonstrated in MCTs after a single dose of SU11654 [15]. Therefore, canine MCTs are an excellent model in which to evaluate the safety and efficacy of novel Kit inhibitors.
Several small-molecule inhibitors of Kit are currently used in the clinical setting, including imatinib [17], sunitinib [18], and dasatinib [19,20]. However, resistance to therapy often develops over time, typically involving mutations that affect drug binding and or circumvent drug activity [21,22]. Therefore, new therapeutic strategies are needed that are less susceptible to development of resistance from additional mutations in the drug target. Heat shock protein 90 (HSP90) is a cellular chaperone responsible for a number of client proteins, including Met, B-Raf, p53, Akt, and Kit [23,24]. While mutations of HSP90 have not yet been identified, higher levels of active HSP90 are often present in cancer cells relative to normal cells [25–27]. Adequate HSP90 function is required by tumor cells to maintain appropriate client protein expression necessary for proliferation and survival [27,28]. Given its critical role, significant effort has been directed at developing HSP90 inhibitors. Geldanamycin, a benzoquinone ansamycin antibiotic and its derivatives, 17-AAG and 17-DMAG, bind HSP90 and prevent stabilization of client proteins, ultimately resulting in their degradation [29–31]. 17-AAG has activity in several murine models of cancer, including breast [32], prostate [33], and melanoma [34], and clinical trials have demonstrated some activity in humans [35–37].
Geldanamycin and its derivatives suffer from limitations, including low solubility and side effects, such as hepatotoxicity. Additionally, they are substrates for the P-glycoprotein export pump important in multidrug resistance. STA-9090 (Synta Pharmaceuticals Corp., Lexington, MA, USA) is a novel triazolone compound unrelated to geldanamycin that is a potent inhibitor of HSP90 and binds in the adenosine triphosphate (ATP)–binding domain of the HSP90 N-terminus. STA-9090 induces the degradation of multiple HSP90 client proteins, the killing of a wide variety of human cancer cell lines at low nanomolar concentrations in vitro, and has shown potent anticancer activity in human xenograft tumor models (unpublished data, Synta). The purpose of this study was to evaluate the activity of STA-9090 against wild-type (WT) and mutant Kit expressing canine malignant mast cell lines and MCTs, and to compare this to the activity of a previously established small-molecule Kit inhibitor, SU11654 [14].
Materials and methods
Reagents
STA-9090 and 17-AAG were provided by Synta Pharmaceuticals Corp. SU11654 was provided by Pfizer, Inc. OSU-03012 was provided by W.C. Kisseberth (College of Veterinary Medicine, Ohio State University, Columbus, OH, USA). Antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA; anti-p-Kit Tyr719, phosphorylated extracellular signal-regulated kinase [ERK], p-Akt Thr308 and Ser473, and total ERK), BD Biosciences (Franklin Lakes, NJ, USA; anti-Akt, anti-p85 phosphoinositide-3 kinase (PI3K), canine CD34, CD117-phycoerythrin [PE] and poly (ADP-ribose) polymerase [PARP]) Santa Cruz Biotechnology (Santa Cruz, CA, β-actin, and HSP90 α/β), Calbiochem (San Diego, CA, USA; anti-Kit), and Stressgen (San Diego, CA, USA; anti-HSP70).
Cell culture
The C2 and BR canine mastocytoma cell lines were maintained as described previously [14]. Canine bone marrow–derived mast cells (BMCMCs) were generated as described previously [38,39]. Primary neoplastic mast cells were collected from three dogs at Ohio State University. Tumor was digested in 1 mL Vercene (Gibco, Carlsbad, CA, USA), undigested connective tissue was removed by filtration, and cells were counted and stained with Wright's-Giemsa. In each sample approximately 70% to 90% of cells were mast cells.
Analysis of cell viability and growth
Canine BMCMCs, fresh neoplastic mast cells, C2 and BR cells were plated in 96-well plates in 150 uL and treated with STA-9090, 17-AAG, or OSU-03012 at various concentrations or SU11654 (100 nM, the established IC50 for Kit [14]) in the presence or absence of 50 ng/mL recombinant canine stem cell factor (rcSCF). Cell viability was evaluated using the WST-1 assay (Roche, Indianapolis, IN, USA) or the CCK-8 kit (Dojindo Molecular Technologies, Gaithers burg, MD, USA).
Analysis of cell cycling and apoptosis
C2 and BR cells (5 × 105) were cultured in complete medium and 1 × 105 canine BMCMCs were cultured in Stemline II with 50 ng/mL rcSCF and left untreated (dimethyl sulfoxide [DMSO]) or treated with STA-9090 or SU11654. Apoptosis was assessed at 24 and 48 hours using the Annexin-V–PE/7-amino-actinomycin D kit (BD Biosciences). Cell cycling was analyzed at 24 and 48 hours using propidium iodide staining as described previously [38]. To measure caspase-3/7 activity, C2 cells (1.5 × 104) were treated with STA-9090 for 24 hours and activity was detected by using SensoLyte AMC caspase-3/7 assay Kit (AnaSpec, San Jose, CA, USA).
Flow cytometry
Cells were collected and incubated in 100 μL BD Cytofix/Cytoperm solution (BD Biosciences) washed in a saponin-containing buffer then incubated with anti-HSP90α/β or anti-PI3K followed by fluorescein isothiocyanate–conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA, USA). Changes in Kit expression following treatment with either STA-9090 or SU11654 for 24 hours were assessed directly by flow cytometry.
Western blotting
C2 and BR cells were serum starved for 2 hours and treated with STA-9090 for 2 or 24 hours, then collected, lysed, and equal amounts of protein were separated by 6 % (Kit) or 10 % (ERK, Akt, PARP, HSP90, and HSP70) sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were blocked in 5% bovine serum albumin/tris-buffered saline Tween-20, then incubated with primary phospho-antibody overnight at 4°C, washed, incubated with secondary antibody and developed using SuperSignal West Dura (Pierce, Rockford, IL, USA). Blots were then stripped, blocked, and reprobed for total protein. To evaluate the association of Kit with HSP90 following STA-9090 treatment, C2 cells (30 × 106) were treated with DMSO or 100 nM STA-9090 for 8 hours. HSP90 was immunoprecipitated (anti-HSP90, Cell Signaling) followed by Trublot anti-rabbit agarose beads (E-Biosciences). After sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE), Western blotting was performed for HSP90 and Kit. Direct Western blotting on protein lysates was also performed to assess total protein levels of Kit and HSP90 in C2 cells after treatment with STA-9090 for 8 hours.
Reverse transcriptase polymerase chain reaction analysis of cKit expression and cKit sequencing
C2 and BR cells were treated with STA-9090 or SU11654 for 2 or 24 hours and RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA). cDNA was made from 2 μg total RNA by Superscript III kit (Invitrogen). cKit and glyceraldehyde phosphate dehydrogenase mRNA were detected and cKit sequencing was performed using primer sets in Table 1.
Table 1. Primers for reverse transcriptase polymerase chain reaction and sequencing.
| Kit primers | Primer sequences |
|---|---|
| PE1 | 5′-CCC ATG TAT GAA GTA CAG TGG AAG-3′ |
| PE2 | 5′ GTT CCC TAA AGT CAT TGT TAC ACG-3′ |
| GAPDH-F | 5′-ACC ACA GTT CCA TGC CAT CAC-3′ |
| GAPDH-R | 5′-TCC ACC ACC CTG TTG CTG TA-3′ |
| P1 | 5′-GAG GAG ATC AAT GGA AAC AAT TAT G-3′ |
| P5 | 5′-CAT GGC CGC ATC CGA CTT AAT CAG-3′ |
| P10 | 5′-GCA ATT ACA CGT GCA CCA AC-3′ |
| cKit 1110R | 5′-CTG ATA TTA CTT TCA TTG TCA G-3′ |
| cKit 582F | 5′-GCA GGA CGG TGC TGT CCA AG-3′ |
| cDNAR2 | 5′-GCT TCA CAC ATC TTc GTG TAC CAG CAG AGG CTG GG-3′ |
Xenograft tumor model
Female severe combined immune-deficient (SCID) mice (Charles River Laboratories, Wilmington, MA, USA) were maintained in an specific pathogen free environment and all procedures were approved by the Institutional Animal Care and Use Committee. C2 cells were suspended at 5 to 10 × 107 cells/mL in medium/50% Matrigel (BD Biosciences), and 0.1 mL was injected into the flanks. Tumors grew to 100 to 200 mm3 (day 8) then animals were randomized into treatment groups so tumor volumes were approximately 150 mm3/group. Tumor volumes (V) were calculated by caliper measurement of width (W), length (L), and thickness (T): V = 0.5236(LWT). STA-9090 stock solution in DMSO was diluted 1:10 with 20% Cremophor RH 40. The final formulation contained 10% DMSO, 18% Cremophor, 3.6% dextrose, 68.4% water, and STA-9090. Animals were treated by tail vein injection at 10 mL/kg and body weights were monitored daily.
Results
HSP90 is not overexpressed in malignant canine mast cells
We evaluated mastocytoma cell lines expressing Kit mutations (BR, point mutation in JM domain; C2, ITD in JM domain) [14], fresh primary malignant mast cells and normal canine BMCMCs for HSP90 expression by intracellular flow cytometry staining. All cells, expressed similar levels of HSP90 (Fig. 1). Based on preliminary RNA microarray data, HSP90 RNA expression was not significantly different among canine BMCMCs, grade I and grade III MCTs (data not shown), suggesting that upregulation of HSP90 expression is not a common feature of malignant MCTs in dogs.
Figure 1.
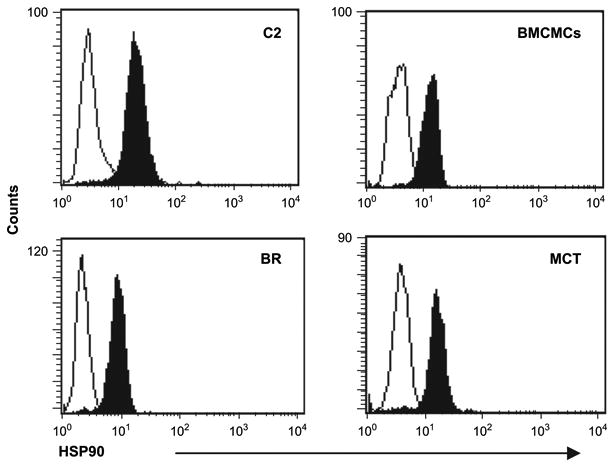
Heat shock protein 90 (HSP90) expression is similar in normal and neoplastic canine mast cells. Canine bone marrow–derived mast cells (BMCMCs), mastocytoma cell lines (C2 and BR), and neoplastic mast cells freshly purified from a spontaneous Mast cell tumor (MCT) were washed, fixed, permeabilized, labeled with a control antibody (open histogram), or labeled with anti-HSP90 antibody (solid histogram) and evaluated by flow cytometry.
STA-9090 induces apoptosis of malignant canine mast cell lines
To evaluate the effects of HSP90 inhibition on canine malignant mast cells, we cultured the C2, BR, and canine BMCMCs with STA-9090 or SU11654 (a small-molecule inhibitor of Kit [14]) and assayed cell viability. Both the C2 line and BMCMCs exhibited significant growth inhibition in the presence of SU11654, while the BR line was somewhat resistant (Fig. 2A). However, C2, BR, and canine BMCMCs were all sensitive to STA-9090 at concentrations as low as 10 nM, with loss of cell viability observed after 24 hours of treatment. Additionally, we compared the activity of STA-9090 to 17-AAG in the C2 and BR cell lines and found that STA-9090 was active at significantly lower concentrations for both (Fig. 2B, STA-9090 IC50 19 nm (C2) and 4 nm (BR) vs 17-AAG IC50 948 nm (C2) and 44 nm (BR)).
Figure 2.

STA-9090 induces growth inhibition and apoptosis of neoplastic mast cells. (A) C2 (1 × 104), BR (1 × 105), and bone marrow–derived mast cell (BMCMC) (1 × 104) cells were washed and plated in 150-uL, 96-well plate along with indicated concentrations of STA-9090 or SU11654 (100 nM) for 1, 2, and 3 days in triplicate. Viability of the cells was determined using the WST-1 assay and the data is shown as a ratio of viable treated cells to viable control cells (treated with 0.01% dimethyl sulfoxide). One of four representative experiments is shown. (B) C2 (2 × 104), and BR (2 × 104) cells were washed and plated in a 96-well plate along with indicated concentrations of STA-9090 or 17-AAG for 72 hours in triplicate. Cell numbers were determined using the CCK-8 assay and the data is shown as a ratio of viable treated cells to viable control cells (treated with 0.01% dimethyl sulfoxide).
To determine whether the growth inhibition was due to induction of cell death, we cultured the C2 and BR lines and canine BMCMCs with STA-9090 or SU11654, and assessed the cells for apoptosis. As shown in Figure 3A, apoptosis was observed in all lines after treatment with 100 nM STA-9090 in a dose- and time-dependent manner. Similar results were obtained when cells were treated with SU11654, although the BR line was less sensitive to kinase inhibition than HSP90 inhibition. These data were confirmed with cell-cycle analysis using propidium iodide staining in which all cells showed significant cell death (sub-G1) following 24 and 48 hours of treatment with STA-9090 or SU11654 (Fig. 3B). The C2 and BR lines were then used to evaluate the ability of STA-9090 to induce caspase-mediated apoptosis. As shown in Figure 3C, cleaved PARP was detected 24 hours following treatment with STA-9090, while untreated cells maintained intact PARP. Cleaved PARP was also detected when C2 cells were treated with SU11654, but was less evident in BR cells treated with SU11654 concordant with our previous finding that BR cell line is somewhat less sensitive to Kit kinase inhibition. To confirm the role of caspase-3/7 in the observed apoptosis caspase activity was directly assessed in the C2 line after 24 hours of STA-9090 treatment (Fig. 3C).
Figure 3.
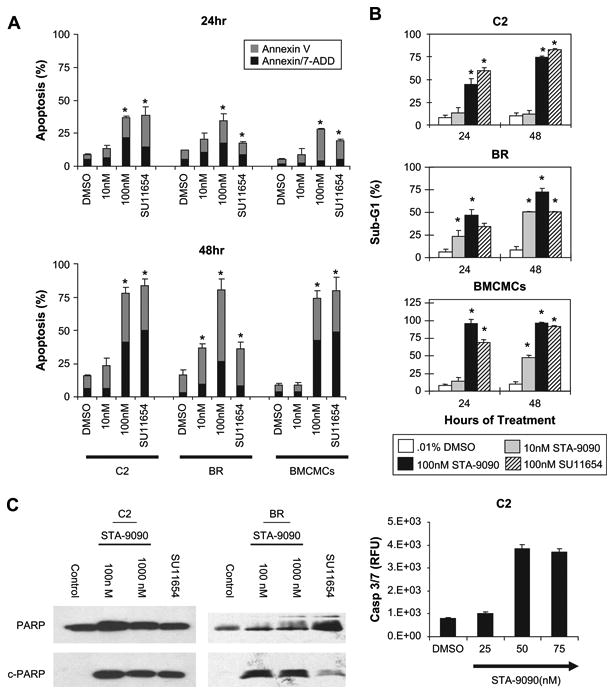
STA-9090 induces caspase-3/7 activity and death of normal and neoplastic mast cells death. (A) C2, BR, and bone marrow–derived mast cell (BMCMC) cells were washed and left untreated or treated with STA-9090 at the indicated concentrations or SU11654 (100 nM) in 24-well plates for 24 and 48 hours. Cells were then collected and labeled with Annexin V-phycoerythrin [PE] /7-amino-actinomycin D (7-AAD) and analyzed by flow cytometry. Percent of cells undergoing apoptosis and dead cells (Annexin-V single-positive and Annexin-V/7-AAD double-positive, respectively) is shown. *p < 0.05, t-test; error bars represent ± standard error of mean (SEM). (B) Cells were treated as in (A), collected, fixed in 70% ethanol, and incubated with propidium iodide staining solution. The percent of sub-G1 cells was assessed by flow cytometry at 24 and 48 hours of culture and is represented graphically. *p < 0.05, t-test; error bars represent ± SEM. (C) C2 and BR cells were cultured in complete medium with 0.01% dimethyl sulfoxide (DMSO), 100 nM or 1000 nM STA-9090, or 100 nM SU11654 for 24 hours. Cells were collected, lysed, and equal amounts of total protein were used for 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Following transfer, Western blotting for poly (ADP-ribose) polymerase (PARP) was performed. Intact PARP is 113 kD and cleavage PARP is approximately 25 kD. To confirm caspase activity, C2 cells were washed and treated with DMSO or STA-9090 for 24 hours and caspase-3/7 activity was assessed using the SensoLyte kit. Data is presented as mean relative fluorescence unit (RFU) and standard deviation.
STA-9090 induces downregulation of Kit, but not PI3K or HSP90
To determine the effects of STA-9090 on WT or mutated Kit, we cultured cells with STA-9090 or SU11654 and evaluated Kit expression. Both WT and mutant Kit were downregulated by 100 nM STA-9090 after 24 hours in all lines treated (Fig. 4A). As expected, cells treated with SU11654 did not lose Kit expression. In contrast, the C2 line and BMCMCs upregulated Kit expression after SU11654 exposure, presumably as a feedback response following inhibition of Kit signaling. No effects on PI3K or HSP90 expression were observed following treatment with STA-9090 or SU11654 (Fig. 4B)
Figure 4.
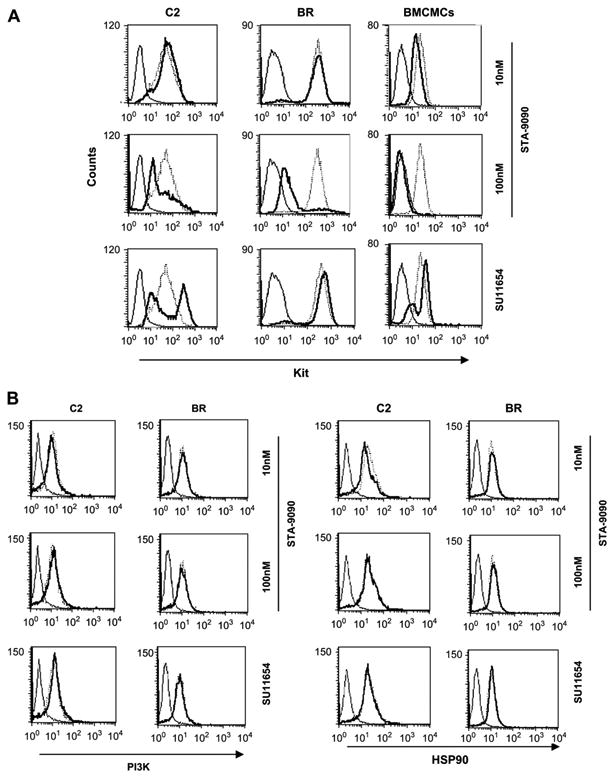
STA-9090 promotes downregulation of Kit, but not total phosphoinositide-3 kinase (PI3K) or heat shock protein 90 (HSP90) expression in normal and neoplastic mast cells. C2, BR, and bone marrow–derived mast cell (BMCMC) cells were washed and treated with or without STA-9090 at the indicated concentrations or SU11654 (100 nM) in 24-well plates for 24 hours. Cells were collected, fixed, permeabilized, and left untreated, incubated with isotype-matched control antibody, or incubated with anti-Kit antibody (A), or anti-p85 PI3K or anti-HSP90 antibodies (B), and the samples were analyzed flow cytometry (solid line, isotype control; dotted line, untreated control; bold line, treated cells).
STA-9090 treatment downregulates Akt expression and upregulates HSP70 expression in malignant mast cell lines
C2 and BR cells were treated with STA-9090 or SU11654 for 2 or 24 hours and the effects on Kit, Akt, Erk, HSP70, and HSP90 were assessed (Fig. 5A). As expected, downregulation of Kit phosphorylation and expression were observed in both the C2 and BR lines after treatment with 100 and 1000 nM STA-9090 at 24 hours, while only modulation of Kit phosphorylation was observed following SU11654 treatment. Interestingly, the C2 line demonstrated more rapid modulation of Kit expression following STA-9090 treatment, in agreement with our flow data. This may reflect a greater sensitivity of Kit possessing an ITD to HSP90 inhibition compared to that possessing a point mutation in the JM domain.
Figure 5.
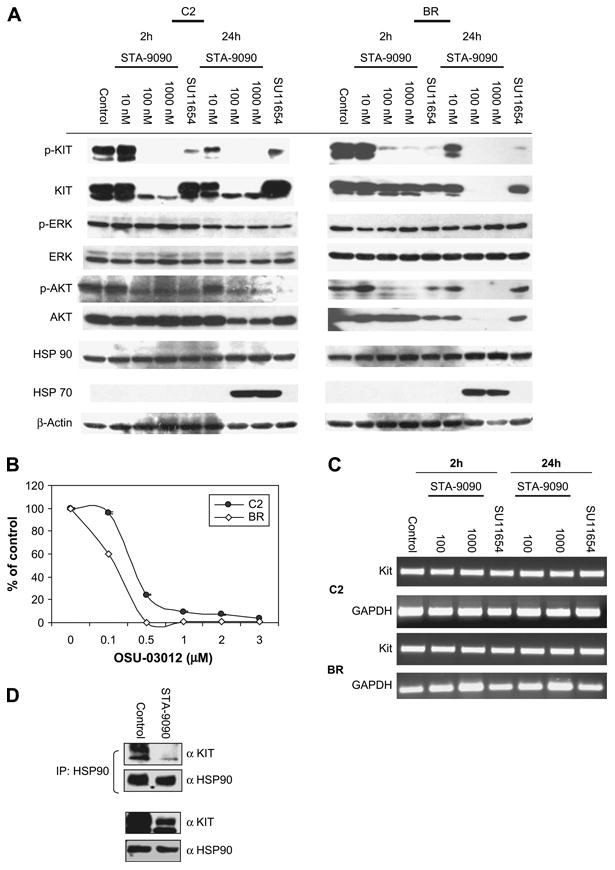
STA-9090 treatment of neoplastic mast cell lines induces downregulation of phosphor-/total Kit, Akt, and upregulation of heat shock protein (HSP) 70. (A) C2 and BR cells were incubated with STA-9090 at the indicated concentrations or SU11654 (100 nM) for 2 or 24 hours. Cells were collected, lysed, protein was quantitated and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using equivalent amounts of lysate. Following transfer, Western blotting for p-Kit, Kit, p-Akt (Thr308 and Ser473), Akt, phosphorylated extracellular signal-regulated kinase [p-ERK], ERK, HSP90, HSP70, and β-actin was performed. (B) C2 (1 × 104) and BR (1 × 105) cells were and plated in a 96-well plate in 150 μL along with indicated concentrations of OSU-03012 for 3 days in triplicate. Viability of the cells was determined using the WST-1 assay and the data is shown as a ratio of viable treated cells to viable control cells (treated with 0.01% dimethyl sulfoxide [DMSO]). (C) C2 and BR cells were incubated with STA-9090 at the indicated concentrations or SU11654 (100 nM) for 2 or 24 hours. Cells were collected, washed, and RNA was generated using Trizol. Reverse transcriptase polymerase chain reaction for c-Kit and glyceraldehyde phosphate dehydrogenase was then performed to assess cells for changes in message. (D) C2 cells were treated with DMSO or 100 nM STA-9090 for 8 hours and HSP90 was immunoprecipitated from cell lysates. Following SDS-PAGE, Western blotting was performed for Kit and HSP90. Western blotting for total Kit and HSP90 was performed following SDS-PAGE of C2 cells treated similarly.
While we did not see modulation of PI3K expression in mast cell lines treated with STA-9090, Akt is known to be a client protein of HSP90. Figure 5A demonstrates that both STA-9090 and SU11654 inhibited phosphorylation of Akt in malignant mast cell line, although this occurred earlier (2 hours) in the SU11654-treated cells. Downregulation of Akt protein expression was observed in both STA-9090–treated cell lines at 24 hours of treatment, but was not seen following SU11654 treatment. In support of the notion that loss of Akt activity can impair malignant mast cell viability, C2 and BR cells treated with the small-molecule Akt inhibitor OSU-03012 [40] demonstrated a dose-dependent loss of cell viability (Fig. 5B). No differences were found in phosphorylated ERK and total ERK expression following treatment of the mast cell lines with either STA-9090 or SU11654. Lastly, upregulation of HSP70 has been reported as a consequence of HSP90 inhibition [41–43]. In agreement with this, we observed induction of HSP70 expression following exposure to STA-9090, but not SU11654 (Fig. 5A).
To determine if modulation of Kit expression was secondary to loss of cKit mRNA, we performed reverse transcriptase polymerase chain reaction for c-Kit following treatment with STA-9090 or SU11654. cKit mRNA was relatively unchanged when compared to controls in both C2 and BR cells at 24 hours post STA-9090 exposure (Fig. 5C), a time point at which Kit protein expression was significantly downregulated (C2) or completely absent (BR). We next investigated whether loss of Kit expression was due to disruption of the association of HSP90 with Kit. As demonstrated in Figure 5D, treatment of C2 cells with STA-9090 induced the loss of Kit binding to HSP90 within 8 hours of drug exposure.
STA-9090 induces downregulation of Kit expression, growth inhibition, and apoptosis in ex vivo fresh malignant canine mast cells
Fresh malignant cells were isolated from two primary cutaneous tumors (MCT1 and MCT3) or following collection of abdominal effusion from a dog with systemic mastocytosis (MCT2). In all cases, >70% of isolated cell populations were neoplastic mast cells. Cells were cultured in complete medium with or without rcSCF, or with 50 ng/mL rcSCF and STA-9090 or SU11654 and cell viability was evaluated at 24, 48, and 72 hours of culture. As shown in Figure 6A, malignant mast cells cultured in the absence of rcSCF remained viable, although their proliferation was enhanced in the presence of rcSCF (MCT2 and MCT3). Dose-dependent growth inhibition was observed in all mast cell tumor samples following treatment with STA-9090. In contrast, there was no effect of SU11654 on viability of the malignant mast cells, suggesting that these tumor cells were not entirely dependent on Kit for their survival. In support of this, c-Kit was sequenced in all tumors and no mutations were identified in the coding sequences, indicating that ligand-independent Kit activation was not present (data not shown).
Figure 6.
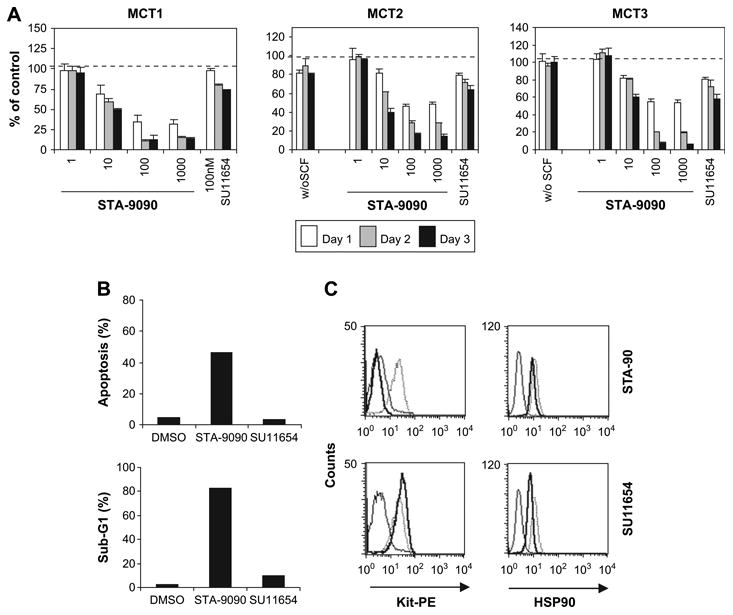
STA-9090 promotes growth inhibition, apoptosis, and Kit downregulation in fresh malignant mast cells treated ex vivo. Neoplastic mast cells were freshly purified from cutaneous mast cell tumors (MCT1 and MCT3) and from the abdominal effusion of a dog with systemic mastocytosis (MCT2), resulting in a population consisting of at least 70% neoplastic mast cells as assessed by Wright-Giemsa staining. (A) Cells (1 × 104/well in 150 uL) were cultured in complete medium alone, or supplied with 50 ng/mL rcSCF and left untreated (0.1% dimethyl sulfoxide [DMSO]), or treated with increasing concentrations of STA-9090 or 100 nM SU11654 in 96-well plates. Viability of the cells was determined using the WST-1 assay and the data is shown as a ratio of viable treated cells to viable control cells (treated with 0.01% DMSO). (B) Neoplastic mast cells from MCT2 were treated with STA-9090 (100 nM) or SU11654 (100 nM) as indicated for 72 hours. Apoptosis and cell cycling were assessed using Annexin-V–phycoerythrin [PE]/7-amino-actinomycin D (7-AAD) (upper) and propidium iodide staining (lower), respectively, as described previously. Data are presented graphically. (C) Neoplastic mast cells from MCT2 were treated with STA-9090 (100 nM) or SU11654 (100 nM) for 24 hours. Kit and heat shock protein 90 (HSP90) expression were determined by the flow cytometry as described previously.
Loss of cell viability in STA-9090–treated cells was secondary to apoptosis, as evidenced by Annexin-V/7-amino-actinomycin D staining and accumulation of Sub-G1 cells following cell-cycle analysis (Fig. 6B). STA-9090 induced downregulation of Kit surface expression in malignant mast cells, an effect that was not seen following SU11654 treatment (Fig. 6C). Similar to the C2 and BR lines, HSP90 expression was not altered in the fresh malignant mast cells after treatment with STA-9090 or SU11654 (Fig. 6C).
STA-9090 inhibits in vivo tumor growth in a malignant mast cell xenograft model
To evaluate STA-9090 in vivo we conducted a xenograft tumor model using the C2 line subcutaneously implanted into the flanks of SCID mice. STA-9090 significantly inhibited tumor growth (Fig. 7) when dosed with two repeating cycles of 25 mg/kg/day for 3 days (p < 0.001), with a %T/C value of 18 [44]. STA-9090 was well-tolerated, with the vehicle and STA-9090 groups having average bodyweight changes relative to the start of the study of +0.3% (±1.4 standard error of mean [SEM]) and −8.1% (±1.6 SEM) on day 17, respectively.
Figure 7.

STA-9090 inhibits tumor growth in the C2 xenograft model. C2 cells were subcutaneously implanted into the flanks of female severe combined immune-deficient (SCID) mice and tumors were permitted to develop to an average starting volume of approximately 150 mm3. Beginning on day 9 after tumor implantation, tumor-bearing animals (eight mice per group) were intravenously injected via the tail vein (arrowheads) with vehicle (10/18 DRD) or 25 mg/kg STA-9090 at one time per day for 3 consecutive days, followed by 2 days with no treatment, and then a second cycle of treatment at one time per day for 3 consecutive days. The average tumor volumes for each group were determined every 1 to 3 days. Treatment with a dose of 25 mg/kg STA-9090 significantly inhibited tumor growth relative to vehicle (*p < 0.001; t-test), with a %T/C value of 18 on day 17. Error bars represent ± standard error of mean.
Discussion
Kit mutations resulting in ligand-independent activation occur frequently in human and canine cancers, including systemic mastocytosis, GISTs, and acute myeloid leukemia [4,5,9,17]. Several small-molecule Kit inhibitors have been developed and these demonstrate significant activity in the clinical setting (imatinib [45], sunitinib [18], and dasatinib [46]). However, they have variable efficacy against particular forms of mutant Kit and resistance to therapy often develops over time [47]. Recently, HSP90 has arisen as a promising target for therapeutic intervention in Kit-dependent tumors. Work with human malignant mast cell and GIST lines demonstrated that the geldanamycin derivative 17-AAG has potent activity against these cells, regardless of the nature of Kit mutation [47–50]. However, the geldanamycins have limitations, including low solubility and hepatotoxicity. The purpose of this study was to evaluate STA-9090 (Synta Pharmaceuticals Corp.), a triazolone HSP90 inhibitor unrelated to geldanamycin, against WT and mutant Kit, using canine malignant mast cells as a model of Kit dysregulation.
STA-9090 inhibited cell proliferation and induced loss of cell viability in normal canine BMCMCs and the C2 and BR malignant mast cell lines that carry activating JM domain mutations that induce sensitivity to the small-molecule inhibitor SU11654 [11,14]. The biologic activity of STA-9090 at 10 nM was similar to that seen with 100 nM SU11654, a drug concentration previously shown to be effective both in vitro and in vivo [14,15]. The activity of STA-9090 against BMCMCs was not unexpected, as our prior studies demonstrated that these cells are dependent on SCF-induced Kit signaling for proliferation and survival in vitro [38]. It is possible that resident tissue mast cells are less sensitive to HSP90 inhibition in vivo as they are typically not actively cycling and may receive survival signals through additional pathways, such as inter-leukin (IL)-3 and IL-9. The BR cell line was slightly more resistant to SU11654 treatment when compared to the C2 line, but exhibited equivalent sensitivity to STA-9090, supporting the notion that HSP90 inhibitors may have a broad spectrum of activity against various Kit mutants.
Downregulation of Kit phosphorylation occurred rapidly following treatment with either STA-9090 or SU11654 in the C2 and BR lines. Work with GIST lines expressing Kit with JM domain mutations demonstrated a similar rapid time course after 17-AAG treatment [22]. Loss of Kit expression was also observed in the human mast cell lines HMC1.1 and HMC1.2 harboring Vla560Gly and Val560Gly/Aps816Val mutations, respectively, in the presence of 17-AAG [48]. However, higher concentrations of 17-AAG were required (500–1000 nM) when compared to those needed to modulate Kit with STA-9090 (100 nM), consistent with our data directly comparing STA-9090 and 17-AAG in the canine lines. Interestingly, alterations in Kit protein expression were variable in GIST lines after 17-AAG exposure [22], indicating that HSP90 may play a more important role in neoplastic mast cells with respect to Kit expression than in GISTs.
Alteration of Akt may explain, in part, the sensitivity of so many different neoplastic cell lines to HSP90 inhibition [48,51]. STA-9090 induced a dose-dependent decrease in Akt phosphorylation and expression in the malignant mast cell lines, while SU11654 altered Akt phosphorylation but not expression. In contrast, significant alteration of Akt expression was not observed in GIST lines following 17-AAG exposure [47]. Although some modulation of Akt was found in the HMC1.1/1.2 cells in the presence of 17-AAG [48], this was less substantial than that observed in the present study, suggesting that STA-9090 may be a more potent inhibitor of HSP90 and thus exhibit a wider range of client protein modulation. In support of this, we observed dose-dependent inhibition of cell viability in C2 and BR cells treated with the Akt inhibitor OSU-03012, suggesting an important role for Akt in malignant mast cell survival.
Recent evidence indicates that chaperone activity is important in maintaining the mitochondrial protective network. HSP90 and a related molecule TRAP-1 have been identified in the mitochondria of tumor cells, but not normal cells [52]. Direct targeting of 17-AAG to the mitochondria of tumor cells resulted in loss of mitochondrial function and cell death. While it is not known if STA-9090 is capable of penetrating the mitochondrial membrane, it possible that at least some of the biologic activity of STA-9090 is secondary to loss of mitochondrial function in the malignant mast cells and studies are ongoing to investigate this possibility. However, this is unlikely to be the primary mechanism of cell death as small molecule inhibitors of Kit induce apoptosis of malignant mast cells within 24 to 72 hours of exposure, supporting the notion that Kit signaling is critical for their survival. Additionally, our data show a rapid loss of Kit association with HSP90 following STA-9090 treatment, indicating a direct effect of STA-9090 on chaperone activity. Lastly, inhibition of Akt activity reduced malignant mast cell viability, suggesting that the ability of STA-9090 to target multiple pathways in these cells contributes to the observed rapid apoptosis of these cells following drug exposure.
We verified our findings in fresh neoplastic mast cells isolated from canine MCTs. These may be more reflective of cancer cells in vivo, having not adapted to in vitro culture conditions. These cells were sensitive to STA-9090 treatment, exhibiting growth inhibition, apoptosis, and downregulation of Kit expression. However, they were not dependent on SCF/Kit signaling for survival, as little effect of SU11654 was observed and no activating mutations in Kit were identified. It is possible that modulation of additional targets of STA-9090 (i.e., Akt) were responsible for the observed biologic effects. Alternatively, Kit expression may be necessary for maintenance of malignant mast cells through its association with additional cell surface proteins.
Although STA-9090 displayed potent cytotoxicity against in vitro, these assays involve prolonged exposure to the compound, which would be expected to poorly mimic the pharmacokinetics of drug exposure in vivo. We therefore confirmed the activity of STA-9090 in the C2 mast cell tumor xenograft model in SCID mice. Similar results have also been obtained in a mouse xenograft model conducted using the Kasumi-1 acute myeloid leukemia cell line expressing activated Kit (Asn822Lys) (K. Foley, unpublished data).
In summary, STA-9090 exhibited significant activity against malignant mast cell lines and fresh malignant mast cells cultured ex vivo, and also inhibited tumor growth in vivo in a mast cell tumor xenograft model. Importantly, STA-9090 was effective on both Kit-dependent and Kit-independent tumor cells, suggesting a broad range of therapeutic activity. These studies serve as the foundation for future clinical trials of STA-9090 in dogs with spontaneous cancers, particularly MCTs, as a prelude to human clinical work.
Acknowledgments
We thank Robert Mihalek and Donald Smith for help with animal experiments. This work was supported by R01 CA93807 (C.L.)
References
- 1.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase—the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338:1307–1315. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 2.Reilly JT. Class III receptor tyrosine kinases: role in leukaemogenesis. Br J Haematol. 2002;116:744–757. doi: 10.1046/j.0007-1048.2001.03294.x. [DOI] [PubMed] [Google Scholar]
- 3.de Silva CM, Reid R. Gastrointestinal stromal tumors (GIST): C-kit mutations, CD117 expression, differential diagnosis and targeted cancer therapy with Imatinib. Pathol Oncol Res. 2003;9:13–19. doi: 10.1007/BF03033708. [DOI] [PubMed] [Google Scholar]
- 4.Fletcher JA, Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev. 2007;17:3–7. doi: 10.1016/j.gde.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Shimada A, Taki T, Tabuchi K, et al. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood. 2006;107:1806–1809. doi: 10.1182/blood-2005-08-3408. [DOI] [PubMed] [Google Scholar]
- 6.Roskoski R., Jr Signaling by Kit protein-tyrosine kinase—the stem cell factor receptor. Biochem Biophys Res Commun. 2005;337:1–13. doi: 10.1016/j.bbrc.2005.08.055. [DOI] [PubMed] [Google Scholar]
- 7.Kuint J, Bielorai B, Gilat D, Birenbaum E, Amariglio N, Rechavi G. C-kit activating mutation in a neonate with in-utero presentation of systemic mastocytosis associated with myeloproliferative disorder. Br J Haematol. 1999;106:838–839. doi: 10.1046/j.1365-2141.1999.01683.x. [DOI] [PubMed] [Google Scholar]
- 8.Sotlar K, Fridrich C, Mall A, et al. Detection of c-kit point mutation Asp-816 → Val in microdissected pooled single mast cells and leukemic cells in a patient with systemic mastocytosis and concomitant chronic myelomonocytic leukemia. Leuk Res. 2002;26:979–984. doi: 10.1016/s0145-2126(02)00041-3. [DOI] [PubMed] [Google Scholar]
- 9.Schnittger S, Kohl TM, Haferlach T, et al. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 2006;107:1791–1799. doi: 10.1182/blood-2005-04-1466. [DOI] [PubMed] [Google Scholar]
- 10.London CA, Kisseberth WC, Galli SJ, Geissler EN, Helfand SC. Expression of stem cell factor receptor (c-kit) by the malignant mast cells from spontaneous canine mast cell tumours. J Comp Pathol. 1006;115:399–414. doi: 10.1016/s0021-9975(96)80074-0. [DOI] [PubMed] [Google Scholar]
- 11.London CA, Galli SJ, Yuuki T, et al. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp Hematol. 1999;27:689–697. doi: 10.1016/s0301-472x(98)00075-7. [DOI] [PubMed] [Google Scholar]
- 12.Reguera MJ, Ferrer L, Rabanal RM. Evaluation of an intron deletion in the c-kit gene of canine mast cell tumors. Am J Vet Res. 2002;63:1257–1261. doi: 10.2460/ajvr.2002.63.1257. [DOI] [PubMed] [Google Scholar]
- 13.Webster JD, Yuzbasiyan-Gurkan V, Kaneene JB, et al. The role of c-KIT in tumorigenesis: evaluation in canine cutaneous mast cell tumors. Neoplasia. 2006;8:104–111. doi: 10.1593/neo.05622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao AT, Chien MB, Shenoy N, et al. Inhibition of constitutively active forms of mutant kit by multitargeted indolinone tyrosine kinase inhibitors. Blood. 2002;100:585–593. doi: 10.1182/blood-2001-12-0350. [DOI] [PubMed] [Google Scholar]
- 15.London CA, Hannah AL, Zadovoskaya R, et al. Phase I dose-escalating study of SU11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin Cancer Res. 2003;9:2755–2768. [PubMed] [Google Scholar]
- 16.Pryer NK, Lee LB, Zadovaskaya R, et al. Proof of target for SU11654: inhibition of KIT phosphorylation in canine mast cell tumors. Clin Cancer Res. 2003;9:5729–5734. [PubMed] [Google Scholar]
- 17.Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103:3222–3225. doi: 10.1182/blood-2003-11-3816. [DOI] [PubMed] [Google Scholar]
- 18.von Mehren M. Beyond imatinib: second generation c-KIT inhibitors for the management of gastrointestinal stromal tumors. Clin Colorectal Cancer. 2006;6(Suppl 1):S30–S34. doi: 10.3816/ccc.2006.s.005. [DOI] [PubMed] [Google Scholar]
- 19.Dizdar O, Dede DS, Bulut N, Altundag K. Dasatinib may also inhibit c-Kit in triple negative breast cancer cell lines. Breast Cancer Res Treat. 2008;107:303. doi: 10.1007/s10549-007-9551-6. [DOI] [PubMed] [Google Scholar]
- 20.Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–481. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 21.Koyama T, Nimura H, Kobayashi K, et al. Recurrent gastrointestinal stromal tumor (GIST) of the stomach associated with a novel c-kit mutation after imatinib treatment. Gastric Cancer. 2006;9:235–239. doi: 10.1007/s10120-006-0368-5. [DOI] [PubMed] [Google Scholar]
- 22.Tamborini E, Gabanti E, Lagonigro MS, et al. KIT/Val654 Ala receptor detected in one imatinib-resistant GIST patient. Cancer Res. 2005;65:1115. author reply 1115. [PubMed] [Google Scholar]
- 23.Wegele H, Muller L, Buchner J. Hsp70 and Hsp90—a relay team for protein folding. Rev Physiol Biochem Pharmacol. 2004;151:1–44. doi: 10.1007/s10254-003-0021-1. [DOI] [PubMed] [Google Scholar]
- 24.Richter K, Hendershot LM, Freeman BC. The cellular world according to Hsp90. Nat Struct Mol Biol. 2007;14:90–94. doi: 10.1038/nsmb0207-90. [DOI] [PubMed] [Google Scholar]
- 25.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 26.Duus J, Bahar HI, Venkataraman G, et al. Analysis of expression of heat shock protein-90 (HSP90) and the effects of HSP90 inhibitor (17-AAG) in multiple myeloma. Leuk Lymphoma. 2006;47:1369–1378. doi: 10.1080/10428190500472123. [DOI] [PubMed] [Google Scholar]
- 27.Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med. 2004;10:47–51. doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 29.Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–1629. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 30.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8(Suppl):S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 31.Vastag B. HSP-90 inhibitors promise to complement cancer therapies. Nat Biotechnol. 2006;24:1307. doi: 10.1038/nbt1106-1307. [DOI] [PubMed] [Google Scholar]
- 32.Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anticancer Drugs. 2004;15:651–662. doi: 10.1097/01.cad.0000136876.11928.be. [DOI] [PubMed] [Google Scholar]
- 33.Solit DB, Scher HI, Rosen N. Hsp90 as a therapeutic target in prostate cancer. Semin Oncol. 2003;30:709–716. doi: 10.1016/s0093-7754(03)00346-4. [DOI] [PubMed] [Google Scholar]
- 34.Smith V, Sausville EA, Camalier RF, Fiebig HH, Burger AM. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models. Cancer Chemother Pharmacol. 2005;56:126–137. doi: 10.1007/s00280-004-0947-2. [DOI] [PubMed] [Google Scholar]
- 35.Goetz MP, Toft D, Reid J, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23:1078–1087. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 36.Solit DB, Ivy SP, Kopil C, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res. 2007;13:1775–1782. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronnen EA, Kondagunta GV, Ishill N, et al. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;24:543–546. doi: 10.1007/s10637-006-9208-z. [DOI] [PubMed] [Google Scholar]
- 38.Lin TY, Rush LJ, London CA. Generation and characterization of bone marrow-derived cultured canine mast cells. Vet Immunol Immunopathol. 2006;113:37–52. doi: 10.1016/j.vetimm.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 39.Lin TY, London CA. A functional comparison of canine and murine bone marrow derived cultured mast cells. Vet Immunol Immunopathol. 2006;114:320–334. doi: 10.1016/j.vetimm.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Huang JW, Tseng PH, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004;64:4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 41.Francis LK, Alsayed Y, Leleu X, et al. Combination mammalian target of rapamycin inhibitor rapamycin and HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin has synergistic activity in multiple myeloma. Clin Cancer Res. 2006;12:6826–6835. doi: 10.1158/1078-0432.CCR-06-1331. [DOI] [PubMed] [Google Scholar]
- 42.Castro JE, Prada CE, Loria O, et al. ZAP-70 is a novel conditional heat shock protein 90 (Hsp90) client: inhibition of Hsp90 leads to ZAP-70 degradation, apoptosis, and impaired signaling in chronic lymphocytic leukemia. Blood. 2005;106:2506–2512. doi: 10.1182/blood-2005-03-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bagatell R, Paine-Murrieta GD, Taylor CW, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6:3312–3318. [PubMed] [Google Scholar]
- 44.Corbett T, Valeriote F, LoRusso P, et al. In vivo methods for screening and preclinical testing: use of rodent solid tumors for drug discovery. In: Teicher BA, editor. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Totowa, NJ: Humana Press; 1997. pp. 75–99. [Google Scholar]
- 45.Droogendijk HJ, Kluin-Nelemans HJ, van Doormaal JJ, Oranje AP, van de Loosdrecht AA, van Daele PL. Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer. 2006;107:345–351. doi: 10.1002/cncr.21996. [DOI] [PubMed] [Google Scholar]
- 46.Shah NP, Lee FY, Luo R, Jiang Y, Donker M, Akin C. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood. 2006;108:286–291. doi: 10.1182/blood-2005-10-3969. [DOI] [PubMed] [Google Scholar]
- 47.Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66:9153–9161. doi: 10.1158/0008-5472.CAN-06-0165. [DOI] [PubMed] [Google Scholar]
- 48.Fumo G, Akin C, Metcalfe DD, Neckers L. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood. 2004;103:1078–1084. doi: 10.1182/blood-2003-07-2477. [DOI] [PubMed] [Google Scholar]
- 49.Radujkovic A, Schad M, Topaly J, et al. Synergistic activity of imatinib and 17-AAG in imatinib-resistant CML cells overexpressing BCR-ABL—inhibition of P-glycoprotein function by 17-AAG. Leukemia. 2005;19:1198–1206. doi: 10.1038/sj.leu.2403764. [DOI] [PubMed] [Google Scholar]
- 50.Yu W, Rao Q, Wang M, et al. The Hsp90 inhibitor 17-allylamide-17-demethoxygeldanamycin induces apoptosis and differentiation of Kasumi-1 harboring the Asn822Lys KIT mutation and down-regulates KIT protein level. Leuk Res. 2006;30:575–582. doi: 10.1016/j.leukres.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 51.Sain N, Krishnan B, Ormerod MG, et al. Potentiation of paclitaxel activity by the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther. 2006;5:1197–1208. doi: 10.1158/1535-7163.MCT-05-0445. [DOI] [PubMed] [Google Scholar]
- 52.Rodina A, Vilenchik M, Moulick K, et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol. 2007;3:498–507. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]