

Background: CaMKII is implicated in both positive and negative regulation of cAMP-response element-binding protein (CREB) activity.
Results: CaMKIIδ increases CREB-Ser142 phosphorylation and inhibits binding of CREB to Sik1 and Rgs2 promoters in vascular smooth muscle (VSM).
Conclusion: In VSM CaMKIIδ negatively regulates CREB activity and target gene transcription by phosphorylating CREB-Ser142.
Significance: Negative regulation of CREB by CaMKIIδ is a mechanism mediating Ca2+ signal-dependent regulation of gene transcription in VSM.
Keywords: Calcium, CaMKII, CREB, Thrombin, Vascular Smooth Muscle Cells, RGS2, SIK1
Abstract
One transcription factor mediator of Ca2+-signals is cAMP response element-binding protein (CREB). CREB expression and/or activity negatively correlates with vascular smooth muscle (VSM) cell proliferation and migration. Multifunctional Ca2+/calmodulin-dependent protein kinases, including CaMKII, have been demonstrated to regulate CREB activity through both positive and negative phosphorylation events in vitro, but the function of CaMKII as a proximal regulator of CREB in intact cell systems, including VSM, is not clear. In this study, we used gain- and loss-of-function approaches to determine the function of CaMKIIδ in regulating CREB phosphorylation, localization, and activity in VSM. Overexpression of constitutively active CaMKIIδ specifically increased CREB phosphorylation on Ser142 and silencing CaMKIIδ expression by siRNA or blocking endogenous CaMKII activity with KN93 abolished thrombin- or ionomycin-induced CREB phosphorylation on Ser142 without affecting Ser133 phosphorylation. CREB-Ser142 phosphorylation correlated with transient nucleocytoplasmic translocation of CREB. Thrombin-induced CREB promoter activity, CREB binding to Sik1 and Rgs2 promoters, and Sik1/Rgs2 transcription were enhanced by a kinase-negative CaMKIIδ2 (K43A) mutant and inhibited by a constitutively active (T287D) mutant. Taken together, these studies establish negative regulation of CREB activity by endogenous CaMKIIδ-dependent CREB-Ser142 phosphorylation and suggest a potential mechanism for CaMKIIδ/CREB signaling in modulating proliferation and migration in VSM cells.
Introduction
Vascular smooth muscle (VSM)2 cells comprise the medial layer of blood vessels providing mechanical support and a means for adjusting vascular wall tone and diameter. However, VSM cells are not terminally differentiated and can switch to a proliferative/synthetic phenotype in response to changes in local environment that accompany a number of chronic vascular diseases as well as acute and chronic vascular injuries (1, 2). Identifying transcriptional and post-transcriptional regulatory processes governing VSM phenotype plasticity are therefore of considerable basic interest and clinical relevance.
One key signal in VSM is free intracellular Ca2+ ([Ca2+]i), which is well known to regulate differentiated contractile function, as well as synthetic phenotype proliferation and migration (3). [Ca2+]i signals also regulate a number of transcription factors, including type II HDAC-MEF2, calcineurin/nuclear factor of activated T cells, and cyclic AMP response element-binding protein (CREB) (4–6). CREB, in addition to its defining regulation by cAMP-dependent protein kinase (PKA), has long been known to be regulated by [Ca2+]i signals via multifunctional Ca2+/calmodulin-dependent protein kinases including CaMKIV and CaMKII (7). Expression of CREB is down-regulated in several vascular diseases, such as hypertension, dyslipidemia, and atherosclerosis, as well as in response to vascular injury (8), suggesting a vasculoprotective effect of CREB (8). Conversely, CREB activation has also been reported to be positively related to thrombin-induced proliferation (9) and TNFα-induced migration in the smooth muscle cells (10).
Several phosphorylation sites in CREB have been demonstrated to regulate transcriptional activity. Ser133 is the primary target of PKA and phosphorylation is required for CREB activation, whereas phosphorylation of Ser142 by CaMKII has been reported to inhibit CREB activity by interfering with CREB dimerization and protein interactions to form an active promoter complex (11). In vitro studies using purified brain CaMKIV and CaMKII have clearly demonstrated that CaMKIV phosphorylates CREB on Ser133, whereas CaMKII has equal affinity for Ser133 and Ser142 (12). These results were confirmed using transient expression assays and constitutively active CaMKIV and CaMKII constructs. In the case of CaMKII overexpression, inhibitory phosphorylation on Ser142 overrides the activating phosphorylation on Ser133 (11, 12). Yet in intact neural cells, CaMKII activation has been positively associated with CREB activation (13–15). Based on these studies, it appears that endogenous CaMKII could be either a positive or negative regulator of CREB, perhaps in a tissue or context-specific manner. Despite the widespread importance of CREB as a transcription factor, there is very little information regarding the importance of negative CREB regulation mediated by CaMKII-dependent Ser142 phosphorylation in any intact cellular system.
Pathways regulating CREB activity in response to Ca2+-dependent stimuli in VSM are incompletely understood, and what little is known has been inferred via indirect pharmacological approaches that suggest a net positive function for CaM kinases in regulating CREB (16–18). In this study, we demonstrate for the first time in VSM cells that stimulation with Ca2+-dependent stimuli thrombin and ionomycin resulted in transient CREB-Ser142 phosphorylation that preceded the activating phosphorylation on Ser133. Multiple gain- and loss-of-function approaches indicated a specific function for endogenous CaMKIIδ in regulating VSM CREB-Ser142 phosphorylation, nucleocytoplasmic translocation, promoter activity, and endogenous target gene CREB binding and transcription. Taken together, these studies definitively establish negative regulation of CREB activity by endogenous CaMKIIδ-dependent phosphorylation of CREB-Ser142 and suggest a potential mechanism for CaMKII/CREB signaling in modulating proliferation and migration in VSM cells.
EXPERIMENTAL PROCEDURES
Materials
The antibody specific for phospho-Ser142-CREB was purchased from GeneScript and the antibodies against total CREB, phospho-Ser133-CREB, histone H3, and α-tubulin were purchased from Cell Signaling. The antibodies against β-actin and GAPDH were purchased from Sigma. Polyclonal antibodies against pan-CaMKII, CaMKIIδ2, and phospho-Thr287-CaMKII were raised in rabbits as described previously (19, 20). Adenoviruses expressing kinase-negative CaMKIIδ2 (K43A), constitutively active CaMKIIδ2 (T287D), and GFP were described previously (21, 22). The plasmid expressing luciferase under a promoter construct containing four CREB binding site repeats was purchased from Affymetrix. Thrombin was purchased from Sigma; and KN93 and ionomycin were purchased from Calbiochem. The cell culture media and all immunoblotting supplies were purchased from Invitrogen and Bio-Rad Laboratories, respectively.
VSM Cell Dispersion and Culture
Primary cultures of VSM cells were obtained from rat aorta as previously described (23). In brief, Sprague-Dawley rats (150–200 g) were sacrificed using CO2, and following brief digestion with collagenase the medial layer of the aorta containing VSM cells was freed from the adventitial layer and intima containing endothelial cells was disrupted mechanically. Medial layer VSM cells were isolated by enzymatic digestion with collagenase and elastase and cultured in DMEM/F-12, supplemented with 10% FBS (fetal bovine serum) and 1% penicillin/streptomycin. Cells were passaged when ∼80% confluence was reached. Experiments were performed with VSM cells from passages 3 to 7. The biochemical and growth properties of these and similar VSM cell cultures have been extensively characterized (5, 19–23). All animal protocols were reviewed and approved by the Albany Medical Center Institutional Animal Care and Use Committee.
Luciferase Reporter Assay
VSM cells (1 × 106) were electroporated with plasmids expressing CREB luciferase reporter (2 μg) and TK-Renilla (0.1 μg) and incubated for a total of 72 h prior to cell lysis. Adenoviruses encoding kinase-negative CaMKIIδ2 (K43A) mutant, which acts as a dominant-negative with respect to kinase activity, or constitutively active CaMKIIδ2 (T287A) mutant (22) were transduced for 48 and 24 h prior to cell lysis, respectively. CREB luciferase reporter activity was stimulated by thrombin (0.5 nm) or forskolin (5 μm) for 6 h prior to lysis. The level of promoter activity was determined by measuring firefly luciferase signal intensity normalized over the internal control TK-Renilla luciferase signal intensity using protocols provided in the Dual Luciferase Assay System (Promega).
Quantitative PCR
VSM cells were seeded onto 35-mm culture dishes (0.5 × 105 cells) and treated with 0.5 μm thrombin and 5 μm forskolin for 6 h. RNA was isolated following the manufacturer's instructions for the RNeasy® mini kit (Qiagen). The concentration of extracted RNA was determined using Nanodrop (Thermo). RNA was reverse transcribed to cDNA using the Quantitech® reverse transcription kit (Qiagen). The quantitative RT-PCR was carried out on an Mx3000P QPCR System using IQTM SYBR® Green Supermix (Bio-Rad). The primers used for Sik1/Kid2, Rgs2, and internal control Gapdh were listed as follows, Sik1 forward, 5′-TCCAAACACCTTCGTTCTCTG-3′, and reverse, 5′-CGATCCCATTACAGCCCAG-3′; Rgs2 forward, 5′-CAAAGTGCCATGTTCCTGGCTG-3′, and reverse, 5′-AAGTAGCTCAAACGGGTCTTC-3′; Gapdh forward, 5′-TCGTCTCATAGACAAGATGGT-3′, and reverse, 5′-GTAGTTGAGGTCAATGAAGGG-3′.
Cell Stimulation and Western Blot
VSM cells were washed twice with pre-warmed Hanks' balanced salt solution and placed on a warm plate (37 °C) for 30 min prior to stimulation with either 50 nm thrombin or 0.5 μm ionomycin. At appropriate times cells were lysed by addition of 3× Laemmli sample buffer. Lysates were heated at 95 °C for 5 min and then resolved using standard 10% SDS-PAGE. Resolved proteins were electrophoretically transferred onto nitrocellulose membranes (GE Healthcare), the membranes blocked with 5% nonfat milk or 5% BSA in Tris-buffered saline supplemented with 1% Tween 20 (TBST) for 1 h, followed by primary antibody incubation for 1 h or overnight at 4 °C and secondary antibody incubation for 1 h. After washing three times with TBST, the membranes were incubated with SuperSignal® chemiluminescent substrate (Thermo) for 5 min. Gel images shown were generated by the Fuji LAS4000 and quantified with Multi Gauge software.
siRNA Electroporation
VMS cells (1 × 106) were harvested and electroporated with 2 μg of siRNA targeting CaMKIIδ gene products (Smart pool siRNA Thermo Fisher, catalog number L-080171-00-0010) or non-targeting siRNA (Thermo Fisher) using the Amaxa Nucleofector system (Lonza) and the VSM cell-specific D33 program (Lonza Amaxa®). After electroporation, the cells were resuspended in 4 ml of cell culture media and reseeded onto four 60-mm culture dishes. Using previously established conditions (5), the cells were used 48 h following electroporation.
Chromatin Immunoprecipitation (ChIP)
To study the interaction between CREB and gene promoters, a ChIP assay was performed using the ChIP-IT® Express Kit from Active Motif (53008). 5–6 × 106 VSM cells were used for each sample. Following cross-linking and recovery of chromatin following kit protocols, chromatin was sheared in 200 μl of kit shearing buffer using a Bioruptor sonicator at medium power for a total time of 16 min (30 s on and 30 s off for 16 cycles). 50 μl of chromatin lysate was immunoprecipitated overnight at 4 °C with anti-CREB antibody and isolated with magnetic protein G beads. Following cross-link reversal and incubation with proteinase K per kit protocols, immunoprecipitated DNA was analyzed by quantitative PCR. Promoter regions were predicted using PromoSer software (24) and CREB-binding elements were predicted with PROMO 3.0 software (25). Primers flanking a predicted CREB binding site (−1020CTGACGTCA−1012) in the rat Sik1/Kid2 promoter were (5′-3′): forward, −1040GCGGCTGCTGAGCCCGGT−1023-3′ and reverse, −962GGCGGTTGCCAAGAGACTGGAG−941. Primers flanking an Rgs2 predicted CREB-binding element (−221GCCTACGTCA−212) were: forward, −243TGTGTGCGCACGAGGATGCGG−223 and reverse, −124ACCTCTAGAGGGCGCGGATTG−104.
Cell Fractionation
Cell fractionation was performed on VSM cells to separate nuclear and cytosolic fractions using a cell fractionation kit and protocols (Active Motif). In brief, cells were washed with pre-cooled PBS containing phosphatase inhibitors (5 ml), collected with a cell scraper and isolated by centrifugation at 500 × g for 5 min at 4 °C. After the resuspension in a kit hypotonic lysis buffer (500 μl), the cells were incubated on ice for 15 min. Detergent (25 μl) was added followed by centrifugation at 14,000 × g for 30 s at 4 °C to pellet a crude fraction containing nuclei. The crude supernatant is considered the “cytosolic” fraction and the washed pellet the “nuclear” fraction. The nuclear marker histone H3 and cytosolic marker tubulin were used to verify the fractionation. Proteins in the fractions were by Western blotting.
Statistical Analysis
Quantitative data are presented as mean ± S.E. Student's unpaired t test was used in the comparisons between two groups of data and one-way ANOVA for simple comparisons between multiple groups. If found significant by ANOVA, Dunnett's Multiple Comparison or Bonferroni post hoc tests were used to identify between group differences. Two-way ANOVA with a Bonferroni post hoc test was used in Fig. 3 to compare time points across siRNA and siControl groups. Statistics were performed using the GraphPad Prism4 program. “n” indicates the number of independent experiments and results were considered to be significantly different if p < 0.05. Statistical significance is presented, *, **, and *** indicate p < 0.05, p < 0.01, and p < 0.001, respectively.
FIGURE 3.

Endogenous CaMKIIδ mediates CREB phosphorylation on Ser142. A, VSM cells were electroporated with non-targeting siRNA (SiControl) or siRNA specifically targeting CaMKIIδ (SiCaMKIIδ). After 3 days cells were stimulated with thrombin (50 nm) for 1 or 10 min. Cell lysates were resolved by SDS-PAGE and immunoblotted for CaMKIIδ, CREB-Ser(P)142, and CREB-Ser(P)133. B, quantification of CREB-Ser(P)142 ECl signals. C, quantification of CREB-Ser(P)133 ECL signals. Data were normalized for loading using GAPDH signals and are expressed as fold-values compared with unstimulated. Values shown are mean ± S.E. n = 3. Two-way ANOVA analysis identified significant differences both across time (p < 0.0001) and between SiControl and SiCaMKIIδ groups (p < 0.0001) in panel B, but only across time in panel C (p < 0.0001). Between groups differences were identified by the Bonferroni post hoc test (***, p < 0.001).
RESULTS
CREB Phosphorylation in VSM
Thrombin has been proven to increase [Ca2+]i via activation of membrane PAR receptors (reviewed in Ref. 26), resulting in activation of both CaMKII (27) and CREB (9). CREB Ser133 and Ser142 are two candidate phosphorylation sites for regulation by Ca2+-dependent pathways with the former leading to CREB activation and the latter to CREB inhibition (12, 28). Addition of the Ca2+ ionophore, ionomycin, was used to selectively induce Ca2+ influx and stimulate activation of CaMKIIδ in VSM cells. CaMKIIδ activation was indexed by immunoblotting lysates with an antibody recognizing phospho-Thr287, the activation-dependent autophosphorylation site (29) (Fig. 1A). Ionomycin (0.5 μm) stimulated CREB phosphorylation 2-fold on both Ser133 and Ser142, suggesting either or both phosphorylation events could be mediated by CaMKIIδ (Fig. 1A). Addition of the physiological stimulus thrombin (50 nm) biphasically activated CaMKII with peaks between 30–60 s and 30 min, consistent with a previous report (27) (Fig. 1B). Thrombin also increased CREB phosphorylation on both Ser133 and Ser142, but with distinct time courses. CREB phosphorylation on the inhibitory site Ser142 peaked in 2 min, slightly lagging the CaMKII activation time course, whereas phosphorylation on the activating site Ser133 lagged considerably with a peak between 5 and 10 min following stimulation. Thus, in VSM cells, activation of CaMKII in response to thrombin correlated temporally with CREB-Ser142 phosphorylation compared with CREB-Ser133 phosphorylation.
FIGURE 1.
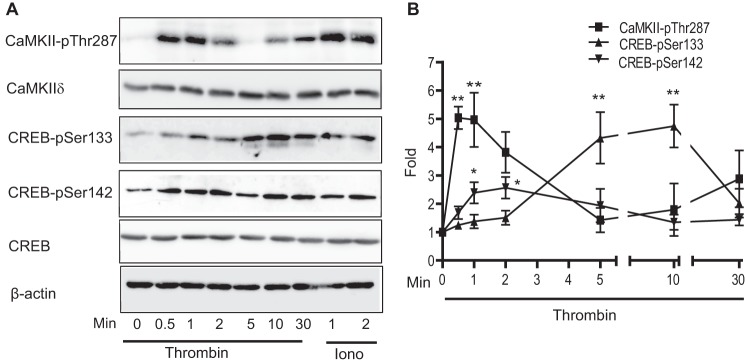
Thrombin-induced phosphorylation of CaMKII and CREB. A, VSM cells were treated with thrombin (50 nm) or ionomycin (Iono; 0. 5 μm). Cell lysates were resolved by SDS-PAGE and immunoblotted for phospho-Thr287-CaMKII (pThr287), total CaMKIIδ, phospho-Ser133-CREB (pSer133), phospho-Ser142-CREB (pSer142), and β-actin. B, ECL signals were quantified with a FujiLAS 4000. The plot quantifies CaMKII-p287 as an index of CaMKII activation, CREB-Ser(P)133 and CREB-Ser(P)142. Data were normalized for loading using β-actin signals and expressed as fold-values compared with unstimulated. Values shown are mean ± S.E., n = 3 and were analyzed by one-way ANOVA. *, p < 0.05 and **, p < 0.01, by Dunnett's Multiple comparison post hoc test.
CaMKIIδ Phosphorylates CREB on Ser142 in VSM
In vitro studies using purified CaMKII from brain have demonstrated that both CREB Ser133 and Ser142 are substrates for the kinase (12). The function of CaMKII in regulating endogenous CREB in VSM has only been inferred through pharmacological approaches that do not distinguish between CaMKII and CaMKIV (30). The principal isoform of CaMKII expressed in VSM cells is CaMKIIδ2 (19), or CaMKIIδC by alternative nomenclature (31). Transduction of VSM cells with an increasing dose of adenovirus expressing a constitutively active CaMKIIδ2 (T287D) mutant (22) significantly enhanced CREB phosphorylation on Ser142 in a dose-dependent manner (Fig. 2, A and B). However, overexpression of the constitutively active mutant did not affect CREB phosphorylation on Ser133 at any of the levels tested. This gain of function experiment indicates a strong specificity of CaMKIIδ for the inhibitory CREB Ser142 site in intact VSM cells.
FIGURE 2.

CaMKIIδ selectively phosphorylates CREB-Ser142. A, adenovirus encoding a constitutively active (CA) CaMKIIδ2 mutant was transduced in VSM cells for 16 h followed by lysis and immunoblotting for CREB-Ser(P)142, CREB-Ser(P)133, total CREB, and HA tag (CaMKII-HA). B, quantification of CREB-Ser(P)142 ECL signals. C, quantification of CREB-Ser(P)133 ECL signals. Data were normalized for loading using GAPDH signals and expressed as fold-values compared with unstimulated. Values shown are mean ± S.E. n = 3 and analyzed by one-way ANOVA. *, p < 0.05 and **, p < 0.01 by Dunnett's post hoc test.
To determine whether endogenous CaMKIIδ regulates CREB phosphorylation on Ser133 or Ser142, we used siRNAs specifically targeting CaMKIIδ isoforms to reduce CaMKIIδ expression by ∼80–90% (Fig. 3A). Consistent with the results in Fig. 1, thrombin stimulated CREB phosphorylation at Ser142 (Fig. 3, A and B) and Ser133 (Fig. 3, A and C) in the presence of control non-targeting siRNA. Suppression of CaMKIIδ expression abolished thrombin-induced CREB phosphorylation on Ser142 (Fig. 3B), but had no significant effect on thrombin-stimulated CREB phosphorylation on Ser133 (Fig. 3C). As an alternative approach, we pharmacologically inhibited CaMKII activation in VSM cells using KN93, a selective CaM kinase inhibitor (32). Pretreatment with KN93 (30 μm) for 30 min inhibited thrombin- or ionomycin-induced CaMKIIδ activation (Fig. 4, A and B) and strongly inhibited CREB phosphorylation on Ser142 induced by either stimulus (Fig. 4C). Taken together with the gain-of-function results (Fig. 2), these loss-of-function results indicate that endogenous CaMKIIδ mediates CREB phosphorylation on Ser142, but not Ser133 in VSM cells.
FIGURE 4.

Inhibition of CaMKII activity blocks CREB phosphorylation on Ser142. A, VSM cells were pretreated with KN93 (30 μm) for 30 min and stimulated with thrombin (50 nm) or ionomycin (0.5 μm) for 1 min. Cell lysates were resolved by SDS-PAGE and immunoblotted for phospho-Thr287-CaMKII (pThr287), phospho-Ser142-CREB (pSer142), and GAPDH. B, quantification of CaMKII-Thr(P)287 ECL signals. C, quantification of CREB-Ser(P)142 ECL signals. Data were normalized for loading using GAPDH signals and expressed as fold-values compared with unstimulated. Values shown are mean ± S.E. n = 3 and analyzed by unpaired two-tailed Student's t test. **, p < 0.01.
Additional studies using the MEK1 inhibitor U0126 (5 μm) indicated that essentially complete inhibition of ERK1/2 activation (Fig. 5, A and B) resulted in a strong inhibition of thrombin- and ionomycin-stimulated CREB-Ser133 phosphorylation (Fig. 5C), confirming a previous study (9). Residual Ser133 phosphorylation in this experiment suggests that another unidentified pathway(s) may also contribute to CREB activation in response to these Ca2+-dependent stimuli.
FIGURE 5.

ERK1/2 mediates thrombin-induced phosphorylation of CREB-Ser133. A, cultured VSM cells were pretreated with the MEK inhibitor U0126 (5 μm) or vehicle control (dimethyl sulfoxide, DMSO) for 30 min prior to stimulation with thrombin (50 nm) or ionomycin (Iono; 0. 5 μm) for the indicated times. Cell lysates were resolved by SDS-PAGE and immunoblotted for phospho-ERK1/2, phospho-Ser133-CREB (pSer133), total CREB, and GAPDH. B, P-ERK ECL signals, and C, CREB-Ser(P)133 signals were quantified with a FujiLAS 4000. Data were normalized for loading using GAPDH signals and expressed as fold-values compared with unstimulated. Values shown are mean ± S.E., n = 3 and were analyzed by one-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001, by Bonferroni post hoc test.
Thrombin Stimulates Nucleocytoplasmic Translocation of Phospho-Ser142-CREB in VSM
Stevenson et al. (33) observed membrane depolarization induced CREB translocation in VSM cells and Garat et al. (34) reported nuclear export of CREB in response to sustained PDGF in pulmonary artery smooth muscle cells. The mechanisms signaling nuclear CREB export are not known. Western blotting of nuclear and cytosolic fractions was carried out to assess localization of CREB. Effective fractionation was verified by nuclear histone H3 and cytoplasmic tubulin content, respectively (Fig. 5A). Under unstimulated conditions, total CREB and resting levels of phospho-Ser142-CREB were detected in both nuclear and cytosolic fractions. Thrombin stimulation transiently decreased nuclear phospho-Ser142-CREB content by over 50% (Fig. 6B), with concomitant increases in the cytosolic fraction (Fig. 6C). Phospho-Ser133-CREB increased but remained localized in the nuclear fraction by 5–10 min (Fig. 6, A and D). Interesting, total CREB also translocated from the nucleus with a time course identical to phospho-Ser142-CREB (Fig. 6, A and B). CaMKIIδ was detected in nuclear fractions and transiently activated with a peak at 2 min, corresponding temporally with nuclear export of phospho-Ser142 and total CREB.
FIGURE 6.

Thrombin-induced CREB translocation. VSM cells stimulated with thrombin (Thr; 50 nm) were fractionated to separate nuclear and cytosolic fractions. A, aliquots of fractions were resolved by SDS-PAGE and immunoblotted for CREB, CREB-Ser(P)142, CREB-Ser(P)133, CaMKIIδ, and CaMKIIδ-Thr(P)287. Histone H3 and -α were used as markers of the nuclear and cytosolic fractions, respectively, and to normalize signals for loading. B, quantification of total CREB signals. C, quantification of phospho-Ser142-CREB (CREB-pSer142) signals. D, quantification of phospho-Ser133-CREB (CREB-pSer133) signal in nuclear fraction. Signals in Cytosol were undetectable. Mean data are expressed as fold of unstimulated at 0 min. Values shown are mean ± S.E., n = 3, and were analyzed by one-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001, by Dunnett's post hoc test.
CaMKIIδ Negatively Regulates CREB Reporter Activity and CREB-regulated Genes
The strong correlation between nuclear export of CREB and Ser142 phosphorylation by CaMKIIδ suggests one potential mechanism for negative regulation of CREB activity by CaMKII in VSM. To evaluate the function of CaMKIIδ in regulating CREB activity in VSM, cells were transfected with a luciferase reporter plasmid containing 4 tandem copies of the CREB binding domain. Forskolin, which is a potent activator of the PKA pathway and strongly stimulates CREB activity (7), was used as a positive control. As expected, forskolin (5 μm) strongly increased CREB reporter activity nearly 6-fold, whereas thrombin significantly activated reporter activity by less than 2-fold (Fig. 7, A and B). Adenoviral overexpression of a kinase-negative CaMKIIδ2 (K43A) mutant, which exerts a dominant-negative effect on endogenous CaMKII activity in situ (19), significantly enhanced subsequent thrombin-induced CREB reporter activity by 3-fold over unstimulated levels (Fig. 7A). Conversely, adenoviral transduction and expression of a constitutively active CaMKIIδ2 (T287D) mutant significantly inhibited thrombin-induced CREB reporter activity to nearly basal levels (Fig. 7B).
FIGURE 7.

CaMKIIδ inhibits CREB reporter activity and CREB-regulated genes. A and B, CREB luciferase reporter stimulated by thrombin (50 nm) or forskolin (5 μm) for 6 h. Adenovirus encoding a dominant-negative CaMKIIδ2 mutant (DN-CaMKII) (A) or constitutively active CaMKIIδ2 mutant (CA-CaMKII) (B) was transduced for 48 and 24 h prior to cell lysis, respectively (see insets for immunoblots (IB) of CaMKIIδ expression). The plotted values are expressed as fold of unstimulated cells transduced with an adenovirus control (Ad-virus). C, mRNA expression of Sik1 (n = 5), and D, Rgs2 (n = 4) was determined by RT-PCR in cells treated as described above. Ct values were normalized to GAPDH. Changes in mRNA in each group are fold-values of the unstimulated control. Values shown are mean ± S.E. and analyzed by one-way ANOVA. *, p < 0.05; **, p < 0.01, by Bonferroni post hoc test.
To investigate whether CaMKIIδ-dependent negative regulation of CREB activity extends to endogenous transcriptional targets for CREB in VSM cells, we examined the effect of CaMKIIδ2 mutants on mRNA expression of Sik1 (salt-induced kinase 1; Fig. 7C) and Rgs2 (regulator of G-protein signaling 2; Fig. 7D). SIK1 is endogenously expressed in VSM and contributes to hypertension through regulation of the plasma membrane Na+,K+-ATPase (35). In addition, SIK1 has been identified as a transcriptional target for CREB in skeletal muscle cells and to interact with and regulate the type 2 HDAC-MEF2 pathway (36). Rgs2 contains a conserved CREB binding site in its promoter (37) and functions in an angiotensin II-dependent feedback loop to regulate blood pressure (38). Similar to the luciferase CREB reporter data above, forskolin treatment dramatically increased mRNA levels of Sik1 and Rgs2 by 30–40-fold, whereas thrombin treatment induced Sik1 and Rgs2 mRNA expression by less than 2-fold. Inhibition of CaMKIIδ activity by overexpressing kinase-negative CaMKIIδ2 significantly enhanced the induction of Sik1 and Rgs2 mRNA expression in response to thrombin. Adenoviral introduction of constitutively active CaMKIIδ2 abolished thrombin-induced Sik1 and Rgs2 mRNA induction. In contrast to previous reports (16–18), these data suggest that Ca2+ signaling mediated by CaMKIIδ inhibits CREB activity in VSM cells.
To directly test the effects CaMKIIδ activity on CREB binding to Sik1 and Rgs2 promoters, chromatin immunoprecipitation (ChIP) assays were performed using CREB antibody to isolate CREB/CREB-binding element complexes (Fig. 8). Control experiments in unstimulated cells confirmed CREB binding to the targeted binding elements in the Sik1 and Rgs2 promoters (Fig. 8A). Thrombin stimulation for 10 min increased CREB binding to the promoters in 3 separate experiments. Because the fold-inductions with thrombin were variable between the experiments (2–35-fold for Sik1; 3–75-fold for Rgs2), effects of overexpressing constitutively active and kinase-negative CaMKIIδ2 were normalized to the thrombin signal. Expression of constitutively active CaMKIIδ2 (Fig. 8B) was reproducibly effective at blocking thrombin-induced CREB binding on both the Sik1 (Fig. 8C) and Rgs2 (Fig. 8D) promoters, whereas overexpression of kinase-negative CaMKIIδ2 reproducibly enhanced CREB binding.
FIGURE 8.
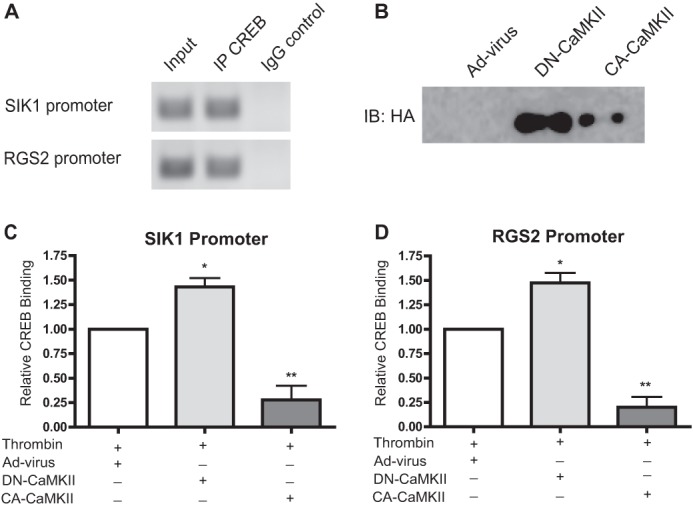
CaMKIIδ inhibits CREB binding to Sik1 and Rgs2 promoters. A, ChIP were performed using anti-CREB antibody on unstimulated VSM cells. CREB-binding elements in Sik1 and Rgs2 promoters were amplified by quantitative PCR and products resolved on an agarose gel as follows: from sheared chromatin prior to IP (input), chromatin immunoprecipitations (IPs) (IP CREB), and IP (IgG) control. B–D, effect of CaMKII mutant expression on thrombin-stimulated CREB binding to SIK1 (C) and RGS2 (D) promoters. VSM cells were transduced with adenovirus encoding dominant-negative (DN)-CaMKII (100 multiplicity of infection), constitutively active CA-CaMKII (50 multiplicity of infection), or adenovirus control (Ad-virus). Thrombin (50 nm) was added 10 min prior to fixation and processing for ChIP. DN-CaMKII and CA-CaMKII constructs were detected by immunoblotting with anti-HA antibodies (immunoblot (IB): HA) in supernatants following ChIP (B). Ct values were normalized to thrombin stimulation alone. Values shown are mean ± S.E., n = 3, and analyzed by one-way ANOVA. *, p < 0.05; **, p < 0.01, by Dunnett's post hoc test.
DISCUSSION
Increasing evidence indicates that Ca2+ signaling via CaMKIIδ positively regulates VSM cell proliferation and migration and contributes to vascular remodeling in response to injury or hypertensive disease (38–40). Here, we present evidence that Ca2+ signaling via CaMKIIδ mediates thrombin-induced CREB phosphorylation selectively on Ser142, correlating with CREB translocation from the nucleus. One outcome of thrombin-induced CaMKII-dependent signaling in VSM is negative regulation of CREB binding to CREB regulatory elements in specific target genes (in this case Sik1 and Rgs2) and negative regulation of CREB-induced transcriptional activity as indicated by a luciferase reporter assay and transcription of Sik1 and Rgs2. We believe the results of this study provide a definitive answer to the function of CaMKIIδ in regulating CREB activity in VSM cells and correct any potential misinterpretations of experiments that rely solely on pharmacological inhibitors of CaMKII or extrapolations of results from other systems such as neural tissues, which have suggested positive regulation of CREB activity by CaMKII (17, 18, 30).
Early studies demonstrated that purified brain CaMKII phosphorylated recombinant CREB at both Ser133 and Ser142 in several cultured cell lines (12, 41). Moreover, Wu and McMurray (11) reported that CaMKII-phosphorylated CREB at both Ser133 and Ser142 in human neuroblastoma and monkey kidney cells. Our experiments, using an adenovirally transduced constitutively active CaMKIIδ2 mutant and multiple molecular and pharmacological loss-of-function approaches demonstrate that in intact VSM cells stimulated by thrombin or a Ca2+ ionophore, CaMKII selectively phosphorylates endogenous CREB on Ser142. Lack of CREB Ser133 phosphorylation downstream of CaMKII in VSM could be due to isoform-specific effects with CaMKIIδ2 the predominant isoform expressed in VSM cells, compared with CaMKIIα and -β isoforms, which are predominant in neural tissue. Alternatively, unknown factors that are properties of intact VSM cells might restrict CaMKIIδ access to CREB-Ser133.
Most of the positive regulation of CREB via Ser133 phosphorylation in this study could be attributed to ERK1/2-dependent signaling as reflected by inhibition of responses by the MEK inhibitor U0126, confirming previous results (9). Although the mechanisms underlying the residual Ser133 phosphorylation are not known, we do not expect that Ser133 phosphorylation is mediated by CaMKIV in VSM cells, because we are unable to reliably detect either CaMKIV mRNA or protein expression.3
Phosphorylation of CREB on Ser142 has been shown to attenuate CREB activity, even if Ser133 is phosphorylated (11). Consistent with this concept, we observed that expression of constitutively active CaMKIIδ2 nearly abolished the thrombin-induced activation of a CREB reporter, whereas, thrombin-stimulated CREB reporter activity was further enhanced by inhibiting CaMKII activity using dominant-negative CaMKIIδ2. Wu and McMurray (11) presented evidence that CREB Ser142 phosphorylation in response to CaMKII activation prevented CREB dimerization and interaction with the CREB-binding protein. There are a few reports of CREB translocation from the nucleus that would also result in decreased activity (33, 34). Membrane depolarization in VSM cells induced elevated intracellular Ca2+ and mediated acute nuclear export of CREB (33) and prolonged stimulation with PDGF-BB-induced nuclear export of CREB and subsequently degradation through the ubiquitination pathway (34). Our studies of CREB localization in VSM cells following acute stimulation with thrombin demonstrate a strong correlation between CaMKII-dependent Ser142 phosphorylation and nuclear export, a mechanism that could also explain depolarization-induced nuclear export. Even with the relatively brief period of Ser142 phosphorylation, the extent of CREB nuclear export is substantial (greater than 50% of total CREB in the nucleus) suggesting a functionally important mode of CREB regulation. However, basal levels of CREB Ser142 phosphorylation appear in the nucleus in unstimulated cells, suggesting that mechanisms in addition to Ser142 phosphorylation may be required for nuclear export.
Although CaMKIIδ2 is the most abundant isoform in VSM and is localized predominantly in the cytosol, we did detect CaMKIIδ in VSM nuclei and activation of the nuclear kinase following thrombin stimulation. This may be explained by co-expression of small amounts of the alternative splice variant CaMKIIδ3 (or CaMKIIδB) that was first cloned from VSM cells (19). CaMKIIδ3 contains a nuclear targeting domain and when co-expressed with CaMKIIδ2 targets the kinase to the nucleus in proportion to composition of hetromultimeric holoenzymes (42). The small fraction of total and active CaMKIIδ in the nuclear fraction of VSM cells is consistent with the relative expression of the δ2 isoform compared with δ3 in VSM cells (19). How much negative CREB regulation is due directly to CaMKIIδ-mediated phosphorylation of Ser142 in the nucleus versus indirectly by CREB-Ser142 phosphorylation and trapping in the cytosol remains to be determined. We would predict that in other systems such as intact differentiated VSM (19) or heart (43–44) where the nuclear targeting isoform CaMKIIδ3 is expressed more abundantly, this mode of regulating CREB may be even more predominant. Alternatively, conditions of prolonged CaMKII activation occurring under conditions with high frequency Ca2+ transients (45) or oxidizing conditions (46–48), may be predicted to promote nuclear CREB export and potentially degradation (34).
Sik1 and Rgs2 have been reported to be regulated by CREB in multiple cell types, including VSM cells (35–37). In this study, ChIP assays confirmed thrombin-induced CREB binding to an element in the RGS2 promoter previously identified using mutagenesis approaches (36). We also confirmed CREB binding to a predicted element in the Sik1 promoter (37). Moreover, we extended knowledge of CREB target regulation by demonstrating CaMKIIδ negatively regulated thrombin-induced CREB binding to Sik1 and Rgs2 promoters, as well as transcription of the respective mRNAs. It has been reported that SIK1 regulates both the activity and expression of Na,K-ATPase in the renal proximal tubule, and in turn influences sodium and fluid reabsorption (49), which may provide a pathway affecting blood pressure regulation. Xie et al. (37) identified a CREB binding site in the RGS2 promoter sequence and demonstrated that angiotensin II-induced CREB activation up-regulated Rgs2 expression in VSM cells with effects on blood pressure homeostasis. CaMKII is also activated in response to angiotensin II stimulation in VSM (5) and is a likely component of angiotensin II-dependent regulation of CREB activity and RGS2 expression.
Our laboratory first reported that CaMKIIδ2 promoted VSM cell migration and proliferation in vitro (50) and exerted a positive effect on neointima formation in vivo (38). Recent studies using global CaMKIIδ gene knock-out mice have confirmed positive roles of CaMKIIδ2 in regulating the VSM cell proliferation and migration (39, 40). Aside from direct regulation of proteins involved in cell cycle control and cell motility, we have reported that CaMKIIδ2 phosphorylates HDAC4/5, resulting in nucleocytoplasmic shuttling of the HDACs and subsequent de-repression of MEF2 transcriptional activity in VSM cells (5). CREB is a well known downstream effector of the cAMP-PKA signaling pathway and has been shown to exert negative regulation of VSM cell migration and proliferation (34, 51). Negative regulation of CREB by CaMKIIδ as defined in the present study, and subsequent relief of its inhibitory effects could be another mechanism whereby CaMKII affects the phenotype of VSM and contributes to increased VSM cell proliferation and migration.
In summary, we have shown that activation of CaMKIIδ2 in response to Ca2+ mobilizing stimuli results in CREB-Ser142 phosphorylation and inhibition of CREB activity and transcriptional responses in vascular smooth muscle. In this system, CaMKII has no effect on the activating CREB-Ser133 phosphorylation event. For the first time we demonstrate a strong correlation between CREB-Ser142 phosphorylation and nuclear export of CREB suggesting one potential mechanism whereby CaMKII negatively regulates CREB activity. Further studies are required to determine the mechanism of CaMKII-dependent nuclear export of CREB and function of the CaMKIIδ2-CREB pathway in VSM cell phenotype control and vascular remodeling contributing to occlusive vascular diseases.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1-HL4942 and RO1-HL092510 from the National Heart, Lung and Blood Institute (to H. A. S.) and the Albany Medical College Candice Weir Fund.
Z. Wang and H. A. Singer, unpublished data.
- VSM
- vascular smooth muscle
- CREB
- cAMP response element-binding protein
- ANOVA
- analysis of variance
- HDAC
- histone deacetylase
- MEF
- myocyte enhancing factor.
REFERENCES
- 1. Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- 2. Gomez D., Owens G. K. (2012) Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 95, 156–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matchkov V. V., Kudryavtseva O., Aalkjaer C. (2012) Intracellular Ca2+ signalling and phenotype of vascular smooth muscle cells. Basic Clin. Pharmacol. Toxicol. 110, 42–48 [DOI] [PubMed] [Google Scholar]
- 4. Barlow C. A., Rose P., Pulver-Kaste R. A., Lounsbury K. M. (2006) Excitation-transcription coupling in smooth muscle. J. Physiol. 570, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ginnan R., Sun L. Y., Schwarz J. J., Singer H. A. (2012) MEF2 is regulated by CaMKIIδ2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem. J. 444, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wamhoff B. R., Bowles D. K., Owens G. K. (2006) Excitation-transcription coupling in arterial smooth muscle. Circ. Res. 98, 868–878 [DOI] [PubMed] [Google Scholar]
- 7. Shaywitz A. J., Greenberg M. E. (1999) CREB. A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861 [DOI] [PubMed] [Google Scholar]
- 8. Schauer I. E., Knaub L. A., Lloyd M., Watson P. A., Gliwa C., Lewis K. E., Chait A., Klemm D. J., Gunter J. M., Bouchard R., McDonald T. O., O'Brien K. D., Reusch J. E. (2010) CREB down-regulation in vascular disease. A common response to cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 30, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tokunou T., Ichiki T., Takeda K., Funakoshi Y., Iino N., Takeshita A. (2001) cAMP response element-binding protein mediates thrombin-induced proliferation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 21, 1764–1769 [DOI] [PubMed] [Google Scholar]
- 10. Ono H., Ichiki T., Fukuyama K., Iino N., Masuda S., Egashira K., Takeshita A. (2004) cAMP-response element-binding protein mediates tumor necrosis factor-α-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 24, 1634–1639 [DOI] [PubMed] [Google Scholar]
- 11. Wu X., McMurray C. T. (2001) Calmodulin kinase II attenuation of gene transcription by preventing cAMP response element-binding protein (CREB) dimerization and binding of the CREB-binding protein. J. Biol. Chem. 276, 1735–1741 [DOI] [PubMed] [Google Scholar]
- 12. Sun P., Enslen H., Myung P. S., Maurer R. A. (1994) Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539 [DOI] [PubMed] [Google Scholar]
- 13. Miyamoto E. (2006) Molecular mechanism of neuronal plasticity. Induction and maintenance of long-term potentiation in the hippocampus. J. Pharmacol. Sci. 100, 433–442 [DOI] [PubMed] [Google Scholar]
- 14. Takeda H., Kitaoka Y., Hayashi Y., Kumai T., Munemasa Y., Fujino H., Kobayashi S., Ueno S. (2007) Calcium/calmodulin-dependent protein kinase II regulates the phosphorylation of CREB in NMDA-induced retinal neurotoxicity. Brain Res. 1184, 306–315 [DOI] [PubMed] [Google Scholar]
- 15. Wheeler D. G., Barrett C. F., Groth R. D., Safa P., Tsien R. W. (2008) CaMKII locally encodes l-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 183, 849–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bell K. F., Bent R. J., Meese-Tamuri S., Ali A., Forder J. P., Aarts M. M. (2013) Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. J. Neurochem. 126, 274–287 [DOI] [PubMed] [Google Scholar]
- 17. Li J., Zhao S. Z., Wang P. P., Yu S. P., Zheng Z., Xu X. (2012) Calcium mediates high glucose-induced HIF-1alpha and VEGF expression in cultured rat retinal Muller cells through CaMKII-CREB pathway. Acta Pharmacol. Sin. 33, 1030–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sánchez-Muñoz I., Sánchez-Franco F., Vallejo M., Fernández A., Palacios N., Fernández M., Cacicedo L. (2010) Activity-dependent somatostatin gene expression is regulated by cAMP-dependent protein kinase and Ca2+-calmodulin kinase pathways. J. Neurosci. Res. 88, 825–836 [DOI] [PubMed] [Google Scholar]
- 19. Schworer C. M., Rothblum L. I., Thekkumkara T. J., Singer H. A. (1993) Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J. Biol. Chem. 268, 14443–14449 [PubMed] [Google Scholar]
- 20. Van Riper D. A., Schworer C. M., Singer H. A. (2000) Ca2+-induced redistribution of Ca2+/calmodulin-dependent protein kinase II associated with an endoplasmic reticulum stress response in vascular smooth muscle. Mol. Cell. Biochem. 213, 83–92 [DOI] [PubMed] [Google Scholar]
- 21. Ginnan R., Pfleiderer P. J., Pumiglia K., Singer H. A. (2004) PKC-δ and CaMKII-δ2 mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am. J. Physiol. Cell Physiol. 286, C1281–1289 [DOI] [PubMed] [Google Scholar]
- 22. Pfleiderer P. J., Lu K. K., Crow M. T., Keller R. S., Singer H. A. (2004) Modulation of vascular smooth muscle cell migration by calcium/calmodulin-dependent protein kinase II-δ 2. Am. J. Physiol. Cell Physiol 286, C1238–1245 [DOI] [PubMed] [Google Scholar]
- 23. Geisterfer A. A., Peach M. J., Owens G. K. (1988) Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 62, 749–756 [DOI] [PubMed] [Google Scholar]
- 24. Halees A. S., Leyfer D., Weng Z. (2003) PromoSer. A large-scale mammalian transcription start site identification service. Nucleic Acids Res. 31, 3554–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Messeguer X., Escudero R., Farré D., Núñez O., Martínez J., Albà M.M. (2002) PROMO. Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 [DOI] [PubMed] [Google Scholar]
- 26. Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 27. Wang Z., Ginnan R., Abdullaev I. F., Trebak M., Vincent P. A., Singer H. A. (2010) Calcium/Calmodulin-dependent protein kinase II δ6 (CaMKIIδ6) and RhoA involvement in thrombin-induced endothelial barrier dysfunction. J. Biol. Chem. 285, 21303–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kornhauser J. M., Cowan C. W., Shaywitz A. J., Dolmetsch R. E., Griffith E. C., Hu L. S., Haddad C., Xia Z., Greenberg M. E. (2002) CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 34, 221–233 [DOI] [PubMed] [Google Scholar]
- 29. Schworer C. M., Colbran R. J., Keefer J. R., Soderling T. R. (1988) Ca2+/calmodulin-dependent protein kinase II. Identification of a regulatory autophosphorylation site adjacent to the inhibitory and calmodulin-binding domains. J. Biol. Chem. 263, 13486–13489 [PubMed] [Google Scholar]
- 30. Cartin L., Lounsbury K. M., Nelson M. T. (2000) Coupling of Ca2+ to CREB activation and gene expression in intact cerebral arteries from mouse. Roles of ryanodine receptors and voltage-dependent Ca2+ channels. Circ. Res. 86, 760–767 [DOI] [PubMed] [Google Scholar]
- 31. Edman C. F., Schulman H. (1994) Identification and characterization of δB-CaM kinase and delta C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim. Biophys. Acta 1221, 89–101 [DOI] [PubMed] [Google Scholar]
- 32. Anderson M. E., Braun A. P., Wu Y., Lu T., Wu Y., Schulman H., Sung R. J. (1998) KN-93, an inhibitor of multifunctional Ca2+/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J. Pharmacol. Exp. Ther. 287, 996–1006 [PubMed] [Google Scholar]
- 33. Stevenson A. S., Cartin L., Wellman T. L., Dick M. H., Nelson M. T., Lounsbury K. M. (2001) Membrane depolarization mediates phosphorylation and nuclear translocation of CREB in vascular smooth muscle cells. Exp. Cell Res. 263, 118–130 [DOI] [PubMed] [Google Scholar]
- 34. Garat C. V., Fankell D., Erickson P. F., Reusch J. E., Bauer N. N., McMurtry I. F., Klemm D. J. (2006) Platelet-derived growth factor BB induces nuclear export and proteasomal degradation of CREB via phosphatidylinositol 3-kinase/Akt signaling in pulmonary artery smooth muscle cells. Mol. Cell. Biol. 26, 4934–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Popov S., Silveira A., Wågsäter D., Takemori H., Oguro R., Matsumoto S., Sugimoto K., Kamide K., Hirose T., Satoh M., Metoki H., Kikuya M., Ohkubo T., Katsuya T., Rakugi H., Imai Y., Sanchez F., Leosdottir M., Syvänen A. C., Hamsten A., Melander O., Bertorello A. M. (2011) Salt-inducible kinase 1 influences Na+,K+-ATPase activity in vascular smooth muscle cells and associates with variations in blood pressure. J. Hypertens. 29, 2395–2403 [DOI] [PubMed] [Google Scholar]
- 36. Berdeaux R., Goebel N., Banaszynski L., Takemori H., Wandless T., Shelton G. D., Montminy M. (2007) SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat. Med. 13, 597–603 [DOI] [PubMed] [Google Scholar]
- 37. Xie Z., Liu D., Liu S., Calderon L., Zhao G., Turk J., Guo Z. (2011) Identification of a cAMP-response element in the regulator of G-protein signaling-2 (RGS2) promoter as a key cis-regulatory element for RGS2 transcriptional regulation by angiotensin II in cultured vascular smooth muscles. J. Biol. Chem. 286, 44646–44658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. House S. J., Singer H. A. (2008) CaMKII-delta isoform regulation of neointima formation after vascular injury. Arterioscler. Thromb. Vasc. Biol. 28, 441–447 [DOI] [PubMed] [Google Scholar]
- 39. Li W., Li H., Sanders P. N., Mohler P. J., Backs J., Olson E. N., Anderson M. E., Grumbach I. M. (2011) The multifunctional Ca2+/calmodulin-dependent kinase II delta (CaMKIIδ) controls neointima formation after carotid ligation and vascular smooth muscle cell proliferation through cell cycle regulation by p21. J. Biol. Chem. 286, 7990–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott J. A., Xie L., Li H., Li W., He J. B., Sanders P. N., Carter A. B., Backs J., Anderson M. E., Grumbach I. M. (2012) The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am. J. Physiol. Heart Circ. Physiol. 302, H1953–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Enslen H., Sun P., Brickey D., Soderling S. H., Klamo E., Soderling T. R. (1994) Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J. Biol. Chem. 269, 15520–15527 [PubMed] [Google Scholar]
- 42. Srinivasan M., Edman C. F., Schulman H. (1994) Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell Biol. 126, 839–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mayer P., Möhlig M., Idlibe D., Pfeiffer A. (1995) Novel and uncommon isoforms of the calcium sensing enzyme calcium/calmodulin dependent protein kinase II in heart tissue. Basic Res. Cardiol. 90, 372–379 [DOI] [PubMed] [Google Scholar]
- 44. Zhang T., Brown J. H. (2004) Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 63, 476–486 [DOI] [PubMed] [Google Scholar]
- 45. Li L., Stefan M. I., Le Novère N. (2012) Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PloS One 7, e43810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Erickson J. R., He B. J., Grumbach I. M., Anderson M. E. (2011) CaMKII in the cardiovascular system. Sensing redox states. Physiol. Rev. 91, 889–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Erickson J. R., Joiner M. L., Guan X., Kutschke W., Yang J., Oddis C. V., Bartlett R. K., Lowe J. S., O'Donnell S. E., Aykin-Burns N., Zimmerman M. C., Zimmerman K., Ham A. J., Weiss R. M., Spitz D. R., Shea M. A., Colbran R. J., Mohler P. J., Anderson M. E. (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shetty P. K., Huang F. L., Huang K. P. (2008) Ischemia-elicited oxidative modulation of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 283, 5389–5401 [DOI] [PubMed] [Google Scholar]
- 49. Taub M., Springate J. E., Cutuli F. (2010) Targeting of renal proximal tubule Na,K-ATPase by salt-inducible kinase. Biochem. Biophys. Res. Commun. 393, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. House S. J., Ginnan R. G., Armstrong S. E., Singer H. A. (2007) Calcium/calmodulin-dependent protein kinase II-δ isoform regulation of vascular smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol 292, C2276–2287 [DOI] [PubMed] [Google Scholar]
- 51. Klemm D. J., Watson P. A., Frid M. G., Dempsey E. C., Schaack J., Colton L. A., Nesterova A., Stenmark K. R., Reusch J. E. (2001) cAMP response element-binding protein content is a molecular determinant of smooth muscle cell proliferation and migration. J. Biol. Chem. 276, 46132–46141 [DOI] [PubMed] [Google Scholar]